Introduction
dTCApFs (Nerofe™, Immune System Key Ltd., Jerusalem,
Israel) is a novel hormone peptide (14 amino acids) with a
demonstrated anticancer activity in the preclinical setting
(personal communication with Dr Devary's laboratory, ISK Ltd.).
dTCApFs is a derivative of the tumor cell apoptosis factor (TCApF),
a 84-amino acid hormone peptide naturally expressed in the thymus,
colon and frontal lobe of the brain (1).
Studies in pancreatic, breast and ovarian cell lines
investigating the mechanism of action (MOA) through which dTCApFs
exerts its anticancer effects, revealed that dTCApFs enters the
cells through the T1/ST2 receptor (a member of the
Toll/interleukin-1 receptor superfamily), and leads to apoptosis of
cancer cells through a novel MOA involving induction of two
opposing effects: Induction of structural changes in the Golgi
apparatus, loss of Golgi function and induction of endoplasmic
reticulum (ER) stress, along with downregulation of the ER stress
repair mechanism (personal communication).
We herein report the results of the first-in-human
study investigating the safety and efficacy of dTCApFs for the
treatment of advanced/metastatic solid tumors.
Patients and methods
Patients
The present study included adult patients (aged ≥18
years) with pathologically confirmed locally advanced and/or
metastatic solid malignancies, who experienced treatment failure or
were unable to tolerate previous standard therapy. The key
inclusion criteria included evaluable/measurable disease and
Eastern Cooperative Oncology Group performance status ≤1. Patients
with liver cancer/hepatic metastases were considered eligible if
liver function met certain criteria, and patients with brain
metastases were considered eligible if radiation therapy was
completed ≥4 weeks prior to enrollment and the patient received ≤4
mg/day of dexamethasone. The key exclusion criteria included
receiving anticancer treatment 14 days prior to the initiation of
the study drug, and a life expectancy of <16 weeks.
Study design
This was a formal open-label phase I dose-escalation
study. The primary objective was to determine the maximum tolerated
dose (MTD) and safety profile of dTCApFs. Assessments included drug
exposure, adverse events (AEs) graded according to the Common
Terminology Criteria for Adverse Events, version 4.0 (https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5×7.pdf),
and characterization of dose-limiting toxicities (DLTs). Other
objectives included assessment of serum levels of angiogenic
factors following dTCApFs administration, pharmacokinetics (PK) and
pharmacodynamics (PD) analyses, as well as assessment of receptor
staining and tumor response.
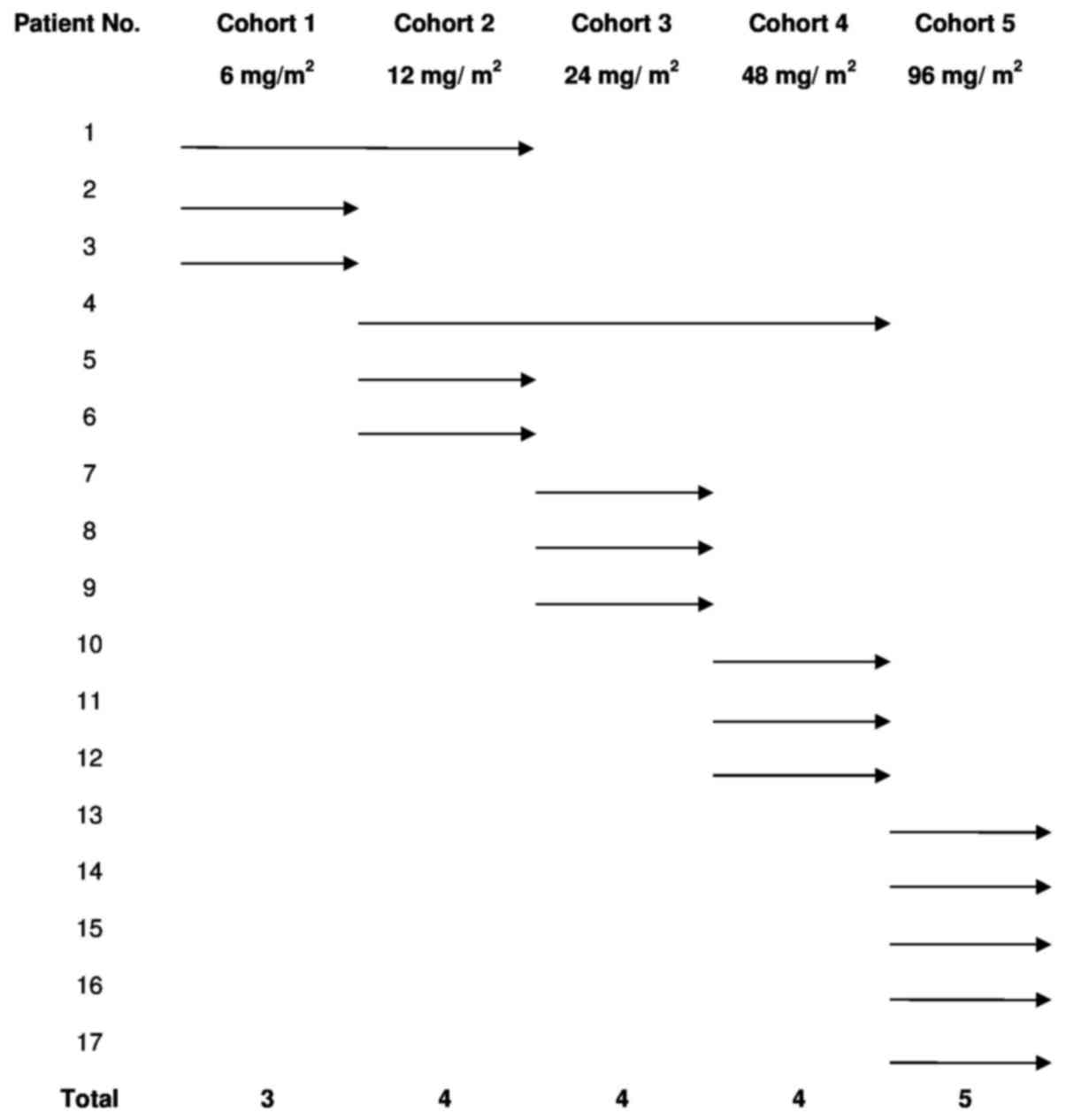
The dose escalation study followed a traditional
‘3+3’ scheme and included doses of 6, 12, 24, 48 and 96
mg/m2 of intravenous (IV) dTCApFs, 3 times/week in
consecutive 28-day cycles. The patient's allocation is presented in
Fig. 1. In all 3-patient cohorts,
there was an interval of 2 weeks between the first dose for the
first and second patients, and ≥1 week for the third patient. New
dose levels started after a follow-up of ≥28 days for the 3
patients at the previous level. MTD was defined as the highest dose
level at which ≥1 of the 3 subjects experienced a DLT during their
first cycle of treatment. Patients who did not complete their first
cycle of treatment for reasons unrelated to AEs were replaced. In
addition, PK parameters, including area under the curve (AUC),
maximal plasma concentration (Cmax) and plasma half-life (t1/2)
were determined. PK parameters were estimated using
non-compartmental models.
The clinical activity of dTCApFs was assessed every
8 weeks by physical examination, computed tomography (CT), or
magnetic resonance imaging techniques (for evaluable disease only),
using the Response Evaluation Criteria In Solid Tumors v1.1
(https://ctep.cancer.gov/protocoldevelopment/docs/recist_guideline.pdf);
where appropriate, informative tumor markers were measured in every
cycle.
This study was approved by the Institutional Review
Board of Rabin Medical Center and the Ministry of Health of Israel,
and was conducted at the Davidoff Center, Rabin Medical Center in
accordance with the principles of the Declaration of Helsinki. All
the patients signed an informed consent prior to enrollment. The
study was registered at ClinicalTrials.gov (NCT01690741).
Biomarker analysis
Blood samples were collected from patients and
placed on ice for 10 min. Serum samples were collected by
centrifugation at 1,000 × g for 10 min at 4°C, kept in separate
vials at ≤-20°C, and shipped to Immune System Key Ltd. at −20°C,
where they were thawed, aliquoted, and stored at ≤-20°C. Repeated
freeze-thaw cycles were avoided.
Immunohistochemistry (IHC) staining was performed
for T1/ST2 receptor using a full-length anti-ST2 antibody (GenMed,
Plymouth, MN, USA). Serum levels of various factors were measured
with enzyme-linked immunosorbent assay (ELISA). The measured
factors included vascular endothelial growth factor (VEGF), VEGF-D,
epidermal growth factor, angiopoietin-1, fibroblast growth factor
(FGF)-1, FGF-2, platelet-derived growth factor (PDGF)-AA, PDGF-BB,
transforming growth factor (TGF)-β (all using ELISA kits by R&D
systems, Abingdon, UK); granulocyte-macrophage colony-stimulating
factor (GM-CSF), interleukin (IL)-2, IL-12p70, IL-21 and tumor
necrosis factor (TNF)-α (Millipore, Billerica, MA, USA); and
glucose-regulated protein 78 (GRP78)/BiP (Enzo, New York, NY,
USA).
Statistical analysis
Descriptive statistics were used for all analyses
and were performed with SAS® software, version 9.1 (SAS
Institute Inc., Cary, NC, USA). Regression analysis was used to
study 2-way correlation between tumor change per month,
administered doses of dTCApF, and levels of the ER-stress biomarker
(BiP). The statistical significance of the correlation was
validated using F-statistics.
Results
Patients
A total of 39 patients were screened, of whom were
17 enrolled and completed the study. The majority of the patients
(64%) were male, and the median age (range) was 65 (51–94) years.
Almost half of the patients (47%) had colorectal cancer and 29% had
pancreatic cancer. Apart from 1 patient, all other patients had
received several lines of anticancer therapy (e.g., chemotherapy,
radiotherapy and biological therapy) prior to enrolment (Table I). The patients received 1–3 cycles
of escalating dTCApFs doses (6, 12, 24, 48 and 96
mg/m2), as detailed in Fig.
1.
 | Table I.Patient demographics and baseline
characteristics. |
Table I.
Patient demographics and baseline
characteristics.
|
| dTCApFs dose,
mg/m2 |
|---|
|
|
|
|---|
| Characteristics | 6 (n=3) | 12 (n=3) | 24 (n=3) | 48 (n=3) | 96 (n=5) |
|---|
| Age, years |
|
|
|
|
|
| Median
(range) | 63 (62–77) | 61 (58–62) | 65 (57–67) | 72 (51–94) | 64 (55–77) |
| Mean
(SE) | 68 (5) | 67 (4) | 67 (2) | 72 (8) | 64 (9) |
| Sex, n
(male/female) | 3/0 | 2/1 | 1/2 | 2/1 | 3/2 |
| Tumor type, n |
|
|
|
|
|
|
Colorectal | 3 | 2 | 0 | 2 | 1 |
|
Pancreatic | 0 | 0 | 1 | 0 | 4 |
|
Othera | 0 | 1 | 2 | 1 | 0 |
| Prior therapies,
n |
|
|
|
|
|
|
Chemotherapy | 3 | 4 | 4 | 1 | 3 |
|
Radiotherapy | 1 | 2 | 1 | 1 | 0 |
|
Surgery | 2 | 2 | 1 | 1 | 2 |
| Treatment
with biological agents | 0 | 0 | 1 | 0 | 0 |
| Treatment
with small molecules, such as TKIs. | 0 | 0 | 0 | 1 | 0 |
Safety and tolerability
The mean number of treatment cycles per patient was
3.2±1.4. No DLTs were observed in any patient up to cohort 5. The
AEs are summarized in Table II.
None were related to the study drug. Hypertension, anemia,
vomiting, diarrhea and abdominal pain were the most frequently
reported grade 2 AEs, and hypertension was the most frequently
reported grade 3 AE. Vomiting was the only grade 4 AE, reported in
1 patient. The majority of the AEs were self-resolving. Overall,
treatment with dTCApFs was well-tolerated, with no cumulative
toxicity. MTD was not reached.
| Table II.Summary of adverse events by dTCApFs
dose group. |
Table II.
Summary of adverse events by dTCApFs
dose group.
|
| dTCApFs dose,
mg/m2 |
|---|
|
|
|
|---|
| Adverse events | 6 (n=3) | 12 (n=3) | 24 (n=3) | 48 (n=3) | 96 (n=5) |
|---|
| Grade 1 |
|
|
|
|
|
| Blood
disorders |
|
|
|
|
|
|
Anemia | 3 | 0 | 0 | 0 | 0 |
|
Increased INR | 0 | 0 | 0 | 1 | 0 |
| GI
disorders |
|
|
|
|
|
|
Abdominal
pain | 0 | 1 | 2 | 0 | 0 |
|
Bowel
obstruction | 1 | 0 | 0 | 0 | 0 |
|
Diarrhea | 0 | 2 | 0 | 0 | 2 |
|
GI hemorrhage | 1 | 0 | 0 | 0 | 0 |
|
Vomiting | 2 | 0 | 0 | 0 | 1 |
| General
disorders |
|
|
|
|
|
|
Dehydration | 0 | 0 | 0 | 0 | 1 |
|
Fatigue | 0 | 1 | 0 | 1 | 0 |
|
Hypertension | 3 | 1 | 1 | 0 | 1 |
| Nervous
system disorders |
|
|
|
|
|
|
Neuropathy | 0 | 1 | 1 | 0 | 0 |
| Grade 2 |
|
|
|
|
|
|
Pain |
|
|
|
|
|
|
Pain, leg | 0 | 2 | 0 | 0 | 0 |
|
Pain, upper
back | 0 | 0 | 0 | 0 | 1 |
|
Respiratory system
disorders |
|
|
|
|
|
|
Cough | 0 | 1 | 0 | 0 | 0 |
| Skin
disorders |
|
|
|
|
|
|
Pruritus | 0 | 0 | 0 | 0 | 1 |
|
Urticaria | 0 | 0 | 0 | 0 | 1 |
| Hepatic
and urinary disorders |
|
|
|
|
|
|
ALT increase | 0 | 0 | 0 | 0 | 1 |
|
AST increase | 0 | 0 | 0 | 0 | 1 |
|
Bilirubin
increase | 0 | 0 | 0 | 1 | 1 |
|
Liver
dysfunction | 0 | 0 | 0 | 0 | 1 |
|
Urinary tract
infection | 0 | 1 | 0 | 0 | 0 |
| Grade 3 |
|
|
|
|
|
| Blood
disorders |
|
|
|
|
|
|
Increased INR | 0 | 0 | 0 | 1 | 0 |
| General
disorders |
|
|
|
|
|
|
Hypertension | 2 | 1 | 1 | 0 | 2 |
| Hepatic
and urinary disorders |
|
|
|
|
|
|
Bilirubin
increase | 0 | 0 | 0 | 1 | 1 |
| GI
disorders |
|
|
|
|
|
|
Bowel
obstruction | 1 | 0 | 0 | 0 | 0 |
|
Diarrhea | 0 | 1 | 0 | 0 | 1 |
|
GI hemorrhage | 1 | 0 | 0 | 0 | 0 |
| Grade 4 |
|
|
|
|
|
| GI
disorders |
|
|
|
|
|
|
Vomiting | 0 | 0 | 0 | 0 | 1 |
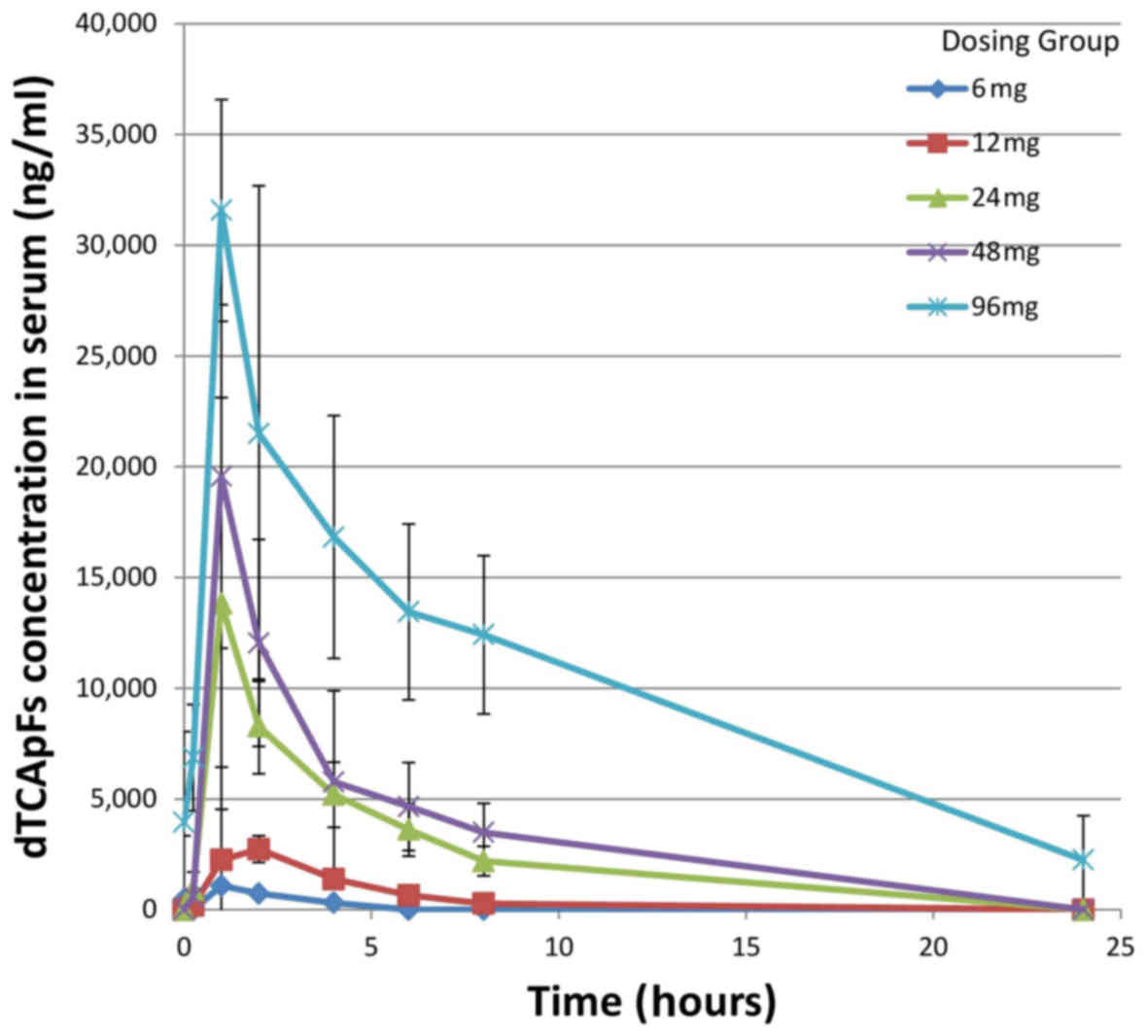
PK results
The PK results for the first day of cycles 1 and 2
are summarized in Table III; t1/2,
Cmax and AUC0 were found to be linearly correlated with dose.
Dose-dependent plasma concentrations of dTCApFs were observed
(Fig. 2).
| Table III.Pharmacokinetics of dTCApFs on the
first day of cycles 1 and 2 (each cycle was 28 days). |
Table III.
Pharmacokinetics of dTCApFs on the
first day of cycles 1 and 2 (each cycle was 28 days).
|
| Cycle 1, day 1 | Cycle 2, day 1 |
|---|
|
|
|
|
|---|
| dTCApFs dose,
mg/m2 | 6 (n=3) | 12 (n=4) | 24 (n=4) | 48 (n=4) | 96 (n=3) | 6 (n=3) | 12 (n=4) | 24 (n=4) | 48 (n=4) | 96 (n=3) |
|---|
| AUC0,
ng·h/ml | 3,813 | 12,905 | 49,630 | 79,935 | 206,742 | 9,719 | 11,452 | 57,069 | 100,093 | 294,682 |
| Cmax,
ng/ml | 1,209 | 6,048 | 14,609 | 18,267 | 32,964 | 1,536 | 6,048 | 14,609 | 22,113 | 32,016 |
| t1/2,
h | 2.3 | 2.1 | 3.2 | 4.9 | 6.0 | 2.8 | 2.0 | 3.7 | 4.6 | 8.5 |
Efficacy
Of the 17 patients who were treated for ≥3 months
(12, 24 and 48 mg/m2), 5 experienced stable disease (SD)
throughout the treatment period. Notably, 1 patient was suffering
from lower back pain, weakness and referred pain in the left
extremity, due to a spinal cord neoplasia compressing the spinal
cord (Ki-67, 30%; ST2-positive staining). Treatment with
painkillers (e.g., tramadol, oxycodone/naloxone, morphine and
pregabalin) was unsuccessful, and the patient used a walker. After
6 months of treatment (12, 24 and 48 mg/m2), the
patient's walking improved without the need for any painkiller
medication. Surgery was performed after 11 months of treatment. At
surgery, no malignancy was observed; however, scar tissue and
bleeding were noted, and the histopathological analysis revealed
strong presence of natural killer (NK) cells and dendritic cells. A
second patient who entered the study with progressive disease after
receiving 4 prior lines of chemotherapy treatment, maintained SD
through 6 cycles of dTCApFs (6 and 12 mg/m2).
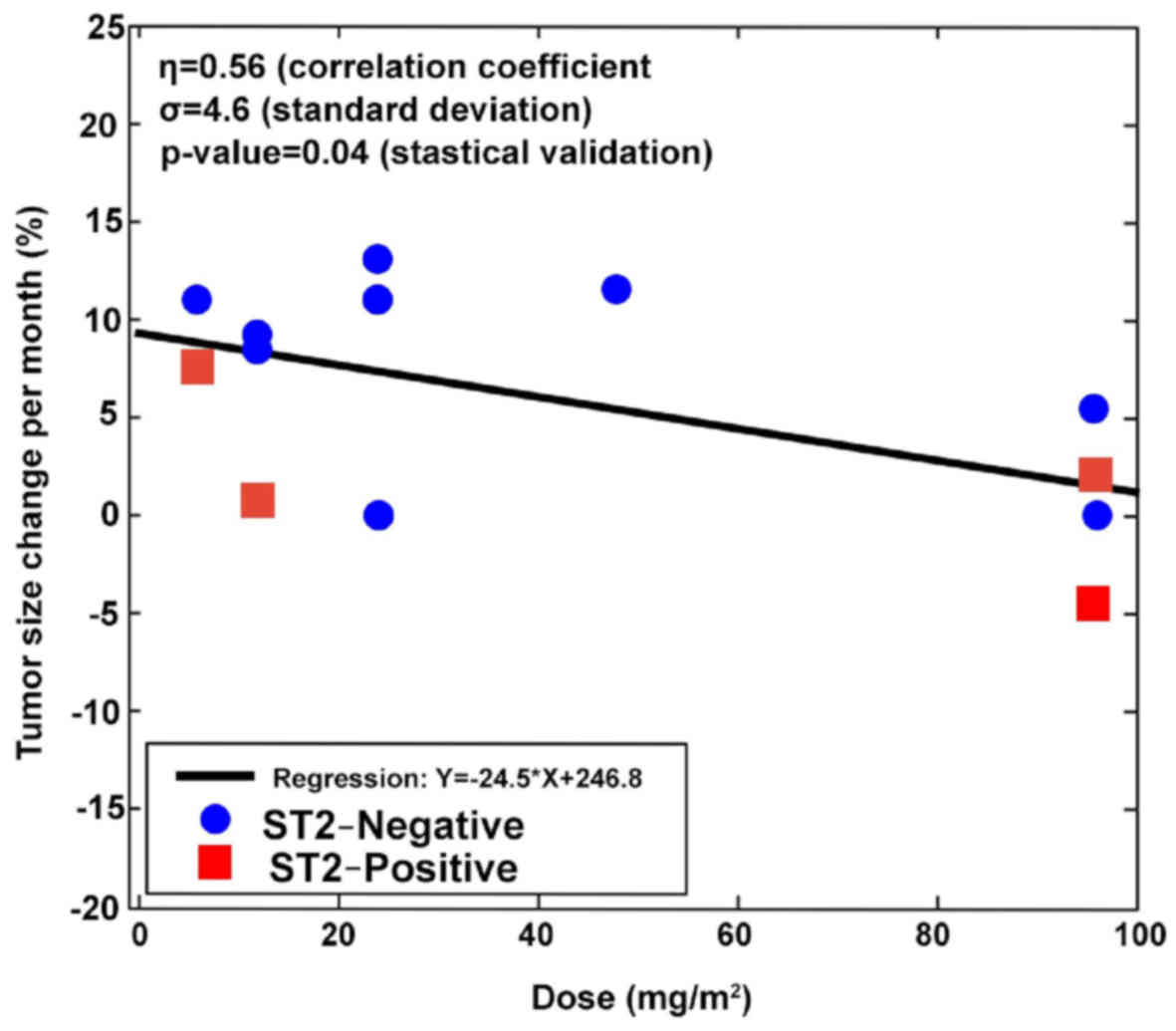
Progression-free survival (PFS) analysis revealed
that 6 patients experienced a longer PFS on dTCApFs compared with
their prior regimen, and 1 patient had a PFS that was comparable to
that on his prior regimen; 1 patient who had not receive prior
treatments was able to remain on the study drug for 330 days
(Table IV). A regression analysis
[robust regression model (2,3)] computing F statistics P-values revealed
a statistically significant correlation between changes in tumor
size and the administered dTCApFs doses (Fig. 3).
| Table IV.PFS on the last regimen before
enrolling the study and on dTCApFs. |
Table IV.
PFS on the last regimen before
enrolling the study and on dTCApFs.
| Patient no. | PFS on the last
regimen pre-enrollment, days | PFS on dTCApFs,
days |
|---|
| 1 |
480 | 53 |
| 2 |
134 | 25 |
|
3 |
110 |
170 |
|
4 |
0 | 330 |
|
5 |
52 |
51 |
| 6 |
384 | 110 |
| 7 |
54 |
90 |
| 8 |
80 | 52 |
| 9 |
375 | 60 |
| 10 | 1,800 | 14 |
| 11 |
41 |
52 |
| 12 |
42 |
50 |
| 13 |
96 | 42 |
| 14 |
365 | 40 |
| 15 |
1 |
80 |
| 16 |
105 | 45 |
| 17 |
564 | 41 |
Biomarker analysis
Treatment with dTCApFs at a dose of 6
mg/m2 led to an increase in serum levels of
angiopoietin-1, FGF-1, FGF-2, PDGF-AA, PDGF-BB, VEGF-D, TGF-β and
VEGF. At doses of 12–48 mg/m2, a decrease in the serum
levels of these factors was observed, and at 96 mg/m2,
an increase in all factors, except for VEGF-D, was noted. In
addition, the serum levels of all anticancer cytokines, such as
GM-CSF, IL2, IL-12p70, IL-21 and TNF-α, increased with dTCApFs
administration in all dose levels (Table
V).
| Table V.Mean change (%) in serum levels of
angiogenic factors and cytokines pre- to post- treatment with
dTCApFs. |
Table V.
Mean change (%) in serum levels of
angiogenic factors and cytokines pre- to post- treatment with
dTCApFs.
|
| dTCApFs dose,
mg/m2 |
|---|
|
|
|
|---|
| Factors | 6 (n=3) | 12 (n=3) | 24 (n=3) | 48 (n=3) | 96 (n=5) |
|---|
| Angiogenic
factors |
|
|
|
|
|
|
Angiopoietin-1 | +960 | −80 | −77 | −50 | +70 |
|
FGF-1 | +120 | −62 | −20 | −27 | +457 |
|
FGF-2 | +199 | −74 | −34 | −13 | +44 |
|
PDGF-AA | +1,379 | −92 | −79 | −73 | +57 |
|
PDGF-BB | +2,271 | −95 | −82 | −78 | +185 |
|
VEGF-A | +265 | −47 | −62 | −72 | −2 |
|
TGF-β1 | +18 | −80 | −59 | −20 | No data |
|
VEGF-D | +117 | −40 | −54 | −63 | +3 |
| Cytokines |
|
|
|
|
|
|
GM-CSF | +2,173 | −97 | +11 | +5,613 | +974 |
|
IL-12-p70 | +469 | −76 | +83 | +477 | +332 |
|
IL-2 | No data | −100 | No data | +242 | +577 |
|
IL-21 | +100 | −61 | +84 | +1,326 | +29 |
|
TNF-α | +4 | −5 | +31 | +74 | +97 |
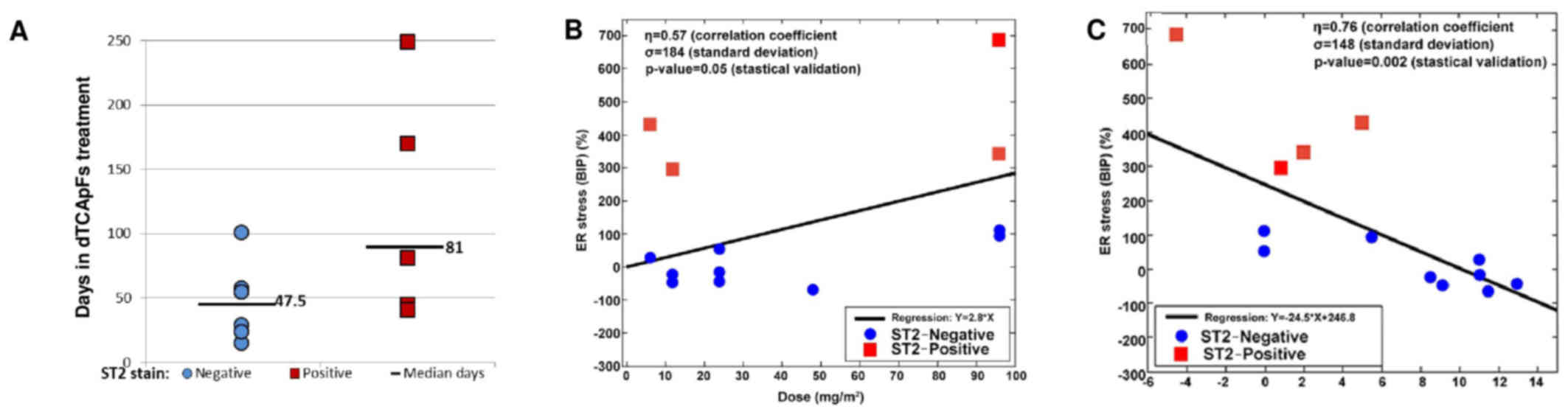
To assess the MOA of dTCApFs, patients were examined
by their T1/ST2 status, as dTCApFs has been shown to enter the
cells through this receptor (personal communication). A total of 14
patients underwent CT at 8 weeks and were evaluable for this
analysis. We observed that patients whose tumors were
T1/ST2-positive (by IHC) remained in the trial longer compared with
those whose tumors were T1/ST2-negative (Fig. 4A) and experienced SD on dTCApFs
treatment. Therefore, the patient population was re-analyzed
(changes in tumor size vs. administered dTCApFs dose), after
excluding T1/ST2-positive patients (n=9). In this re-analysis, the
correlation coefficient increased from 0.56 to 0.76, and the
standard deviation for tumor size decreased from 4.6 to 2.6
(P=0.02). A separate statistically valid regression analysis for
the subpopulation of T1/ST2-positive patients could not be
performed due to the small sample size (n=4). The serum levels of
the GRP78/BiP protein (ER stress biomarker) were also measured
prior to the initiation of dTCApFs treatment and after 29 days of
treatment. A statistically significant correlation was observed
between administered dTCApFs doses and change in serum GRP78/BiP
levels (P≤0.05), as well as between changes in tumor size and
change in serum GRP78/BiP levels (P≤0.002), suggesting that dTCApFs
induced ER stress (Fig. 4B and C).
These correlation analyses were then repeated after excluding
T1/ST2-negative patients, and an increase in the correlation
coefficients (for dTCApFs vs. change in GRP78/BiP levels, from 0.57
to 0.75; for change in GRP78/BiP levels vs. changes in tumor size,
from 0.79 to 0.83) were observed, along with a decrease in the
standard deviation for GRP78/BiP changes (from 184 to 67 and from
148 to 36, respectively). These changes were statistically
significant (P≤0.01).
Discussion
The aim of the present phase I dose-escalation study
was to investigate dTCApFs, a novel hormone peptide, whose activity
is driven by its interaction with the T1/ST2 receptor and its
anticancer activity is exerted through several MOAs, including a
unique MOA involving ER stress induction and downregulation of the
ER stress repair mechanism. Intravenous dTCApFs was found to be
safe, well-tolerated and potentially efficacious in treating
advanced/metastatic solid tumors. Furthermore, the PK examinations
revealed that t1/2, Cmax, AUC0 and plasma concentrations of dTCApFs
were linearly correlated with dose. In addition, that dTCApFs was
found to have anti-angiogenic activity, as well as the ability to
induce ER stress and expression of anticancer cytokines.
The present phase I study provides insight into the
MOA by which dTCApFs exerts its anticancer effects. dTCApFs enters
the cells through the T1/ST2 receptor (personal communication).
T1/ST2 is a member of the IL-1 receptor family and IL-33, which
regulate the Th1/Th2 immune responses in autoimmune and
inflammatory conditions. Lipopolysaccharides have been shown to
stimulate T1/ST2 expression in monocytes, muscle cells and
splenocytes, both in vitro and in vivo (4). The T1/ST2 receptor is expressed in
macrophages, dendritic cells, as well as in mast cells. This
receptor is a stable marker of Th2 polarized thymocytes (but not of
Th1 polarized thymocytes) and is important in the response of Th2
to viral antigens and allergens (5–7). T1/ST2
has been shown to play an important role in various diseases,
including cancer, Alzheimer's disease, inflammatory diseases,
trauma, sepsis, cardiovascular diseases and idiopathic pulmonary
fibrosis (6–14). Knocking out this receptor in BALB/c
mice bearing mammary carcinoma attenuated tumor growth and
metastasis. In these knockout mice (compared with wild-type mice),
the serum levels of IL-17, interferon-γ and TNF-α increased, along
with higher ex vivo cytotoxic activity of splenocytes, NK
cells and CD8+ T cells (1). In the present study, dTCApFs treatment
led to increased serum levels of anticancer cytokines (e.g.,
GM-CSF, IL-12p70, IL-2, IL-21 and TNF-α), likely due to the
downregulation of the T1/ST2 receptor. In addition, a correlation
between the antitumor activity of dTCApFs and T1/ST2 expression
status in the tumors was observed. A direct correlation was found
between T1/ST2 positivity, tumor size changes and induction of ER
stress. These findings are consistent with preclinical studies,
where treating ST2 gene knockout OV-90 cells with dTCApFs did not
result in ER stress. Taken together, these observations suggest
that the T1/ST2 receptor may serve as a biomarker to select
T1/ST2-positive patients who are more likely to respond to dTCApFs.
Additionally, the biomarker analysis revealed that dTCApFs
treatment increased the levels of IL-21 and IL12p70, which are
known activators of NK cells (15,16), as
well as the levels of GM-CSF and IL-2, which are known activators
of dendritic cells (17,18). Furthermore, histopathological
analysis of a post-treatment surgical specimen from a patient who
achieved a complete response revealed strong presence of NK and
dendritic cells. These findings suggest that dTCApFs may activate
the innate immune response, consistent with prior studies showing
such response with ST2 activation (19,20).
Drugs with MOAs involving ST2 activation are currently being
investigated (21,22). dTCApFs was also found to have broad
anti-angiogenic properties (it reduced the expression of multiple
angiogenic factors at levels of 12–48 mg/m2). Targeting
angiogenesis is a well-established MOA in anticancer drugs, with
commercially available and investigational drugs targeting factors
such as VEGF and FGF receptors (5,23–25). It
should be noted that, despite an observed increase in the levels of
angiogenic factors with dTCApFs treatment at the highest
investigated dose (96 mg/m2), the cytotoxic activity of
dTCApFs was, in fact, enhanced at this dose level, possibly due to
another MOA of dTCApFs.
Notably, the findings of dTCApFs-induced ER stress
are consistent with our preclinical studies showing a novel
mechanism involving two opposing effects of dTCApFs that together
result in apoptosis: ER stress induction, and downregulation of the
ER stress repair mechanism. Specifically, dTCApFs molecules enter
the cells through the ST2 receptor. Subsequently, they bind to the
sST2 soluble T1/ST2 receptor, enter the Golgi apparatus, and induce
structural changes that lead to destruction of the Golgi apparatus
and loss of Golgi function. This, in turn, leads to accumulation of
proteins in the ER, resulting in ER stress. dTCApFs also
downregulates sXBP1 and, thus, inhibits the ER stress repair
mechanism, leading to apoptosis. Interestingly, over the last
decade, the interaction between ER stress and tissue
vascularization has been intensively investigated and a clear
interaction between the stress-response mechanism and VEGF was
observed in cancer, diabetic retinopathy, atherosclerosis and
ischaemic renal disease (26).
GRP78/BiP is as an ER stress marker, and its upregulation following
anti-angiogenic therapy has been demonstrated in multiple studies.
For example, Han et al, demonstrated that sunitinib
treatment, which inhibits PDGF and vascular VEGFR receptors,
induced hypoxia in Caki-1 xenografts, that was followed by elevated
expression of GRP78/BiP in the treated group compared with the
control group (27). It may be
hypothesized that dTCApFS interrupts angiogenesis, thereby causing
accumulation of unfolded proteins in the ER of the cancer cells,
resulting in ER stress, leading to apoptosis.
In conclusion, treatment with intravenous dTCApFs
(6–96 mg/m2, 3 times/week, in consecutive 28-day cycles)
in locally advanced or metastatic solid tumors was found to be safe
and well-tolerated, with a dose-dependent, linear PK. dTCApFs
suppressed angiogenic factors, induced anticancer cytokine
production and ER stress, which likely led to the clinical outcome
observed in some of our patients. Positive T1/ST2 staining may
serve as a predictive marker for response to dTCApFs. Further
studies on the efficacy of dTCApFs in advanced malignancies
expressing high levels of T1/ST2 are warranted.
Acknowledgements
The present study was supported by ISK and Israel
Chief Scientist grants (grant no. 54811). Silverman MH, Sandler U,
Oren-Apoteker P, Ohana J and Devary Y are employed by ISK.
References
|
1
|
Sandler U, Devary O, Braitbard O, Ohana J,
Kass G, Rubinstein AM, Friedman ZY and Devary Y: NEROFE-a novel
human hormone-peptide with anti-cancer activity. J Exp Ther Oncol.
8:327–339. 2010.PubMed/NCBI
|
|
2
|
Marazzi A: Algorithms, routines and S
functions for robust statistics: The FORTRAN library ROBETH with an
interface to S-PLUS. Chapman and Hall; New York City, NY: 1993
|
|
3
|
Fox J: Applied regression analysis, linear
models, and related methods. Sage Publications, Inc.; London,
England: 1997
|
|
4
|
Saccani S, Polentarutti N, Penton-Rol G,
Sims JE and Mantovani A: Divergent effects of LPS on expression of
IL-1 receptor family members in mononuclear phagocytes in vitro and
in vivo. Cytokine. 10:773–780. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gutheil JC, Campbell TN, Pierce PR,
Watkins JD, Huse WD, Bodkin DJ and Cheresh DA: Targeted
antiangiogenic therapy for cancer using Vitaxin: A humanized
monoclonal antibody to the integrin alphavbeta3. Clin Cancer Res.
6:3056–3061. 2000.PubMed/NCBI
|
|
6
|
Lu DP, Zhou XY, Yao LT, Liu CG, Ma W, Jin
F and Wu YF: Serum soluble ST2 is associated with ER-positive
breast cancer. BMC Cancer. 14:1982014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
O'Donnell C, Mahmoud A, Keane J, Murphy C,
White D, Carey S, O'Riordain M, Bennett MW, Brint E and Houston A:
An antitumorigenic role for the IL-33 receptor, ST2L, in colon
cancer. Br J Cancer. 114:37–43. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lambrecht BN, De Veerman M, Coyle AJ,
Gutierrez-Ramos JC, Thielemans K and Pauwels RA: Myeloid dendritic
cells induce Th2 responses to inhaled antigen, leading to
eosinophilic airway inflammation. J Clin Invest. 106:551–559. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Walzl G, Matthews S, Kendall S,
Gutierrez-Ramos JC, Coyle AJ, Openshaw PJ and Hussell T: Inhibition
of T1/ST2 during respiratory syncytial virus infection prevents T
helper cell type 2 (Th2)- but not Th1-driven immunopathology. J Exp
Med. 193:785–792. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Coyle AJ, Lloyd C, Tian J, Nguyen T,
Erikkson C, Wang L, Ottoson P, Persson P, Delaney T, Lehar S, et
al: Crucial role of the interleukin 1 receptor family member T1/ST2
in T helper cell type 2-mediated lung mucosal immune responses. J
Exp Med. 190:895–902. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Meisel C, Bonhagen K, Löhning M, Coyle AJ,
Gutierrez-Ramos JC, Radbruch A and Kamradt T: Regulation and
function of T1/ST2 expression on CD4+ T cells: Induction of type 2
cytokine production by T1/ST2 cross-linking. J Immunol.
166:3143–3150. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu D, Chan WL, Leung BP, Huang FP, Wheeler
R, Piedrafita D, Robinson JH and Liew FY: Selective expression of a
stable cell surface molecule on type 2 but not type 1 helper T
cells. J Exp Med. 187:787–794. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Townsend MJ, Fallon PG, Matthews DJ, Jolin
HE and McKenzie AN: T1/ST2-deficient mice demonstrate the
importance of T1/ST2 in developing primary T helper cell type 2
responses. J Exp Med. 191:1069–1076. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xiong Z, Thangavel R, Kempuraj D, Yang E,
Zaheer S and Zaheer A: Alzheimer's disease: Evidence for the
expression of interleukin-33 and its receptor ST2 in the brain. J
Alzheimers Dis. 40:297–308. 2014.PubMed/NCBI
|
|
15
|
Van Elssen CH, Vanderlocht J, Frings PW,
Senden-Gijsbers BL, Schnijderberg MC, van Gelder M, Meek B, Libon
C, Ferlazzo G, Germeraad WT and Bos GM: Klebsiella
pneumoniae-triggered DC recruit human NK cells in a CCR5-dependent
manner leading to increased CCL19-responsiveness and activation of
NK cells. Eur J Immunol. 40:3138–3149. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Frederiksen KS, Lundsgaard D, Freeman JA,
Hughes SD, Holm TL, Skrumsager BK, Petri A, Hansen LT, McArthur GA,
Davis ID and Skak K: IL-21 induces in vivo immune activation of NK
cells and CD8(+) T cells in patients with metastatic melanoma and
renal cell carcinoma. Cancer Immunol Immunother. 57:1439–1449.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wuest SC, Edwan JH, Martin JF, Han S,
Perry JS, Cartagena CM, Matsuura E, Maric D, Waldmann TA and
Bielekova B: A role for interleukin-2 trans-presentation in
dendritic cell-mediated T cell activation in humans, as revealed by
daclizumab therapy. Nat Med. 17:604–609. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
van de Laar L, Coffer PJ and Woltman AM:
Regulation of dendritic cell development by GM-CSF: Molecular
control and implications for immune homeostasis and therapy. Blood.
119:3383–3393. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rank MA, Kobayashi T, Kozaki H, Bartemes
KR, Squillace DL and Kita H: IL-33-activated dendritic cells induce
an atypical TH2-type response. J Allergy Clin Immunol.
123:1047–1054. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nabekura T, Girard JP and Lanier LL: IL-33
receptor ST2 amplifies the expansion of NK cells and enhances host
defense during mouse cytomegalovirus infection. J Immunol.
194:5948–5952. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
ClinicalTrials.gov, . Description of a
trial investigating Interleukin-12 in ovarian epithelial cancer or
primary peritoneal cancer. https://clinicaltrials.gov/ct2/show/NCT00016289?term=IL12p70&rank=7January
11–2017
|
|
22
|
ClinicalTrials.gov, . Description of the
recombinant interleukin-21 in metastatic melanoma and kidney
cancer. https://clinicaltrials.gov/ct2/show/NCT00095108?term=IL-21&rank=7January
11–2017
|
|
23
|
Vasudev NS and Reynolds AR:
Anti-angiogenic therapy for cancer: Current progress, unresolved
questions and future directions. Angiogenesis. 17:471–494. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Boehm T, Folkman J, Browder T and O'Reilly
MS: Antiangiogenic therapy of experimental cancer does not induce
acquired drug resistance. Nature. 390:404–407. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jain RK: Antiangiogenic therapy for
cancer: Current and emerging concepts. Oncology (Williston Park).
19 4 Suppl 3:S7–S16. 2005.
|
|
26
|
Tahergorabi Z and Khazaei M: Imbalance of
angiogenesis in diabetic complications: The mechanisms. Int J Prev
Med. 3:827–838. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Han KS, Li N, Raven PA, Fazli L, Frees S,
Ettinger S, Park KC, Hong SJ, Gleave ME and So AI: Inhibition of
endoplasmic reticulum chaperone protein glucose-regulated protein
78 potentiates anti-angiogenic therapy in renal cell carcinoma
through inactivation of the PERK/eIF2α pathway. Oncotarget.
6:34818–34830. 2015.PubMed/NCBI
|