Introduction
Acute lymphoblastic leukemia (ALL) is the most
common hematologic malignancy in the pediatric population, at least
in the USA (1). The typical
presentation includes signs and symptoms associated with bone
marrow failure, including fever, infection, fatigue and excessive
bruising. Liver involvement in ALL is a frequent phenomenon, but
hyperbilirubinemia is not a frequently presenting symptom in ALL.
Abnormal laboratory findings and their degree reflect the leukemic
cell burden. However, there are few reports in the literature
describing cases of ALL presenting with acute hepatic failure in a
child or young adult (2–5). Acute hepatic failure is a relatively
rare disease as an initial presentation of ALL in an otherwise
healthy adolescent. This case report describes a case of an
adolescent with ALL presenting with acute hepatic failure.
Case report
Six days prior to presentation, a previously healthy
15-year-old male student began experiencing tiredness, inappetence,
nausea, vomiting, and jaundice (yellow sclera and dark urine).
Vomitus was nonbilious without hematemesis. There was no associated
fever, cough, expectoration, abdominal pain, diarrhea, or other
symptoms. The patient received a 3-day period of intravenous
therapy at a Dingzhou People's Hospital (Dingzhou, China) between
6th of February and the 9th of February 2018, and he became
increasingly jaundiced. He was then admitted to a primary care
center for evaluation of his hyperbilirubinemia. The initial
laboratory findings showed an alanine aminotransferase (ALT) of 492
IU/l, aspartate aminotransferase (AST) of 477 IU/l, total bilirubin
(TBIL) in serum of 11.7 mg/dl, direct bilirubin (DBIL) of 7.4
mg/dl, and lactic dehydrogenase (LDH) of 1,255 U/l. The prothrombin
time (PT) was 30.2 sec and the prothrombin activity (PTA) was
13.4%. The blood ammonia was 38.8 µmol/l. The routine blood test
showed that the patient's white blood cell count (WBC) was
2.41×109/l and the platelet (PLT) count was
49×109/l. The patient's serology was negative for viral
hepatitis. The patient underwent a 2-day transfusion treatment to
protect the liver and lower transaminase levels. However, his
physical condition progressively deteriorated and he was
immediately transferred to the Third Hospital of Hebei Medical
University (Shijiazhuang, China) on the 9th February 2018.
The patient was a student with sports talent.
Further history of illness revealed that he had received a
transfusion treatment 1 month previously due to a common cold.
There was no history of hepatotoxic drugs or any other toxic
agents. He had been healthy with no history of medical disorders or
surgical procedures, and no history of taking medications. His
family history was unremarkable for metabolic diseases or any other
hereditary conditions. On physical examination, he had
lymphadenectasis and splenectasis, and his vital signs were normal.
No spider angioma or palmar erythema of chronic liver disease were
present.
The laboratory data revealed a white blood cell
count of 3.18×109/l (33.6% neutrophils, 43.1%
lymphocytes, 23.0% monocytes, and 0.3% eosinophils), hemoglobin
level of 14.1 g/dl, hematocrit of 41.9%, and PLT count of
52×109/l. The metabolic panel revealed an ALT of 3,261
IU/l, AST of 2,110 IU/l, TBIL of 15.0 mg/dl, DBIL of 11.1 mg/dl,
LDH of 1,767 U/l, and alkaline phosphatase (ALP) of 175 IU/l. The
international normalized ratio was 2.66, PTA was 24.3%, fibrinogen
(FIB) was 0.86 g/l, PT was 31.2 sec, and activated partial
thromboplastin time (APTT) was 40.1 sec. Urinalysis revealed urine
was bilirubin-positive. A routine stool test and toxicological
analysis were negative. A viral assessment for hepatitis A virus,
hepatitis B virus (HBV), hepatitis C virus, hepatitis E virus,
human immunodeficiency virus, Epstein-Barr virus, cytomegalovirus,
Treponemapallidum and Salmonella typhi returned
normal. An abdominal ultrasound with Doppler revealed splenomegaly,
and the chest computed tomography scan was normal. Liver biopsy was
not performed due to increased bleeding risk. Due to the evidence
of decreased WBC and PLT count, lymphadenectasis and splenomegaly,
a peripheral smear was obtained immediately. Atypical cells were
observed by a hematologic pathologist as being consistent with
hematologic malignancy. A bone marrow biopsy was then performed.
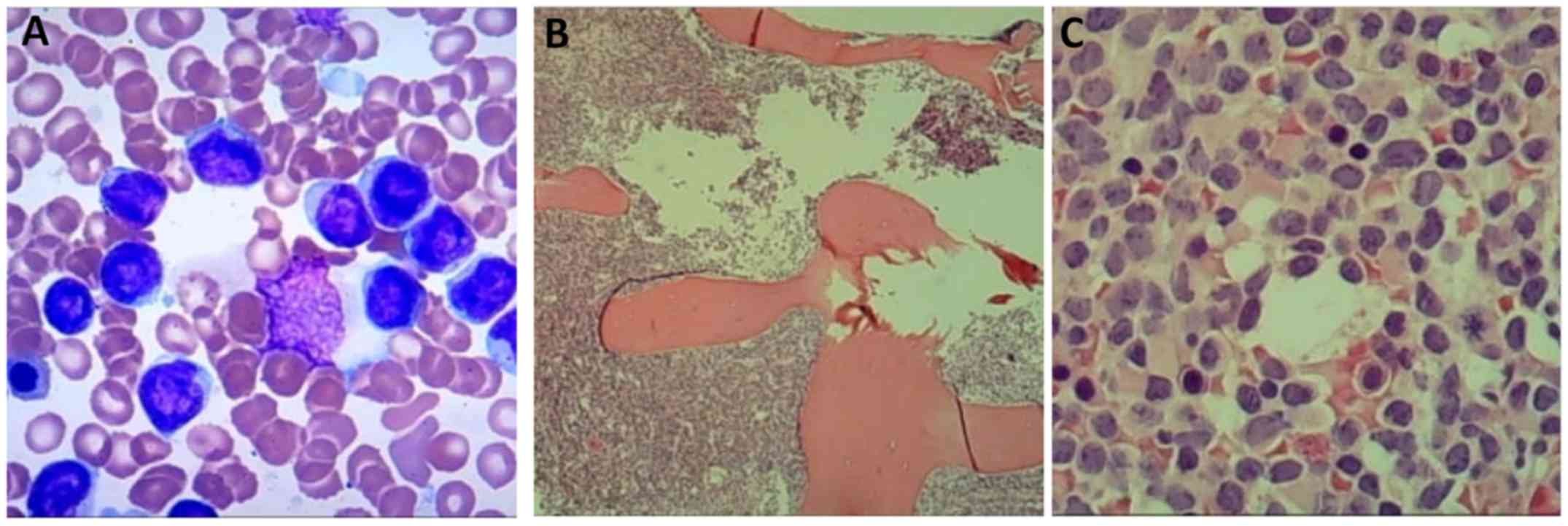
The bone marrow cell morphology indicated hypercellularity, and the
usual hematopoietic cells had been almost completely replaced by
amonotonous lymphoid population composed mainly of lymphoblasts
(Fig. 1A). The bone marrow core
biopsy revealed large aggregates of blasts (Fig. 1B and C). Immunohistochemistry
revealed the following:terminal deoxynucleotidyl transferase (TdT)
(+), paired box 5 (+), CD34 (−), CD20 (−), CD3 (−), CD117 (−),
myeloperoxidase (MPO) (−), and lysozyme (−). The cells did not show
myeloperoxidase activity on cytochemical analysis. Bone marrow was
submitted for flow cytometry, which revealed a predominant B-cell
population (68.15%) expressing CD19, cCD79a, CD33, CD38, CD123, CD9
and CD38, partially expressing CD10 and TdT, and showing weak
expression of CD22, with no expression of CD117, CD34, CD7, CD15,
CD64, CD11b, CD20, MPO, cCD3, mCD3, CD56, kappa or lambda. The
diagnosis of early precursor B-cell (pro-B) ALL was established. In
retrospect, the atypical cells of the initial peripheral blood
smear were actually lymphoblasts, although the blood smear was not
available for re-review following the diagnosis of ALL.
The patient underwent transfusion treatment at the
Third Hospital of Hebei Medical University to protect the liver,
including reduced glutathione injection (1.8 g per day for a total
of 14 days), ornithine aspartate injection (2.0 g per day for a
total of 14 days), compound diisopropylamine dichloroacetate
injection (120 mg per day for a total of 14 days), and ademetionine
1,4-butanedisulfonate injection (1.0 g per day for a total of 14
days), and low-dose dexamethasone (10 mg per day for a total of 5
days). The levels of ALT, AST, LDH and TBIL continued to decrease,
and the PTA and fibrinogen recovered (Table I). The patient presented with
improvement of symptoms. Admission laboratory data revealed a WBC
of 2.82×109/l (55% neutrophils, 33% lymphocytes, 11%
monocytes, 0.4% eosinophils and 0.6% basophils), hemoglobin of 91.3
g/l, TBIL of 6.9 mg/dl, ALT of 95 IU/l, AST of 46 IU/l, and LDH of
124 U/l. Due to persistent improvement in the patient's liver
function tests, a liver biopsy was not necessary. Bone marrow cell
morphology, flow cytometry and biopsy revealed young blasts with
markers consistent with B-cell ALL. Cytogenetic examination
indicated no abnormal chromosomes (46, XY) or fusion genes,
including breakpoint cluster region/ABL proto-oncogene 1,
non-receptor tyrosine kinase andmixed lineage leukemia. The
adolescent was transferred to the Department of Hematology, where
he was started on induction chemotherapy with pediatric-based
protocols.
 | Table I.Liver function and blood coagulation
analyses. |
Table I.
Liver function and blood coagulation
analyses.
| Laboratory
parameter | Normal level | Admission (Day
1) | Day 3 | Day 5 | Day 10 | Day 12 | Day 18 |
|---|
| ALT | 9–50 U/l | 3,261 U/l | 1,506 U/l | 694 U/l | 171 U/l | 120 U/l | 95 U/l |
| AST | 15–40 U/l | 2,110 U/l | 880 U/l | 310 U/l | 35 U/l | 31 U/l | 46 U/l |
| LDH | 120–250 U/l | 1,767 U/l | 794 U/l | 606 U/l | 157 U/l | 123 U/l | 124 U/l |
| TBIL | 3–20 µmol/l | 256 µmol/l | 274 µmol/l | 312 µmol/l | 196 µmol/l | 198 µmol/l | 118 µmol/l |
| DBIL | 2–6 µmol/l | – | 205 µmol/l | 246 µmol/l | 164 µmol/l | 169 µmol/l | 101 µmol/l |
| PT | 10–14 sec | 31.2 sec | 33.8 sec | 24.5 sec | 15.4 sec | 11.8 sec | 11.1 sec |
| PTA | 80–150% | 24.3% | 22.1% | 32.0% | 61.0% | 106% | 100% |
| FIB | 2–4 g/l | 0.86 g/l | 0.61 g/l | 0.83 g/l | 0.60 g/l | 1.29 g/l | 3.47 g/l |
Discussion
ALL is the most common hematological neoplasm in the
pediatric population, with a frequency peak for children between 2
and 5 years of age. The overall survival rate of ALL in childhood
is approaching 90%. By contrast, ALL is less common in adolescents
and adults, with estimated survival rates between 20 and 40%
(1,2). The common presenting signs and symptoms
of ALL, although non-specific, include fever and infection caused
by neutropenia, fatigue and pallor due to anemia, and bruising and
hemorrhage secondary to thrombocytopenia. Other manifestations
involve lymphadenectasis, hepatomegaly, splenomegaly, sternal
tenderness, and joint pain. Hepatomegaly is a common feature of
leukemia, however, acute hepatic failure as the initial
presentation is uncommon. There are limited case reports in the
literature describing ALL presenting as acute hepatic failure in an
adolescent (3–5). Although rare, it is important to
consider ALL in the differential diagnosis of multi-organ
dysfunction, including acute liver failure, particularly in
previously otherwise healthy individuals.
Several mechanisms have been proposed to explain the
etiopathogenesis of acute hepatic failure in leukemia, including
comorbidity with viral infections, particularly HBV, sepsis,
autoimmune hepatitis, or hypoxia and ischemia caused by leukemic
cell infiltration that results in the obstruction of hepatic blood
flow. In the case described herein, on awaiting the results of bone
marrow biopsy, hepatoprotective treatment was administered and the
ALT, AST and TBIL levels slowly decreased. The biopsy revealed
large aggregates of blasts, following which low-dose dexamethasone
was initiated and serum levels of ALT, AST and TBIL showed
accelerated decrease with marked improvement in the patient's
physical condition. A limitation was that histological examination
was not performed on the patient's liver. Therefore, it was not
possible to demonstrate whether the elevated liver enzymes,
hyperbilirubinemia, and hepatic failure were secondary to hepatic
necrosis produced by leukemic infiltration. However, the early
application of glucocorticoids was more effective than the hepatic
protectant, which suggested that hepatic injury was likely caused
by the infiltration of leukemic cells.
The majority of previous reports describing liver
disease in patients with ALL are in the pediatric population. In
one retrospective pediatric study which examined hepatitis at the
time of ALL diagnosis, approximately one third of patients had
increased aminotransferase levels, with normal bilirubin and ALP
and no evidence of viral hepatitis. ALL-induced hepatitis was found
to be more common in patients with T-cell ALL and in patients with
higher WBC, uric acid, and LDH levels, suggesting leukemic cell
infiltration as the underlying etiology (6). The mechanism of hepatic injury remains
to be fully elucidated, but is likely to be either a paraneoplastic
syndrome or due to lymphocyte infiltration of the liver. The
patient's abnormal liver function tests in the case study reported
normalized following the early application of dexamethasone. This
indicated that lymphoblast infiltration had led to the hepatic
failure. The patient received induction chemotherapy according to
pediatric-based protocols and without any dose adjustment due to
liver dysfunction. This most likely resulted in an improvement in
long-term survival rate (7). Taken
together, it is important to consider ALL in the initial
presentation of acute hepatic failure. A peripheral blood smear is
useful for the differential diagnosis of hematological neoplasms,
and bone marrow or liver biopsy is required as soon as possible for
early diagnosis.
Acknowledgements
The authors would like to thank Dr Fei-yan Ma
(Department of Ophthalmology, The Second Hospital of Hebei Medical
University, Shijiazhuang, China) for her assistance in language
editing.
Funding
No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
JY conceived and designed the study and collected
clinical data. RLG, MX and JS collected clinical data and edited
the figure. JY and RLG wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The case report was approved by Institutional Review
Board of the Third Affiliated Hospital of Hebei Medical University
for chart review and data analysis.
Patient consent for publication
Patient consent was obtained for publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Inaba H, Greaves M and Mullighan CG: Acute
lymphoblastic leukaemia. Lancet. 381:1943–1955. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vallacha A, Haider G, Raja W and Kumar D:
Remission rate of acute lymphoblastic leukemia (ALL) in adolescents
and young adults (AYA). J Coll Physicians Surg Pak. 28:118–121.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Litten JB, Rodríguez MM and Maniaci V:
Acute lymphoblastic leukemia presenting in fulminant hepatic
failure. Pediatr Blood Cancer. 47:842–845. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kader A, Vara R, Egberongbe Y, Height S
and Dhawan A: Leukaemia presenting with fulminant hepatic failure
in a child. Eur J Pediatr. 163:628–629. 2004.PubMed/NCBI
|
|
5
|
Heincelman M, Karakala N and Rockey DC:
Acute lymphoblastic leukemia in a young adult presenting as
hepatitis and acute kidney injury. J Investig Med High Impact Case
Rep. Sep 22–2016.(Epub ahead of print). doi:
10.1177/2324709616665866. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Segal I, Rassekh SR, Bond MC, Senger C and
Schreiber RA: Abnormal liver transaminases and conjugated
hyperbilirubinemia at presentation of acute lymphoblastic leukemia.
Pediatr Blood Cancer. 434–439. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ribera JM, Ribera J and Genescà E:
Treatment of adolescent and young adults with acute lymphoblastic
leukemia. Mediterr J Hematol Infect Dis. 6:e20140522014. View Article : Google Scholar : PubMed/NCBI
|