Introduction
Myelodysplastic syndromes (MDS) comprise a
heterogeneous group of hematological disorders that are
characterized by impaired hematopoiesis, resulting in cytopenias of
different grades (1). Examination of
the bone marrow (aspirate, biopsy, cytogenetics and flow cytometry)
and the peripheral blood should reveal the morphological features
of the disease and exclude from other conditions (2).
The World Health Organization (WHO) classifies MDS
as refractory cytopenias with unilineage dysplasia, refractory
anemia with ring sideroblasts, refractory anemia with excess
blasts-1, refractory anemia with excess blasts-2, myelodysplastic
syndrome-unclassified and myelodysplastic syndrome associated with
isolated del 5q (3). In addition, the
WHO classification recognizes a group of myeloid neoplasms with
overlapping features of myelodysplastic syndromes (MDS) and
myeloproliferative neoplasms (MPN) and places them under a separate
category of MDS/MPN. Within this category, refractory anemia with
ring sideroblasts with marked thrombocytosis (RARS) and MDS/MPN,
unclassifiable (MDS/MPN-U) are also included (4).
The incidence of MDS is ~3.8 cases/100,000 habitants
each year. It is rare in people <40 years old (0.14/100,000
habitants) and it increases according to aging (5). With regards to Brazil, the
epidemiological data are restricted. In a retrospective study of
315 MDS patients, an incidence of 7% was reported. They were
classified as RARS, RA, RAEB and MDS/MPD-U (6).
Accurate prediction of a patient prognosis is useful
to define the risk posed by the disease and the treatment options
that are most appropriate. Since its publication in 1997, the
International Prognostic Scoring System (IPSS) has been the tool
used to stratify risk in these patients (7). In 2012, the Revised International
Prognostic Scoring System (IPSS-R) included the same major
categories as the IPSS, but with significant improvements (7–9).
The treatment varies according to the level of risk.
For low-risk patients the treatment consists of cytopenia
correction. For anemia, the treatment consists of transfusion and
erythropoietin (10). Lenalidomide
was useful for patients with 5q-syndrome (11–13).
Granulocyte-colony stimulating factor improves the neutropenia
(14,15). The hypomethylating agents, decitabine
and azacitidine, can be used in low-risk patients who are
transfusion dependent, and who have failed or are not candidates
for the use of lenalidomide or growth factors (14,15). In
patients with high- and intermediate risk 2, the treatment focuses
on changing the natural history of the disease, due to the high
risk of progression to acute myeloid leukemia (AML) and reduced
survival. Therapeutic options include hypomethylating, targeted
chemotherapy regimens for AML and allogeneic stem cell
transplantation, which is the only curative treatment option.
Allogeneic transplantation is reserved for patients <65 and
human leukocyte antigen (HLA)-matched donor (1,5,13).
Thrombocytosis may be observed in certain MDS cases,
mainly in cases of refractory anemia with ringed sideroblasts.
However, in cases with the absence of ring sideroblasts, the MDS
diagnosis is challenging and it may confound with
myeloproliferative disorders (MPD), mainly if there is a Janus
kinase 2 gene (JAK2) V617F mutation (16). This abnormality is much more common in
this group of disorders compared to MDS. The present study
describes an uncommon case of MDS that was initially classified as
essential thrombocytosis (ET).
Case report
A 78-year-old woman presented with anemia and
thrombocytosis for the last 6 months. The patient did not
experience paresthesias, weight loss, pruritus, fever, headaches or
arthralgia, and had not received transfusions. The initial
laboratory tests revealed macrocytic anemia (mean corpuscular
volume=102 fl), with a hemoglobin concentration of 10.8 g/dl, a
leukocyte count of 6.5×109/l without detected
circulating blasts, and a platelet count of 812×109/l.
The renal and hepatic functions were within normal limits and
lactate dehydrogenase (LDH) was found to be elevated to 419 IU/l
(normal range is ≤250 IU/l). The ferritin and vitamin B12 levels
were 167 ng/ml and 686 pg/ml, respectively (within the normal
range). Ultrasonography revealed mild splenomegaly (12.8 cm; normal
size is ≤12 cm) and portal vein thrombosis. Bone marrow aspiration
revealed megakaryocytic hyperplasia and dysmegakaryopoiesis,
without alterations of the erythroid or granulocytic lineages. A
bone marrow biopsy revealed 70% hematopoietic cellularity with
trilineage hematopoiesis, a normal ratio of myeloid to erythroid
cells, megakaryocytic hyperplasia, dysmegakaryopoiesis, and no
deposition of reticulin fibers (Fig.
1A). A cytogenetic investigation was not performed at this
stage however, molecular analysis performed on a peripheral blood
sample detected a JAK2 mutation (V617F). The diagnosis was
ET, according to the criteria proposed by the Polycythemia Vera
Group (17). Therapy with hydroxyurea
and aspirin were initiated. Two years later, the patient presented
with weakness, fatigue, epistaxis and progressive dyspnea. Physical
examination revealed tachycardia, tachypnea, fever and
splenomegaly. The laboratory tests revealed normocytic anemia with
a hemoglobin concentration of 6.0 g/dl, as well as thrombocytopenia
(61×109/l) and leukocytosis (12×109/l) with
circulating blasts. The therapy with hydroxyurea was interrupted
and the patient received therapeutic blood transfusions. The renal
and hepatic functions were normal, but LDH was elevated to 2,080
IU/l. The examination of bone marrow morphology revealed
myelodysplasia: As shown in Fig. 1B,
a bone marrow biopsy revealed that the marrow was markedly
hypercellular (85%) with atypical localization of immature
precursors (ALIP), megakaryocytic hyperplasia with
dysmegakaryopoiesis and grade 1 fibrosis (Fig. 1C). Immunophenotypic study identified
dysplasia of the granulocytic and monocytic lineages, an increase
in blasts positive for CD34, CD33 and CD117, and monocytosis, with
aberrant expression of CD56, anti-HLA DR (dim) and partial
expression of CD15. Perls' staining was performed and was negative
for ring sideroblasts (results not shown). Banding cytogenetics
analysis of the bone marrow revealed an aberrant karyotype
(Fig. 1D), which was further
characterized by molecular cytogenetics (Fig. 1E) using whole-chromosome painting
probes for chromosomes 3, 5 and 7 as follows: 45, XX, der(3) t(3;7) (p12;p13), del(5)(q12q33), −7, del(12)(p12)[18]/46, XX[2]. Tazocin treatment
was initiated, but was interrupted on day 9, due to the patient's
progressively altered mental status, as well as development of a
pulmonary infection. Soon after, the patient succumbed to septic
shock.
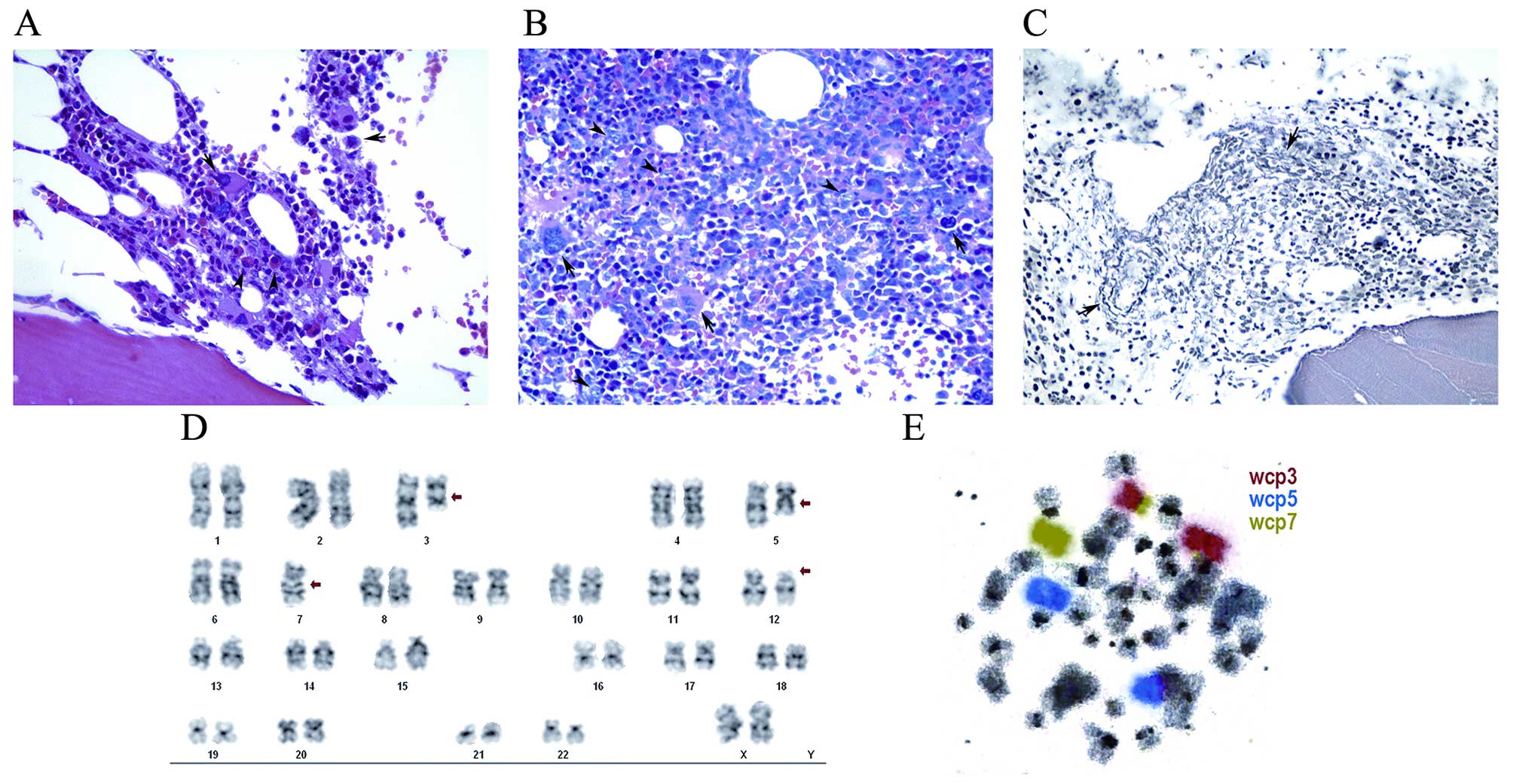 | Figure 1.Pathology and cytogenetics findings.
(A) Bone marrow biopsy at diagnosis [hematoxylin and eosin
(H&E) staining; magnification, ×400] showing fragmented bone
marrow represented by three intertrabecular spaces, displaying
~60–70% overall cellularity. Three hematopoietic lineages were
present. The granulocyte:erythroid ratio was 5:1. The granulocytic
series displayed mature form however, there were common elements
with left maturation. The normoblastic erythroid series were found
in a small proportion (arrowheads). The megakaryocytes were
increased in number, with moderate dysmorphism (small and
hypolobulated; arrows). (B) Bone marrow biopsy in evolution
(H&E staining; magnification, ×400). Bone marrow exhibiting
~80% overall cellularity. Three hematopoietic series were present.
The granulocyte: Erythroid ratio was 5:1. The granulocytic series
at various maturation stages exhibited blasts (arrowheads) in the
center of the medullary cavity, corresponding to ~15%, with
atypical localization of immature precursors. Megakaryocytes were
present at an increased proportion, exhibiting dysmorphism, often
in the form of small and hypolobulated cells and occasional
microcytic forms, usually located in the center of the medullary
cavity, predominantly isolated (arrows). Ferric pigment deposits
were also present. (C) Bone marrow biopsy in evolution (reticulin
staining; magnification, ×400). A network of reticulin with
numerous intersections was observed, particularly in the
perivascular areas (arrows), defined as grade 1 myelofibrosis. (D)
Trypsin-Giemsa banding. (E) Chromosome painting. |
Discussion
After the patient's death, the case was reclassified
as MDS based on a comprehensive histopathological,
immunophenotypic, molecular and cytogenetics review. At diagnosis,
the patient presented with mild anemia and increased platelet
count, and the results of the ultrasound examination were
compatible with thrombosis. These characteristics may be common
between ET and MDS, which may be misleading (18–20). The
spleen size was within normal limits on ultrasonography. The World
Health Organization characterized certain disorders as
myelodysplastic/myeloproliferative neoplasms (MDS/MPN) in 2008
(5,19). This group includes clonal myeloid
diseases that exhibit an overlap between MDS and MPN. At the time
of diagnosis, these patients exhibit clinical, laboratory, or
morphological characteristics supporting the diagnosis of these two
groups. Chronic myelomonocytic leukemia, juvenile myelomonocytic
leukemia, atypical BCR-ABL1-negative chronic myeloid leukemia,
MDS/MPN unclassifiable, refractory anemia with ring sideroblasts
and thrombocytosis (RARS-T) are included (19). The patient in the present case was
carrier of a JAK2 (V617F) mutation, which is more common in
ET compared with MDS patients (3,16,19–22). The
JAK2 (V617F) mutation is described in >95% of
polycythemia vera and 50–60% of ET and myelofibrosis patients.
However, in MDS, the JAK2 (V617F) mutation is found mainly
in patients with RARS-T (refractory anemia with ring sideroblasts),
which was excluded by Perls' staining (23–25). The
histopathological and immunophenotypic studies as well as the
presence of micromegakaryocytes and ALIP favored the diagnosis of
MDS. In ET the presence of micromegakaryocytes are uncommon
(Fig. 1C). Immunophenotypic studies
also support this hypothesis, as described in the present study.
Cytogenetics was also valuable for the diagnosis in this case.
Chromosome banding combined with molecular cytogenetics was useful
and indicative of the presence of a myeloid disorder. The presence
of chromosomal abnormalities, such as abnormalities of 3q or
monosomy 7, is a known indicator of an adverse clinical outcome
(9). The classical risk factor
associated with MDS is ageing. However, to learn more about this
case, the son of the patient consented to being interviewed after
her death: He reported that the deceased patient and her family had
no previous history of malignant disease. The patient was a
non-smoker and did not regularly consume alcoholic beverage. She
was a housewife, but had lived in close proximity to a leather
factory for 35 years, which may have contributed to the onset of
her disease, since carcinogenic chemical substances are associated
with this type of industry (24,25).
In conclusion, due to its heterogeneity, MDS may be
misdiagnosed as ET and inappropriate treatment may be initiated.
Thus, it is crucial to carefully combine all available data on
morphology and immunophenotype, and to conduct the necessary
molecular, cytogenetic and molecular cytogenetic analyses, in order
to reach the correct diagnosis and administer the optimal
treatment.
The present study was approved by the Ethical Board
of the Pedro Ernesto University Hospital (HUPE); registration
number, 0010.0.228.000-07. The patient's son signed an informed
consent regarding the publication of the case details.
References
|
1
|
Garcia-Manero G: Myelodysplastic
syndromes: 2015 Update on diagnosis, risk-stratification and
management. Am J Hematol. 90:831–841. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nimer SD: Myelodysplastic syndromes.
Blood. 111:4841–4851. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brunning RD, Orazi A, Germing U, Le Beau
Porwit, Baumann I, Vardiman JW and Hellstrom-Lindberg E:
Myelodysplastic syndromes/neoplasms, overview. WHO Classification
of Tumours of Haematopoietic and Lymphoid Tissues. Swerdlow SH,
Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J and
Vardiman JW: (4th). IARC Press. (Lyon). 87–93. 2008.
|
|
4
|
Foucar K:
Myelodysplastic/myeloproliferative neoplasms. Am J Clin Pathol.
132:281–289. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sekeres MA: The epidemiology of
myelodysplastic syndromes. Hematol Oncol Clin North Am. 24:287–294.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cabello AI, Collado R, Ruiz MA, Martínez
J, Navarro I, Ferrer R, Sosa AM and Carbonell F: A retrospective
analysis of myelodysplastic syndromes with thrombocytosis:
Reclassification of the cases by WHO proposals. Leuk Res.
29:365–370. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Greenberg P, Cox C, LeBeau MM, Fenaux P,
Morel P, Sanz G, Sanz M, Vallespi T, Hamblin T, Oscier D, et al:
International prognostic scoring system for evaluating prognosis in
myelodysplastic syndromes. Blood. 89:2079–2088. 1997.PubMed/NCBI
|
|
8
|
Greenberg PL, Tuechler H, Schanz J, Sanz
G, Garcia-Manero G, Solé F, Bennett JM, Bowen D, Fenaux P, Dreyfus
F, et al: Revised international prognosis scoring system for
myelodysplastic syndromes. Blood. 120:2454–2465. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Haase D, Germing U, Schanz J, Pfeilstöcker
M, Nösslinger T, Hildebrandt B, Kundgen A, Lübbert M, Kunzmann R,
Giagounidis AA, et al: New insights into the prognostic impact of
the karyotype in MDS and correlation with subtypes: Evidence from a
core dataset of 2124 patients. Blood. 110:4385–4395. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Moyo V, Lefebvre P, Duh MS, Yektashenas B
and Mundle S: Erythropoiesis-stimulating agents in the treatment of
anemia in myelodysplastic syndromes: A meta-analysis. Ann Hematol.
87:527–536. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
List A, Dewald G, Bennett J, Giagounidis
A, Raza A, Feldman E, Powell B, Greenberg P, Thomas D, Stone R, et
al: Lenalidomide in the myelodysplastic syndrome with chromosome 5q
deletion. N Engl J Med. 355:1456–1465. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fenaux P, Giagounidis A, Selleslag D,
Beyne-Rauzy O, Mufti G, Mittelman M, Muus P, Te Boekhorst P, Sanz
G, Del Cañizo C, et al: A randomized phase 3 study of lenalidomide
versus placebo in RBC transfusion-dependent patients with
low-/intermediate-1 risk myelodysplastic syndromes with del5q.
Blood. 118:3765–3776. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fenaux P and Adès L: How we treat
lower-risk myelodysplastc syndromes. Blood. 121:4280–4286. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fenaux P, Haase D, Sanz GF, Santini V and
Buske ESMO: Guidelines Working Group: Myelodysplastic syndromes:
ESMO clinical practice guidelines for diagnosis, treatment and
follow-up. Ann Oncol. 25(Suppl 3): iii57–iii69. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Garcia-Manero G: Myelodysplastic
syndromes: 2014 update on diagnosis, risk-stratification, and
management. Am J Hematol. 89:97–108. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jeromin S, Haferlach T, Weissmann S,
Meggendorfer M, Eder C, Nadarajah N, Alpermann T, Kohlmann A, Kern
W, Haferlach C and Schnittger S: Refractory anemia with ring
sideroblasts and marked thrombocytosis cases harbor mutations in
SF3B1 or other spliceosome genes accompanied by JAK2V617F and ASXL1
mutations. Haematologica. 100:e125–e127. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Murphy S, Peterson P, Iland H and Laszlo
J: Experience of the Polycythemia Vera study group with essential
thrombocythemia: A final report on diagnostic criteria, survival,
and leukemic transition by treatment. Semin Hematol. 34:29–39.
1997.PubMed/NCBI
|
|
18
|
Steensma DP: JAK2 V617F in myeloid
disorders: Molecular diagnostic techniques and their clinical
utility: A paper from the 2005 William Beaumont Hospital Symposium
on Molecular Pathology. J Mol Diagn. 8:397–411. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vardiman JBR, Bennett JM, Bain BJ, Baumann
I, Thiele J and Orazi A: Myelodysplastic/myeloproliferative
neoplasm, unclassifiable. WHO Classification of Tumours of
Haematopoietic and Lymphoid Tissues. Swerdlow SH, Campo E, Harris
NL, Jaffe ES, Pileri SA, Stein H, Thiele J and Vardiman JW: IARC
Press. (Lyon). 85–86. 2008.
|
|
20
|
Orazi A and Germing U: The
myelodysplastic/myeloproliferative neoplasms: Myeloproliferative
diseases with dysplastic features. Leukemia. 22:1308–1319. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Szpurka H, Tiu R, Murugesan G, Aboudola S,
Hsi ED, Theil KS, Sekeres MA and Maciejewski JP: Refractory anemia
with ringed sideroblasts associated with marked thrombocytosis
(RARS-T), another myeloproliferative condition characterized by
JAK2 V617F mutation. Blood. 108:2173–2181. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Malcovati L and Cazzola M: Refractory
anemia with ring sideroblasts. Best Pract Res Clin Haematol.
26:377–385. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Patnaik MM and Tefferi A: Refractory
anemia with ring sideroblasts and RARS with thrombocytosis. Am J
Hematol. 90:549–559. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mikoczy Z, Schütz A and Hagmar L: Cancer
incidence and mortality among Swedish leather tanners. Occup
Environ Med. 51:530–535. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mikoczy Z, Schütz A, Strömberg U and
Hagmar L: Cancer incidence and specific occupational exposures in
the Swedish leather tanning industry: A cohort based case-control
study. Occup Environ Med. 53:463–467. 1996. View Article : Google Scholar : PubMed/NCBI
|














