Introduction
A multitude of aberrant signaling pathways have been
implicated in meningioma tumorigenesis. Potentially responsible
signaling pathways include RB/p53, MAPK, PI3K/Akt, Wnt, and many
others (1). Many genes involved in
cell-cycle control, cell growth and proliferation have been
reported to be mutated or aberrantly expressed and often associated
with high grade meningioma (2,3).
However, the molecular etiology and pathogenesis of this tumor
still seek many answers. The p53 pathway is involved in and often
dysregulated in anaplastic meningiomas. The involvement of p53
pathway in meningiomas was first observed by marked loss of
expression of p53 protein (4). Tumor
suppressor p53, encoded by the TP53 gene, is a key protein
involved in many major cellular tumor suppression processes such as
the control of cell cycle and apoptosis. In the present study we
wanted to investigate presence of mutations of TP53's exon 4
relevant for intracranial meningioma. The exons 2–4 encode for the
two transactivation domains of p53 protein (amino acids 1 through
42 and 43–62) (5). Moreover, exons
4–8 code for the so called core domain of p53, an important domain
by which the protein binds to DNA.
In spite of the fact that tumor suppressor gene
TP53 is the most frequently mutated gene in human cancers
overall, the findings on TP53 in meningioma are scarce and
controversial. In order to understand the role of p53 in the
etiology and pathogenesis of meningioma thorough investigation is
still necessary (6). Past studies
have examined alterations in TP53 tumor suppressor gene in
meningiomas and it is generally considered that mutations of
TP53 are not frequent and thus not important for meningioma
biology (7–11). However, this is a misconception,
because when examining literary reports in more detail it becomes
obvious that only several meningioma cases were reported and few
exons sequenced. Opposing studies relevant to our work provide
findings on p53 pathway in meningioma development (12). For example, the correlation of p53
protein expression with histological tumor grade and meningioma
recurrence (13,14) and the loss of detectable MDM2 protein
in high grade meningiomas (15,16) are
all in favor of p53's involvement. Recently, a meningioma candidate
tumor suppressor gene called maternally expressed gene 3 (MEG3) has
been identified to transactivate TP53 (17). Loss of MEG3 expression and its
allelic loss were associated with higher meningioma grades. MEG3, a
noncoding RNA with antiproliferative functions, is strongly
expressed in normal arachnoidal cells, but absent in meningioma
cell lines.
Cancer genome sequencing of 12 different tumor types
has shown that 42% of investigated cases carry mutant TP53
genes (18). Yet, different tumors
displayed quite different frequencies of TP53 mutations,
some harboring only 2.2%, while others even 95%. It is well known
that p53 has many functions. It acts as a transcription factor
activated upon sensing cellular stress of different kinds. Its
accumulation affects the cell cycle, DNA repair, apoptosis,
senescence and cell differentiation. In normal cells p53 protein
levels are kept low by the following mechanism: p53 upregulates its
negative regulator, E3 ubiquitin-protein ligase MDM2 which causes
rapid ubiquitination and degradation of p53 by the proteasome
(18,19).
Meningiomas account for approximately 30% of primary
intracranial and intraspinal neoplasms. It is believed nowadays
that the tumor arises from the arachnoidal cap cells of the
leptomeninges. Most meningiomas are characterized as benign, slowly
growing tumors with long survival time and are classified as WHO
grade I (20,21). However, meningiomas can show an
aggressive nature. In between benign and malignant types are
atypical meningiomas characterized by increased mitotic activity,
brain invasion and a higher risk for recurrence. They are WHO grade
II tumors and represent about 5–7% of all meningioma cases. Grade
III tumors-anaplastic meningiomas, are typically associated with
brain invasion and recurrence. They exhibit true malignancy and
represent 3% of all meningioma cases with the low overall 10-year
survival rate of 14.2% (22,23). Meningiomas as a whole can display a
broad spectrum of clinical, histological and cytogenetic features.
Hence, 15 different subtypes are described in the current
pathohistological classification of meningiomas. The considerable
variability in the biological behavior could be observed even
within the same WHO grade. So, histologically distinct subtypes of
benign meningiomas may exhibit high risk of recurrence, or even
evolve into atypical and anaplastic subtypes (24–26). Our
understanding of the genetic profiles of sporadic meningiomas only
recently started to uncover owing to large-scale genomic analyses
(27–29). However, relevant genetic events for
atypical and anaplastic cases as well as molecular mechanisms of
meningioma progression still need to be fully understood. It has
been well documented that the neurofibromatosis type 2 gene (NF2)
located on 22q is inactivated through mutation and loss of
heterozygosity (LOH) in the majority of meningiomas (30). Consequent loss of NF2 encoded protein
merlin, is a consistent finding in all neurofibromatosis type 2
associated meningiomas and in about half of sporadic benign cases
(31).
Our investigation that focused on exon 4, that codes
for the functionally important domains for p53 protein,
demonstrated that mutations that impact the p53's transactivation
and DNA binding domains are present in our group of collected
meningiomas.
Patients and methods
Tissue samples
We conducted our study by collecting 48 paired
meningioma tumors and autologous blood from the Departments of
Neurosurgery and Pathology, Hospital Center ‘Sisters of Charity’,
Zagreb, Croatia, following patients' consents. Collected tumor
tissues were frozen in liquid nitrogen and were kept at −80°C. The
peripheral blood samples were collected in EDTA and processed
immediately. Using the magnetic resonance imaging (MRI) tumor
lesions were diagnosed in different cerebral regions, with the
surrounding zone of perifocal edema. During the operative procedure
the tumor was maximally reduced using a microneurosurgical
technique. No family history of brain tumors was recorded in the
collected patients and they did not undergo chemotherapy or
radiotherapy prior to surgery. All meningioma samples were first
evaluated after resection and then studied by neuropathologists who
classified them according to the WHO criteria (21). There were 17 meningothelial
meningiomas, 7 fibrous (fibroblastic), 15 transitional (mixed), 1
psammomatous, 4 angiomatous, 2 atypical and 2 anaplastic. Clinical
and pathological data are shown in Table
I. Twenty-six patients were females and 22 males. The age of
patients varied from 32 to 79 (mean age=63.19, median 66 years).
The mean age at diagnosis for females was 65.84, and for males
60.05 years. All procedures involving human participants were
performed in accordance with the ethical standards of the
Institutional and National Research Committee, and with the
Declaration of Helsinki. Ethical approval was obtained from the
Ethical Committee of School of Medicine University of Zagreb
(approval no. 380-59-10106-14-55/147; Class: 641-01/14-02/01,1. 07.
2014) and of the University Hospital Center ‘Sisters of Mercy’
(approval no. EP-7426/14-9,11. 06. 2014). Verbal informed consent
was obtained from all patients included in the present study.
 | Table I.Nucleotide alterations of TP53
exon 4 and the pathohistological type, grade and demographic
variables of the collected meningioma samples. |
Table I.
Nucleotide alterations of TP53
exon 4 and the pathohistological type, grade and demographic
variables of the collected meningioma samples.
| Patient | TP53 e4
nucleotide alterations | Meningioma
type | Age (years) | Sex |
|---|
| 1 | c.215G>C,
p.R72P, Mi; c.186delA, p.E62del, Fr | Meningothelial
(I) | 72 | M |
| 2 | c.186delA,
p.E62del, Fr; c.209_210G, p.A70ins, Fr | Meningothelial
(I) | 70 | F |
| 3 | No mutation | Meningothelial
(I) | 62 | F |
| 4 | No mutation | Meningothelial
(I) | 64 | M |
| 5 | c.190C>G,
p.P64A, Mi; c.215G>C, p.R72P, Mi | Meningothelial
(I) | 65 | M |
| 6 | No mutation | Meningothelial
(I) | 35 | M |
| 7 | c.186delA,
p.E62del, Fr | Meningothelial
(I) | 55 | F |
| 8 | c.213C>A,
p.P71P, S | Meningothelial
(I) | 47 | F |
| 9 | No mutation | Meningothelial
(I) | 70 | M |
| 10 | No mutation | Meningothelial
(I) | 54 | M |
| 11 | c.186delA,
p.E62del, Fr; c.215G>C, p.R72P, Mi; c.276C>A, p.P92P, S | Meningothelial
(I) | 62 | F |
| 12 | c.217G>A,
p.V73M, Mi; c.300G>T, p.Q100H, Mi; c.312G>A, p.Q104Q, S;
c.315C>T, p.G105G, S | Meningothelial
(I) | 71 | M |
| 13 | c.215G>C,
p.R72P, Mi; c.229C>T, p.P77S, Mi; c.245C>T, p.P82L, Mi;
c.263C>G, p.A88G, Mi; c.271T>C, p.W91R, Mi | Meningothelial
(I) | 40 | M |
| 14 | c.186delA,
p.E62del, Fr; c.215G>C, p.R72P, Mi; c.271T>C, p.W91R, Mi;
c.312G>A, p.Q104Q, S | Meningothelial
(I) | 63 | F |
| 15 | c.215G>C,
p.R72P, Mi; c.322G>C, p.G108R, Mi | Meningothelial
(I) | 62 | M |
| 16 | No mutation | Meningothelial
(I) | 75 | F |
| 17 | c.186delA,
p.E62del, Fr; c.209_210G, p.A70ins, Fr | Meningothelial
(I) | 67 | F |
| 18 | No mutation | Fibrous (I) | 54 | M |
| 19 | c.186delA,
p.E62del, Fr; c.190C>G, p.P64A, Mi; c.209_210G, p.A70ins,
Fr | Fibrous (I) | 63 | F |
| 20 | No mutation | Fibrous (I) | 45 | M |
| 21 | No mutation | Fibrous (I) | 51 | F |
| 22 | No mutation | Fibrous (I) | 73 | F |
| 23 | No mutation | Fibrous (I) | 66 | F |
| 24 | c.215G>C,
p.R72P, Mi | Fibrous (I) | 74 | F |
| 25 | No mutation | Transitional
(I) | 56 | F |
| 26 | No mutation | Transitional
(I) | 61 | M |
| 27 | c.243_244insA,
p.P82ins, Fr; c.300G>T, p.Q100H, Mi | Transitional
(I) | 45 | M |
| 28 | No mutation | Transitional
(I) | 72 | F |
| 29 | c.186delA,
p.E62del, Fr; c.315C>T, p.G105G, S | Transitional
(I) | 74 | F |
| 30 | c.215G>C,
p.R72P, Mi | Transitional
(I) | 75 | M |
| 31 | c.215G>C,
p.R72P, Mi | Transitional
(I) | 32 | M |
| 32 | c.202_203insT,
p.E68ins, Fr | Transitional
(I) | 77 | M |
| 33 | c.186delA,
p.E62del, Fr | Transitional
(I) | 71 | F |
| 34 | c.186delA,
p.E62del, Fr; c.315C>T, p.G105G, S | Transitional
(I) | 64 | F |
| 35 | c.215G>C,
p.R72P, Mi c.217G>A, p.V73M, Mi; c.312G>A, p.Q104Q, S;
c.315C>T, p.G105G, S | Transitional
(I) | 66 | F |
| 36 | c.186delA,
p.E62del, Fr | Transitional
(I) | 73 | F |
| 37 | c.186delA,
p.E62del, Fr; c.215G>C, p.R72P, Mi | Transitional
(I) | 67 | F |
| 38 | c.186delA,
p.E62del, Fr | Transitional
(I) | 79 | F |
| 39 | c.315C>T,
p.G105G, S; c.322G>C, p.G108R, Mi | Transitional
(I) | 61 | F |
| 40 | c.190C>G,
p.P64A, Mi; c.215G>C, p.R72P, Mi | Psammomatous
(I) | 60 | F |
| 41 | No mutation | Angiomatous
(I) | 66 | M |
| 42 | No mutation | Angiomatous
(I) | 39 | M |
| 43 | c.215G>C,
p.R72P, Mi | Angiomatous
(I) | 70 | F |
| 44 | c.209_210insG,
p.A70ins, Fr | Angiomatous
(I) | 78 | M |
| 45 | No mutation | Atypical (II) | 76 | M |
| 46 | No mutation | Atypical (II) | 73 | M |
| 47 | c.213C>A,
p.P71P, S; c.245delC, p.P82del, Fr | Anaplastic
(III) | 71 | F |
| 48 | No mutation | Anaplastic
(III) | 67 | M |
DNA extraction
We obtained tumor DNA by homogenizing approximately
0.5 g of each specimen tumor tissue with 1 ml extraction buffer (10
mM Tris HCl; 0.1 M EDTA; 0.5% sodium dodecyl sulfate, pH 8.0) and
incubated with proteinase K (100 mg/ml; Sigma; overnight at 37°C).
Phenol/chloroform extraction and ethanol precipitation followed.
Leukocyte DNA was extracted from the blood samples. Three ml of
blood was lysed with 7 ml distilled water and centrifuged (15
min/5.000 g). After that, DNA extraction was the same as for the
tissue samples. The concentration of double-stranded DNA from the
tumors and blood was quantified with Nanodrop.
Polymerase chain reaction, DNA
sequencing reactions
We investigated mutational status of exon 4 of the
TP53 gene in our collected meningioma sample by direct
Sanger sequencing of this exon. Each PCR reaction was performed in
a total reaction volume of 25 µl with the following final optimized
concentrations: 0.2 mM of each dNTP, 3 mM MgCl2, 0.2 µM
of each primer (Sigma-Aldrich), 1× Flexi buffer and 0.5 U of
GoTaq® G2 Hot Start Polymerase (both Promega). Reaction
conditions were as follows: 96°C/5 min (one cycle), 45 cycles of
96°C/30 sec, 51°C/45 sec, 72°C/30+1 sec, and the final step of
72°C/10 min. Primers used for the TP53's exon 4 were
5′-GATGCTGTCCGCGGACGATAT-3′, 5′-CGTGCAAGTCACAGACTTGGC-3′. The PCR
products of the target sequence were 247-bp long and resolved on 2%
agarose gels.
Nucleotide variations were discovered by Sanger
sequencing using the Big DyeTerminator v3.1 Cycle Sequencing kit.
An enzymatic clean-up using ExoI and FastAP from ThermoFisher
Scientific was performed prior to sequencing. The BigDye Terminator
3.1 Sequencing Buffer for the set-up of the reactions was used and
the plate was purified via a Sephadex plate after PCR. DNA
sequencing reactions were analyzed on the Applied Biosystems 3730XL
(Applied Biosystems) with 96 capillaries (Source BioScience GmbH).
All samples were sequenced twice with independent PCR reactions.
Sequence analysis was carried out using NCBI Nucleotide Blast
(https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch).
Reference sequence was used from GeneBank accession number
U94788.1. The evaluation of pathogenicity was performed with the
PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and ClinVar
(https://www.ncbi.nlm.nih.gov/clinvar/) tools (32) and the prediction whether an amino
acid substitution affects protein function with SIFT (Sorting
Intolerant from Tolerant algorithm http://sift.jcvi.org/) and Mutation Taster2
(http://www.mutationtaster.org/) tools
(33,34).
Immunohistochemistry
Forty-six meningiomas were available for this
analysis. The sections were immunostained using
streptavidin-horseradish peroxidase/DAB (EnVisionTM, Dako REALTM)
as previously described (2). The
primary antibodies used for p53 detection were mouse anti-human
monoclonal antibodies (clone DO-7; DAKO; diluted 1:25).
Statistical Analysis
All statistical evaluations were carried out using
SPSS statistical package version 14.0 (SPSS Inc.). Following
variables were tested for all patients: Meningioma histological
subtype and malignancy grade, p53 mutational type, protein
presence, sex and age. The normality of data was tested using the
Shapiro-Wilk test. Pearson Chi-Square and Spearman's correlation
were employed to test the relationships between the various
parameters. P<0.05 was considered to indicate a statistically
significant difference.
Results
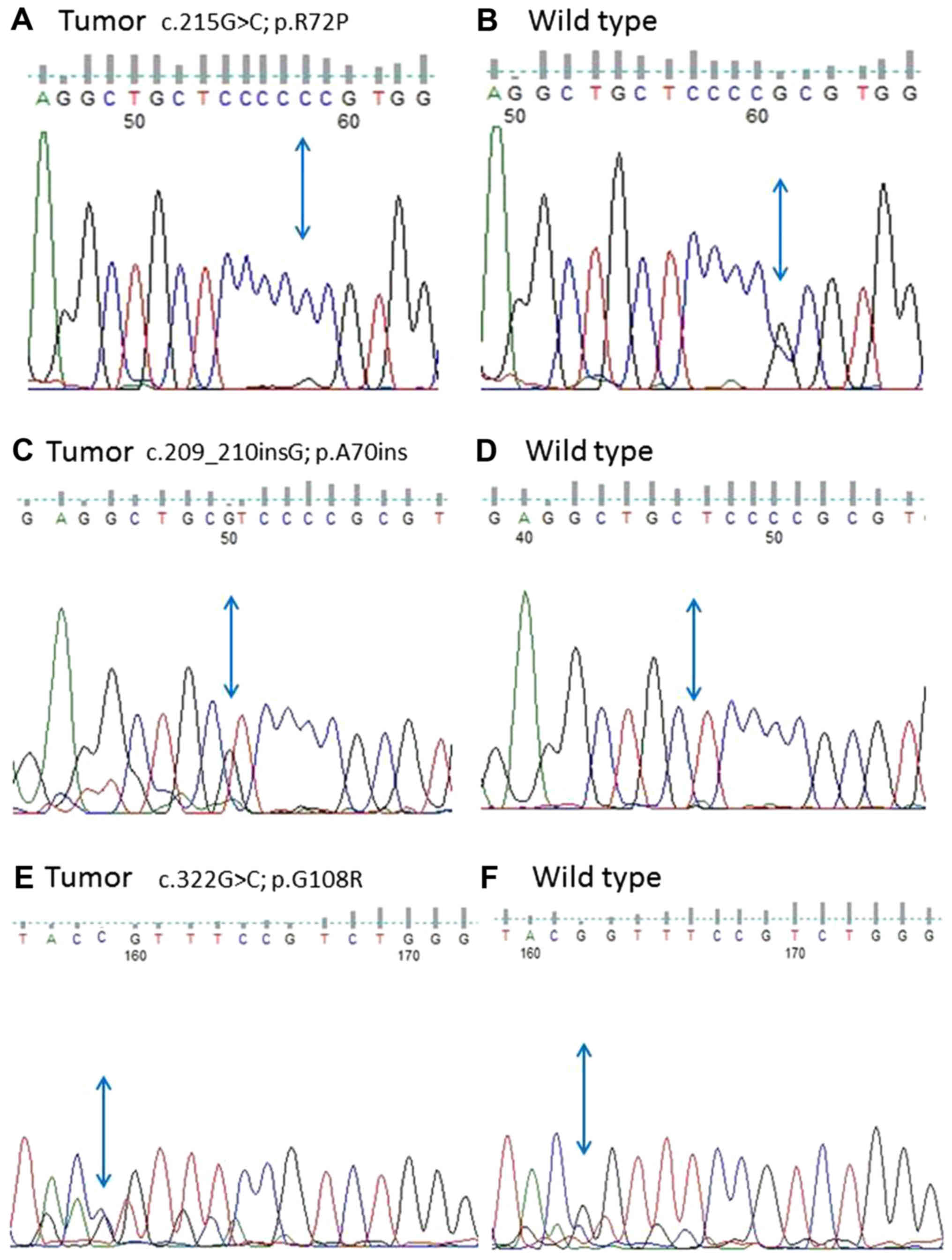
The frequency and type of
mutations
Mutations of exon 4 of TP53 gene were very
frequent and altogether detected in 29 meningioma patients
(60.42%). Several patients harbored more than one mutation. There
were altogether 18 different mutations across our meningioma
sample. Meningiomas with nucleotide alterations are listed in
Table I. Sequencing results are
shown in Fig. 1. Several changes
have been characterized as polymorphisms in the literature and
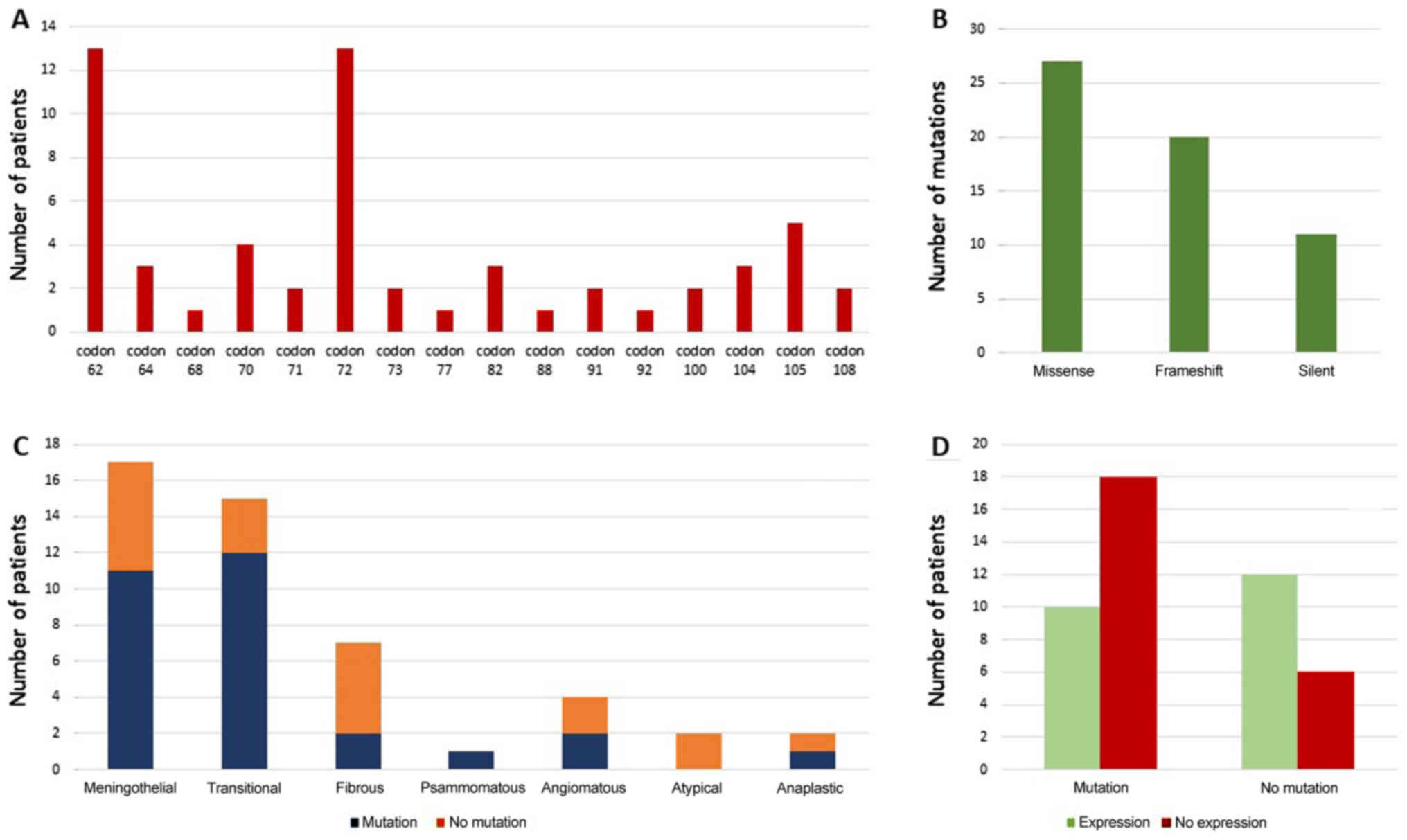
databases. Interestingly 13 observed alterations were appearing
more than once, and some were repeatedly found in several
meningioma patients. For example, out of 29 samples showing
nucleotide alterations, 13 meningiomas had changes in codon 72
(44.8%) when compared to blood DNA and database sequences (IARC T53
Database http://p53.iarc.fr/TP53GermlineMutations.aspx), making
this change highly represented in meningioma. This frequent change
is a well-known polymorphism CGC to CCC, that was recently revised
and categorized as a missense, resulting in amino acid change of
ARG to PRO frequently found in different tumor types (c.215G>C;
p.R72P; g.7676154C>G, Variant ID: 0000487638). The mutational
frequency distributed across codons, their consequences and
distribution to meningioma subtypes, are illustrated in Fig. 2.
Another highly represented mutation was at codon 62
(c.186delA; p.E62del; Variant ID: 0000487618, g.7579501del) where
meningiomas showed deletion of nucleotide A when compared to normal
sequence reported in databases (GAA coding for Glu). This mutation
was also represented in 44.8% of total number of mutations found
introducing a potential frameshift in meningioma patients. Other
nucleotide substitutions that we identified and listed by the
observed frequency included: Five substitutions on codon 105 (GGC
to GGT; c.315C>T; p.G105G; g.7676054G>A, Variant ID:
0000487634) making this a silent mutation; four insertions on codon
70 (G was inserted in tumor sequence; c.209_210insG; p.A70ins;
g.7676159_7676160ins1, Variant ID: 0000497667) introducing
frameshift; three missense on codon 64 (CCC to GCC; c.190C>G;
p.P64A; g.7676179G>C; Variant ID: 0000487614); three changes on
codon 82 (one missense (245C>T; p.P82L, g.7676124G>A; Variant
ID: 0000487640), one deletion (245delC; p.P82del, g.7676124del1;
Variant ID: 0000497669) and one insertion (243_244insA; p.P82ins;
g.7579444dup; Variant ID: 0000489041)); three silent on codon 104
(c.312G>A; p.Q104Q; g.7676057C>T, Variant ID: 0000487633),
two missense on codon 108 (GGT to CGT; c.322G>C; p.G108R;
g.7676047C>G, Variant ID: 0000487662), two silent on codon 71
(CCC to CCA, c.213C>A; p.P71P; g.7676156G>T, Variant ID:
0000487619), two missense on codon 73 (GTG to ATG, c.217G>A;
p.V73M; g.7676152C>T, Variant ID: 0000487629), codon 91 (TGG to
CGG, c.271T>C; p.W91R; g.7676098A>G, Variant ID: 0000487646)
and codon 100 (CAG to CAT, c.300G>T; p.Q100H; g.7676069C>A,
Variant ID: 0000487631). Finally, codons 68 (insertion of T
nucleotide, GAG to GTA; c.202_203insT; p.E68ins, frameshift,
Variant ID: 0000489055), 77 (CCA to TCA, c.229C>T; p.P77S,
missense; g.7676140G>A, Variant ID: 0000487639), 88 (GCC to GGC;
c.263C>G; p.A88G, missense; g.7676106G>C, Variant ID:
0000487644) and 92 (TGG to CGG, c.276C>A; p.P92P, silent;
g.7676093G>T, Variant ID: 0000487622) were changed only once
(Fig. 2A). We submitted our variants
to LOVD (Leiden Open Variation Database) and can be viewed at the
following URL: https://databases.lovd.nl/shared/users/03344. All
nucleotide alterations that we found were already listed in the
TP53 mutation database (http://p53.iarc.fr/TP53GeneVariations.aspx) but not in
connection to meningioma except for codon 72.
There were altogether 27/58 (46.6%) nucleotide
alterations representing missense mutations, 20/58 (34.5%)
frameshift mutations and 11/58 (19%) silent mutations (Fig. 2B). We defined frameshifts as
truncating mutations, that may lead to non-functional p53, based on
previous studies (35–37).
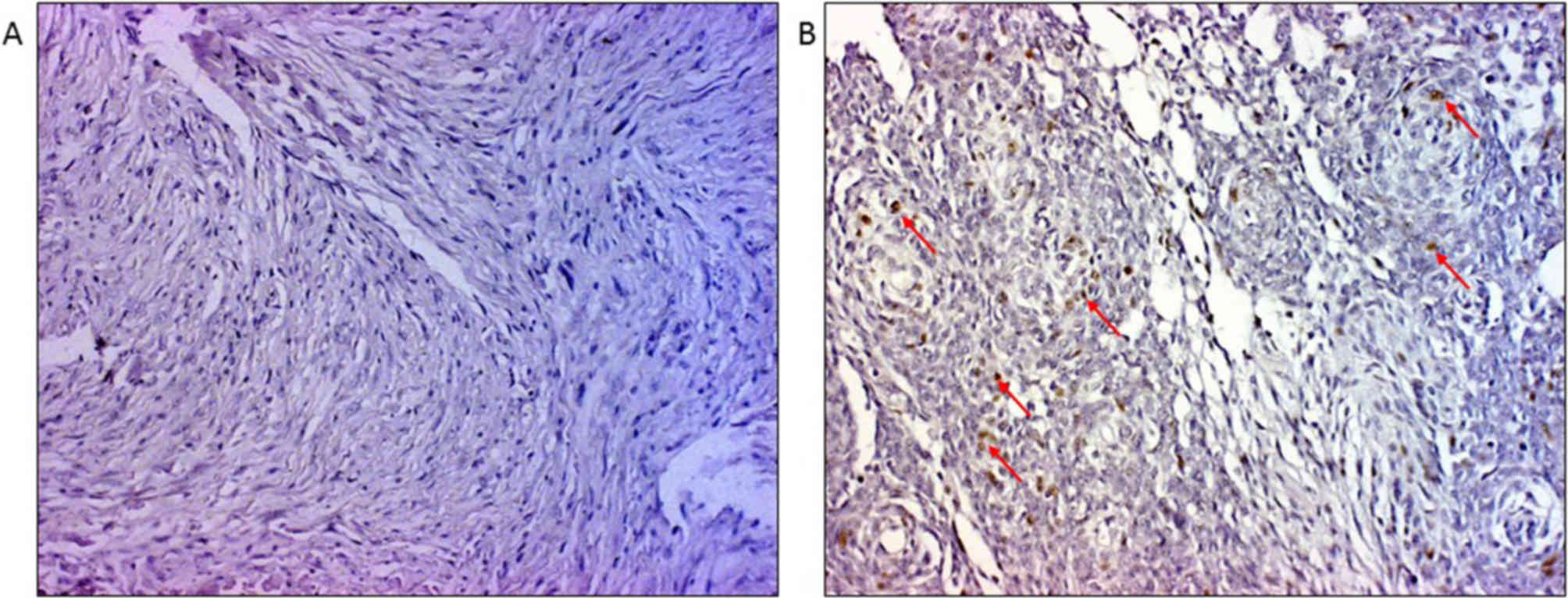
Immunohistochemistry brought additional data on the
influence of TP53 mutational status to protein levels. There
were altogether 47.8% of samples with p53 positive immunostain
(Fig. 3A and B). However, when the
positivity was distributed we have found that the group of samples
harboring mutations expressed significantly less protein (P=0.04;
Fig. 2D). We further inspected if
mutations of specific codons influenced p53 positivity and found
that codon 105 was significantly associated to the lack of p53
(P=0.031) while codon 62 showed the same trend (P=0.065). Finally,
we observed that 63% of meningiomas harboring missense and
frameshift mutations did not express p53 protein.
Association to histopathology, sex and
age
We also wanted to test if the specific type of
nucleotide alterations were associated to a specific meningioma
histopathology or WHO grades. The results of statistical analysis
obtained on our total sample demonstrate that significant
differences in mutations were not associated to any specific
meningioma subtype or grade (Chi-Square=1.022; P=0.336). Although
meningothelial and transitional subtypes showed the highest numbers
of both, codon 72 and 62 changes, and transitional subtype harbored
the highest number of codon 105 changes, these values were not
statistically significant (Fig.
2C).
Furthermore, possible associations to sex or age
patterns were also inspected. The difference in overall mutational
frequencies was statistically significantly associated to females.
Women exhibited significantly higher frequency of alterations when
compared to males (Chi square=3.802; sig=0.049). When analyzing the
most frequently affected codons separately, it has been shown that
females had significantly more alterations of codon 62 than male
patients (Chi square=10.447; sig=0.001). We further compiled two
large age categories, one consisting of individuals 63 years or
younger (n=20), and the other of individuals older than 63 years
(n=28), but observed that nucleotide alterations were not
statistically associated to any specific age group.
In silico prediction of mutational
consequences
In order to be able to explain the possible
consequences of the observed mutations we performed the evaluation
of pathogenicity with the PolyPhen-2 and ClinVar and the prediction
if amino acid substitutions affect protein function with SIFT and
Mutation Taster2 softwares (33,38). The
obtained results were similar between the databases with few
exceptions. The majority of alterations were not validated (87.5%).
Transcriptional activity was classified as partially functional in
15.5% and functional in 8.6% of total mutations observed. There
were 44.4% of alterations classified as benign and also 44.4% as
deleterious and possibly damaging while 11.2% were
undetermined.
The alteration at codon 72 (c.215G>C; p.R72P) was
validated and characterized by PolyPhen-2 and Mutation Taster2 as
benign, but IARC p53 database characterizes this change as a
missense and potentially disease causing. Alterations at codons 62
(c.186delA; p.E62del) and 71 (c.213C>A; p.P71P) were
nonvalidated and with unknown functional consequences. The changes
at codons 64 (c.190C>G; p.P64A), 100 (c.300G>T; p.Q100H), 77
(c.229C>T; p.P77S); 88 (c.263C>G; p.A88G); 73 (c.217G>A;
p.V73M) and 68 (c.202_203insT; p.E68ins) were all nonvalidated,
transcriptionally functional and benign. Codon 108 (c.322G>C;
p.G108R) harbored nonvalidated, transcriptionally partially
functional, but deleterious disease causing alteration found by
SIFT, PolyPhen-2 and Mutation Taster2, while codon 91 (c.271T>C;
p.W91R) harbored possibly damaging alteration found by both
PolyPhen-2 and Mutation Taster2. Mutation Taster2 furthermore
characterized mutations on codons 104 (c312G>A); 105
(c.315C>T; p.G105G); 70 (c.209_210insG; p.A70ins) and 92
(c.276C>A; p.P92P) as possibly disease causing. Of the three
changes on codon 82, the missense (245C>T; p.P82L) was
nonvalidated, transcriptionally functional and benign, while the
deletion (245delC; p.P82del) was predicted as disease causing by
Mutation Taster2 and insertion (243_244insA; p.P82ins) was
characterized as possibly damaging by PolyPhen-2.
Discussion
Loss of p53 suppressor function is a common event in
the development of a wide variety of human tumors and contributes
to an increase in genetic instability and metastatic potential. It
is implicated in many types of cancer and its mutations are present
in substantial number of all sporadic human cancers examined
(18,36,39).
However, its role in intracranial meningiomas has not been satiable
explained.
In the present study we have demonstrated the
constant and relatively high frequency of TP53's mutations
in its exon 4. It has been reported that the most common
TP53 mutations are missense mutations representing 80% of
all mutations reported and this was also confirmed with our study.
Most missense TP53 mutations disrupt p53 DNA binding. The
strong selection for TP53 missense mutations can be
explained with the production of mutant p53 proteins. It has been
shown that such mutant proteins have lost wild-type p53 tumor
suppression functions and acquired the oncogenic gain-of-function
that enables them to inactivate other p53 family members, for
instance tumor proteins p63 and p73 (40) and promote tumor progression and
metastasis. It is also important to take into account that a
fraction of TP53 mutations represent frameshift mutations
that ultimately lead to nonsense that will produce truncated p53
proteins with impaired wild-type function (18).
Mutation analysis of exon 4 of TP53 gene that
we conducted provided valuable information on p53 protein behavior
in intracranial meningioma. Our investigation demonstrates that
mutations that impact the p53's transactivation domain and DNA
binding domain are present in our group of tumors, which represents
novel result for meningioma and further confirmation on the
importance of this gene region. DNA binding domain (DBD, residues
102–292) is an important domain by which the protein binds to DNA
is located in the central part of the protein and is partially
coded by exon 4. Nevertheless, exon 4 also codes for the
transactivation domain (AD2, residues 43–92) of p53 protein, in
which functionally important proline rich segments lie (PRD,
residues 64–92) (5,41). The reason for choosing to study this
exon is primarily because it shows high frequency of mutations in
other cancer types, and because it is the only TP53's exon
that partially codes for 3 different functionally important domains
of p53 protein and also possesses an internal promoter (41). Furthermore, Verheijen et al
(42) indicated the involvement of
exon 4 in meningioma since different migration patterns were
evidenced in 10 out of 17 investigated cases by PCR-SSCP. We found
nucleotide alterations in 60.42% of investigated meningiomas. This
relatively high frequency was primarily distributed to codons 72
and 62. A total of 32.5% of meningioma investigated by our study
showed nucleotide alterations in codons 72 and 62 (each making
44.8% of total mutations found) when compared to blood DNA and
database sequences (IARC T53 Database http://p53.iarc.fr/TP53GeneVariations.aspx; p53
Knowledge base http://p53.bii.a-star.edu.sg/index.php), making these
changes highly represented in meningioma. The codon 72 is a known
single nucleotide polymorphism reviewed by expert panel and
validated, which results in Arg72Pro amino acid change
(c.215G>C; p.R72P; rs1042522) and frequently differs in tumors
(43). The codon is coding within
the polyproline domain of p53 protein well known for harboring many
tumor-associated mutations. The resulting structural variant of the
protein differs in molecular functions since it has been shown that
this proline-rich region is important for p53's apoptotic role and
the Arg variant has stronger ability to induce apoptosis. Also, it
has been proposed that the Arg variant binds and inactivates p73
more efficiently thus improving p53's suppressive functions. Of
note is that we have found 62% meningioma with Pro variant and 38%
with Arg variant. Many studies indicate that this polymorphism
could be linked to cancer susceptibility to lung, breast, colon and
gastric cancers and this change was recently reevaluated and
classified as missense and potentially linked to disease by IARC
database. Its differences in ethnicity and response to
chemotherapeutic treatments has also been documented (44,45).
Chang et al (16) also report
on the variability of the codon 72 found among meningioma cases
with allelic frequencies of 45.6% (Arg72/Arg72), 44.6%
(Arg72/Pro72), and 9.8% (Pro72/Pro72).
The deletion of nucleotide A at codon 62
(c.186A>del; p.E62) that we observed in 27.1% of patients, and
which represented 44.8% of total mutational burden, could introduce
a potential frameshift and truncation of the protein in meningioma
patients. We confirmed this finding at the protein level where the
majority of patients with this deletion lacked p53 protein
expression. Other nucleotide substitutions were found on codon 105
in 10.42% of meningiomas (c.315C>T; silent mutation); 8.3%
insertions on codon 70 (c.209_210insG) introducing frameshift;
6.25% on codons 64 (c.190C>G; missense); 82 (245C>T; 245delC;
and 243_244insA) and 104 (c.312G>A) each; 4.2% on codons 108
(c.322G>C), 71 (c.213C>A), 73 (c.217G>A;), 91
(c.271T>C) and 100 (c.300G>T) each. Codons that harbored only
one nucleotide change (2.1%) were codons 68 (c.202_203insT), 77
(c.229C>T), 88 (c.263C>G) and 92 (c.276C>A). Although when
we compared our findings with the different databases: IARC
TP53 mutation database, (http://p53.iarc.fr/TP53GeneVariations.aspx), Ensamble
(https://www.ensembl.org/Homo_sapiens/Gene/Sequence?db=core;g=ENSG00000141510;r=17:7661779-7687550)
and COSMIC (https://cancer.sanger.ac.uk/cosmic) we observed that
the alterations were already reported but (except for the codon 72)
not for meningiomas. Altogether we have found 46.6% of missense
alterations, 34.5% of frameshift and 19% of silent mutations.
Variants novel for meningioma we reported to Leiden Open Variation
Database (LOVD).
The evaluation of pathogenicity and protein function
showed that the majority of alterations were not validated.
However, further investigation on functional predictions showed
that seven alterations (in codons 82, 91, 108, 104, 105, 70 and 92)
were characterized as deleterious and possibly damaging by
PolyPhen-2 and Mutation Taster2 tools amounting to 44.4% of total
mutations. Predicted benign alterations also numbered 44.4%. There
were 24.1% mutations whose transcriptional activity was evaluated
as functional or partially functional.
Since mutations of TP53 (17q) have been
reported to be rare in meningiomas, a general view emerged that
other molecular regulators of the p53 pathway are the ones
mutationally targeted and responsible (20,46).
Some authors propose that p53 aberrations in meningiomas are
probably related to mechanisms controlling p53 rather than
affecting the gene itself (47).
Thus most probable candidates being tumor suppressors
CDKN2A/p16INKa (encoding p16), p14ARF (encoding p14), and
CDKN2B/p15ARF (encoding p15). p14 is involved with regulating cell
apoptosis through modulation of the p53 pathway, and p16 and p15
control cell cycle progress through the G1/S-phase checkpoint
(48). However, there are opposing
papers reporting on constantly found point mutations (9,13,49) and
COSMIC reports on three missense substitutions in TP53 in
meningioma. Cho et al (12)
investigated exons 5 to 8 of the TP53 gene by SSCP and found
7 mutations (38.9%). Two meningioma cases had mutations on exon 5,
three on exon 7 and two on exon 8. Based on the present sudy we
believe that the number of cases studied specifically for
TP53 mutations across literature is rather low targeting
only several exons and therefore account for such conclusions.
Several studies collectively showed that p53
positivity is correlated with the meningioma grade (12,13,24,25,47,50–52).
Positive p53 protein expression was found in 77% of grade III
meningiomas (53) and meningiomas
with higher malignancy grades displayed higher frequency of nuclear
p53 positivity (26). Similarly,
studies by Amatya et al (15)
and Csonka et al (54) found
a higher staining intensity in the high-grade tumors which
suggested that the p53 activation might be associated to tumor
progression. Our own previous analyses showed that p53 positivity
was significantly associated with higher meningioma grades. We
speculated that the positive nuclear signal was indicative of
mutant p53 proteins because it has been shown that they are more
stable and generally highly expressed in cancer (6). However, present mutational analysis
demonstrated that the group of samples harboring mutations
expressed significantly less protein (P=0.04; Fig. 2D) from the group without mutations,
indicating that mutational gain-of-function could not be attributed
to meningioma, but rather the classical behavior of tumour
suppressor loss. In the present study codons 72 and 62 were most
frequently changed in both meningothelial and transitional
subtypes, while codon 105 in the transitional subtype, but the
observed frequencies were not statistically significantly
associated to meningioma histopathology, nor to their grades,
probably because of small number of meningiomas with grades II and
III.
Our previous results clearly demonstrated that the
inactivation of tumor suppressor gene TP53 in meningioma was
not achieved through allelic loss (55). Although usual mode of action for
tumor suppressor genes is the loss of heterozygosity followed by
mutations, action mechanisms should not be generalized for all
suppressor genes and all tumors (38,56).
The p53's fundamental tumor suppressive role, as
well as its oncogenic mechanisms are still incompletely explained.
Mutations of TP53 and the consequent heterotetramerization
of wild-type and mutant p53 often stabilize the complex leading to
abrogation of normal p53 function in a cell with both mutant and
wild type alleles (12,24,25,47,50,51).
Moreover, isoforms of p53 family have been found in human arising
through alternative splicing, use of alternative translation site
or alternative promoter (19,57). The
prevailing base substitutions of TP53 and the resultant
inactivation of p53 allow evasion of apoptosis and rapid
carcinogenesis. Nevertheless, occurring variants may be substrate
for specific cancer risk.
Sex has been known to be an intrinsic risk factor
for meningeoma. The twice as high incidence observed in women as
compared to men is related to female sex hormones especially during
the reproductive life. We have found that women exhibited
significantly higher frequency of total nucleotide alterations when
compared to males (Chi square=3.802; sig=0.049). The analysis of
specific codon changes demonstrated that females had significantly
more alterations of codon 62 than male patients (Chi square=10.447;
sig=0.001). Although it has been demonstrated that the protective
function of the p53 protein declines with age (58), our results could not establish such a
connection, TP53 nucleotide alterations were not
significantly associated to age.
Although highly mutated in cancer, the spectrum and
the frequency of TP53 mutations vary between tumor types.
There are also views that each tumor-associated TP53
missense amino acid change will have a unique effect on p53
structure, its binding to the DNA and its other interactions. Thus,
mutant p53 proteins can be regarded as a heterogeneous group of
proteins with various degrees of normal tumor suppressor function
loss and various degrees of gain of oncogenic properties (18).
In conclusion, the present study shows that
nucleotide alterations found in TP53's exon 4 are much more
frequent than previously reported. This exon partially codes for 3
functionally important domains of p53 protein and highly
represented changed codons could be responsible but need to be
functionally validated. Our results contribute to meningioma
genetic profile.
Acknowledgements
Not applicable.
Funding
The present study was funded by the Croatian Science
Foundation (grant no. 6625) and the Scientific Centre of Excellence
for Basic, Clinical and Translational Neuroscience (project
‘Experimental and clinical research of hypoxic-ischemic damage in
perinatal and adult brain’; grant no. GA KK01.1.1.01.0007; funded
by the European Union through the European Regional Development
Fund).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
AB and NPŠ designed the current study. AB, AK, RH,
TV and NPŠ acquired the data. AB, TV and NPŠ analyzed the data. AB,
RH, TV and NPŠ interpreted the data. AB and AK performed the
experiments. AB, AK, RH and NPŠ wrote the manuscript. AB, AK, RH,
TV and NPŠ revised the manuscript for intellectual content. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All procedures of the current study involving human
participants were performed in accordance with the ethical
standards of the Institutional and National Research Committee and
with the Declaration of Helsinki. Ethical approval was received
from the Ethical Committee of the School of Medicine, University
Zagreb (approval no. 380-59-10106-14-55/147; Class,
641-01/14-02/01,1. 07. 2014) and the University Hospital Center
‘Sisters of Charity’ (approval no. EP-7426/14-9,11. 06. 2014).
Informed consent was obtained from all individual participants
included in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Domingues P, González-Tablas M, Otero Á,
Pascual D, Ruiz L, Miranda D, Sousa P, Gonçalves JM, Lopes MC,
Orfao A and Tabernero MD: Genetic/molecular alterations of
meningiomas and the signaling pathways targeted. Oncotarget.
6:10671–10688. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pećina-Šlaus N, Nikuseva Martic T, Tomas
D, Beros V, Zeljko M and Cupic H: Meningiomas exhibit loss of
heterozygosity of the APC gene. J Neurooncol. 87:63–70. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
El-Habr EA, Levidou G, Trigka EA,
Sakalidou J, Piperi C, Chatziandreou I, Spyropoulou A, Soldatos R,
Tomara G, Petraki K, et al: Complex interactions between the
components of the PI3K/AKT/mTOR pathway, and with components of
MAPK, JAK/STAT and Notch-1 pathways, indicate their involvement in
meningioma development. Virchows Arch. 465:473–485. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Al-Khalaf HH, Lach B, Allam A, Lakhani A,
Alrokayan SA and Aboussekhra A: The p53/p21 DNA damage-signaling
pathway is defective in most meningioma cells. J Neurooncol.
83:9–15. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sorrell AD, Espenschied CR, Culver JO and
Weitzel JN: Tumor protein p53 (TP53) testing and Li-Fraumeni
syndrome: Current status of clinical applications and future
directions. Mol Diagn Ther. 17:31–47. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Muller PA and Vousden KH: Mutant p53 in
cancer: New functions and therapeutic opportunities. Cancer Cell.
25:304–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ohgaki H, Eibl RH, Schwab M, Reichel MB,
Mariani L, Gehring M, Petersen I, Höll T, Wiestler OD and Kleihues
P: Mutations of the p53 tumor suppressor gene in neoplasms of the
human nervous system. Mol Carcinog. 8:74–80. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ellison DW, Lunec J, Gallagher PJ, Steart
PV, Jaros E and Gatter KC: Accumulation of wild-type p53 in
meningiomas. Neuropathol Appl Neurobiol. 21:136–142. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Joachim T, Ram Z, Rappaport ZH, Simon M,
Schramm J, Wiestler OD and von Deimling A: Comparative analysis of
the NF2, TP53, PTEN, KRAS, NRAS and HRAS genes in sporadic and
radiation-induced human meningiomas. Int J Cancer. 94:218–221.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Das A, Tan WL and Smith DR: p53 point
mutation is rare in meningiomas from Singaporean patients. Asian J
Surg. 28:7–10. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pykett MJ, Landers J and George DL:
Expression patterns of the p53 tumor suppressor gene and the mdm2
proto-oncogene in human meningiomas. J Neurooncol. 32:39–44. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cho H, Ha SY, Park SH, Park K and Chae YS:
Role of p53 gene mutation in tumor aggressiveness of intracranial
meningiomas. J Korean Med Sci. 14:199–205. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang JL, Zhang ZJ, Hartman M, Smits A,
Westermark B, Muhr C and Nistér M: Detection of TP53 gene mutation
in human meningiomas: A study using immunohistochemistry,
polymerase chain reaction/single-strand conformation polymorphism
and DNA sequencing techniques on paraffin-embedded samples. Int J
Cancer. 64:223–228. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ohkoudo M, Sawa H, Hara M, Saruta K, Aiso
T, Ohki R, Yamamoto H, Maemura E, Shiina Y, Fujii M and Saito I:
Expression of p53, MDM2 protein and Ki-67 antigen in recurrent
meningiomas. J Neurooncol. 38:41–49. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Amatya VJ, Takeshima Y and Inai K:
Methylation of p14(ARF) gene in meningiomas and its correlation to
the p53 expression and mutation. Mod Pathol. 17:705–710. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chang Z, Guo CL, Ahronowitz I,
Stemmer-Rachamimov AO, MacCollin M and Nunes FP: A role for the p53
pathway in the pathology of meningiomas with NF2 loss. J
Neurooncol. 91:265–270. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang X, Gejman R, Mahta A, Zhong Y, Rice
KA, Zhou Y, Cheunsuchon P, Louis DN and Klibanski A: Maternally
expressed gene 3, an imprinted noncoding RNA gene, is associated
with meningioma pathogenesis and progression. Cancer Res.
70:2350–2358. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bykov VJN, Eriksson SE, Bianchi J and
Wiman KG: Targeting mutant p53 for efficient cancer therapy. Nat
Rev Cancer. 18:89–102. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Marcel V, Nguyen Van Long F and Diaz JJ:
40 years of research put p53 in translation. Cancers (Basel).
10:E1522018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Riemenschneider MJ, Perry A and
Reifenberger G: Histological classification and molecular genetics
of meningiomas. Lancet Neurol. 5:1045–1054. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Louis DN, Perry A, Reifenberger G, von
Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD,
Kleihues P and Ellison DW: The 2016 World Health Organization
classification of tumors of the central nervous system: A summary.
Acta Neuropathol. 131:803–820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fathi AR and Roelcke U: Meningioma. Curr
Neurol Neurosci Rep. 13:3372013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
He S, Pham MH, Pease M, Zada G, Giannotta
SL, Wang K and Mack WJ: A review of epigenetic and gene expression
alterations associated with intracranial meningiomas. Neurosurg
Focus. 35:E52013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pavelin S, Becic K, Forempoher G, Mrklic
I, Pogorelic Z, Titlic M and Andelinovic S: Expression of Ki-67 and
p53 in meningiomas. Neoplasma. 60:480–485. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Narla S, Uppin MS, Saradhi MV, Sahu BP,
Purohit AK and Sundaram C: Assessment of expression of epidermal
growth factor receptor and p53 in meningiomas. Neurol India.
62:37–41. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Trott G, Pereira-Lima JF, Leães CG,
Ferreira NP, Barbosa-Coutinho LM and Oliveira MC: Abundant
immunohistochemical expression of dopamine D2 receptor and p53
protein in meningiomas: Follow-up, relation to gender, age, tumor
grade, and recurrence. Braz J Med Biol Res. 48:415–419. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Preusser M, Brastianos PK and Mawrin C:
Advances in meningioma genetics: Novel therapeutic opportunities.
Nat Rev Neurol. 14:106–115. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang N and Osswald M: Meningiomas:
Overview and new directions in therapy. Semin Neurol. 38:112–120.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pereira BJA, Oba-Shinjo SM, de Almeida AN
and Marie SKN: Molecular alterations in meningiomas: Literature
review. Clin Neurol Neurosurg. 176:89–96. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pećina-Šlaus N: Merlin the NF2 gene
product. Pathol Oncol Res. 19:365–373. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pećina-Šlaus N, Kafka A and Lechpammer M:
Molecular genetics of intracranial meningiomas with emphasis on
canonical Wnt signalling. Cancers (Basel). 8:E672016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sim NL, Kumar P, Hu J, Henikoff S,
Schneider G and Ng PC: SIFT web server: Predicting effects of amino
acid substitutions on proteins. Nucleic Acids Res. 40((Web Server
Issue)): W452–W457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Olivier M, Langerød A, Carrieri P, Bergh
J, Klaar S, Eyfjord J, Theillet C, Rodriguez C, Lidereau R, Bièche
I, et al: The clinical value of somatic TP53 gene mutations in
1,794 patients with breast cancer. Clin Cancer Res. 12:1157–1167.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lindenbergh-van der Plas M, Brakenhoff RH,
Kuik DJ, Buijze M, Bloemena E, Snijders PJ, Leemans CR and
Braakhuis BJ: Prognostic significance of truncating TP53 mutations
in head and neck squamous cell carcinoma. Clin Cancer Res.
17:3733–3741. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Omura G, Ando M, Ebihara Y, Saito Y,
Kobayashi K, Fukuoka O, Akashi K, Yoshida M, Asakage T and Yamasoba
T: The prognostic value of TP53 mutations in hypopharyngeal
squamous cell carcinoma. BMC Cancer. 17:8982017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bouaoun L, Sonkin D, Ardin M, Hollstein M,
Byrnes G, Zavadil J and Olivier M: TP53 variations in human
cancers: New Lessons from the IARC TP53 Database and Genomics Data.
Hum Mutat. 37:865–876. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bieging KT, Mello SS and Attardi LD:
Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev
Cancer. 14:359–730. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Walerych D, Lisek K and Del Sal G: Mutant
p53: One, no one, and one hundred thousand. Front Oncol. 5:2892015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Harms KL and Chen X: The functional
domains in p53 family proteins exhibit both common and distinct
properties. Cell Death Differ. 13:890–897. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Verheijen FM, Sprong M, Kloosterman JM,
Blaauw G, Thijssen JH and Blankenstein MA: TP53 mutations in human
meningiomas. Int J Biol Markers. 17:42–48. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Faria MH, Neves Filho EH, Alves MK,
Burbano RM, de Moraes Filho MO and Rabenhorst SH: TP53 mutations in
astrocytic gliomas: An association with histological grade, TP53
codon 72 polymorphism and p53 expression. APMIS. 120:882–889. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Grochola LF, Zeron-Medina J, Mériaux S and
Bond GL: Single-nucleotide polymorphisms in the p53 signaling
pathway. Cold Spring Harb Perspect Biol. 2:a0010322010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Naccarati A, Polakova V, Pardini B,
Vodickova L, Hemminki K, Kumar R and Vodicka P: Mutations and
polymorphisms in TP53 gene-an overview on the role in colorectal
cancer. Mutagenesis. 27:211–218. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Choy W, Kim W, Nagasawa D, Stramotas S,
Yew A, Gopen Q, Parsa AT and Yang I: The molecular genetics and
tumor pathogenesis of meningiomas and the future directions of
meningioma treatments. Neurosurg Focus. 30:E62011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Terzi A, Saglam EA, Barak A and
Soylemezoglu F: The significance of immunohistochemical expression
of Ki-67, p53, p21, and p16 in meningiomas tissue arrays. Pathol
Res Pract. 204:305–314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Boström J, Meyer-Puttlitz B, Wolter M,
Blaschke B, Weber RG, Lichter P, Ichimura K, Collins VP and
Reifenberger G: Alterations of the tumor suppressor genes CDKN2A
(p16(INK4a)), p14(ARF), CDKN2B (p15(INK4b)), and CDKN2C
(p18(INK4c)) in atypical and anaplastic meningiomas. Am J Pathol.
159:661–669. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mashiyama S, Murakami Y, Yoshimoto T,
Sekiya T and Hayashi K: Detection of p53 gene mutations in human
brain tumors by single-strand conformation polymorphism analysis of
polymerase chain reaction products. Oncogene. 6:1313–1318.
1991.PubMed/NCBI
|
|
50
|
Matsuno A, Nagashima T, Matsuura R, Tanaka
H, Hirakawa M, Murakami M, Tamura A and Kirino T: Correlation
between MIB-1 staining index and the immunoreactivity of p53
protein in recurrent and non-recurrent meningiomas. Am J Clin
Pathol. 106:776–781. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Nagashima G, Aoyagi M, Yamamoto M,
Yamamoto S, Wakimoto H, Ohno K, Yamamoto K and Hirakawa K: P53
overexpression and proliferative potential in malignant
meningiomas. Acta Neurochir (Wien). 141:53–61. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ozen O, Demirhan B and Altinörs N:
Correlation between histological grade and MIB-1 and p53
immunoreactivity in meningiomas. Clin Neuropathol. 24:219–224.
2005.PubMed/NCBI
|
|
53
|
Karamitopoulou E, Perentes E, Tolnay M and
Probst A: Prognostic significance of MIB-1, p53, and bcl-2
immunoreactivity in meningiomas. Hum Pathol. 29:140–145. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Csonka T, Murnyák B, Szepesi R, Kurucz A,
Klekner Á and Hortobágyi T: Poly(ADP-ribose) polymerase-1 (PARP1)
and p53 labelling index correlates with tumour grade in
meningiomas. Folia Neuropathol. 52:111–120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Pećina-Šlaus N, Kafka A, Vladušić T, Tomas
D, Logara M, Skoko J and Hrašćan R: Loss of p53 expression is
accompanied with upregulation of beta-catenin in meningiomas: A
concomitant reciprocal expression. Int J Exp Pathol. 97:159–169.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Leroy B, Fournier JL, Ishioka C, Monti P,
Inga A, Fronza G and Soussi T: The TP53 website: An integrative
resource centre for the TP53 mutation database and TP53 mutant
analysis. Nucleic Acids Res. 41((Database Issue)): D962–D969. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Marcel V and Hainaut P: p53 isoforms-a
conspiracy to kidnap p53 tumor suppressor activity? Cell Mol Life
Sci. 66:391–406. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Feng Z, Hu W, Teresky AK, Hernando E,
Cordon-Cardo C and Levine AJ: Declining p53 function in the aging
process: A possible mechanism for the increased tumor incidence in
older populations. Proc Natl Acad Sci USA. 104:16633–16638. 2007.
View Article : Google Scholar : PubMed/NCBI
|