Introduction
Colorectal cancer (CRC) is the third most common
cancer in men and the second in women, with 1.4 million estimated
cases worldwide, and 700,000 estimated deaths (1,2).
Familial adenomatous polyposis (FAP) is an autosomal dominant
pre-cancerous syndrome characterized by the development of hundreds
to thousands of colorectal adenomatous polyps that, if untreated,
lead to CRC in the third to fourth decade of life (3-5).
According to the European Medicines Agency, in 2009, there were
3-10/100,000 new cases in the European Union (6).
Germline pathogenic variants in the tumor suppressor
adenomatous polyposis coli (APC) gene are responsible for
FAP (7). De novo
pathogenetic variants in APC are also found in the majority
of sporadic cases of FAP (8,9). The
APC gene is located on chromosome 5q and encodes a 312 kDa
protein that is involved in several cellular processes, such as
cell migration, adhesion and cell cycle regulation, as well as
chromosome segregation, signal transduction and apoptosis (10,11).
The tumour suppressing activity of APC depends on its capacity to
regulate β-catenin levels in the nucleus. In fact, in the absence
of Wnt signalling, APC induces β-catenin degradation. If the
extracellular Wnt signal is absent, APC-induced β-catenin
degradation is inhibited, β-catenin accumulates in the nucleus and
modulates gene transcription. Pathogenic variants that disrupt APC
interaction with β-catenin are oncogenic (6-11).
The classical symptoms of FAP, including bleeding,
diarrhoea and abdominal pain, are generally diagnosed at around
20-25 years of age. Genetic testing is performed to verify the
clinical diagnosis and to identify asymptomatic carriers in
affected families. Early FAP detection by genetic screening in
at-risk families is crucial in order to implement effective
prophylaxis strategies. Moreover, the results of genotyping should
be considered together with clinical data to decide the type and
time of surgery (12).
Various symptomatic young patients with a family
history of FAP have been reported, however we describe a case of
severe paediatric FAP caused by a de novo pathogenic
variant.
Case report
The patient described herein is a ten-year-old boy
clinically diagnosed with FAP at the age of nine and recruited to
the Ambulatorio di Pediatria of AOU Federico II of Naples in
December 2016. The anamnesis revealed that symptoms, i.e. diarrhoea
and rectal bleeding, first appeared at the age of two and became
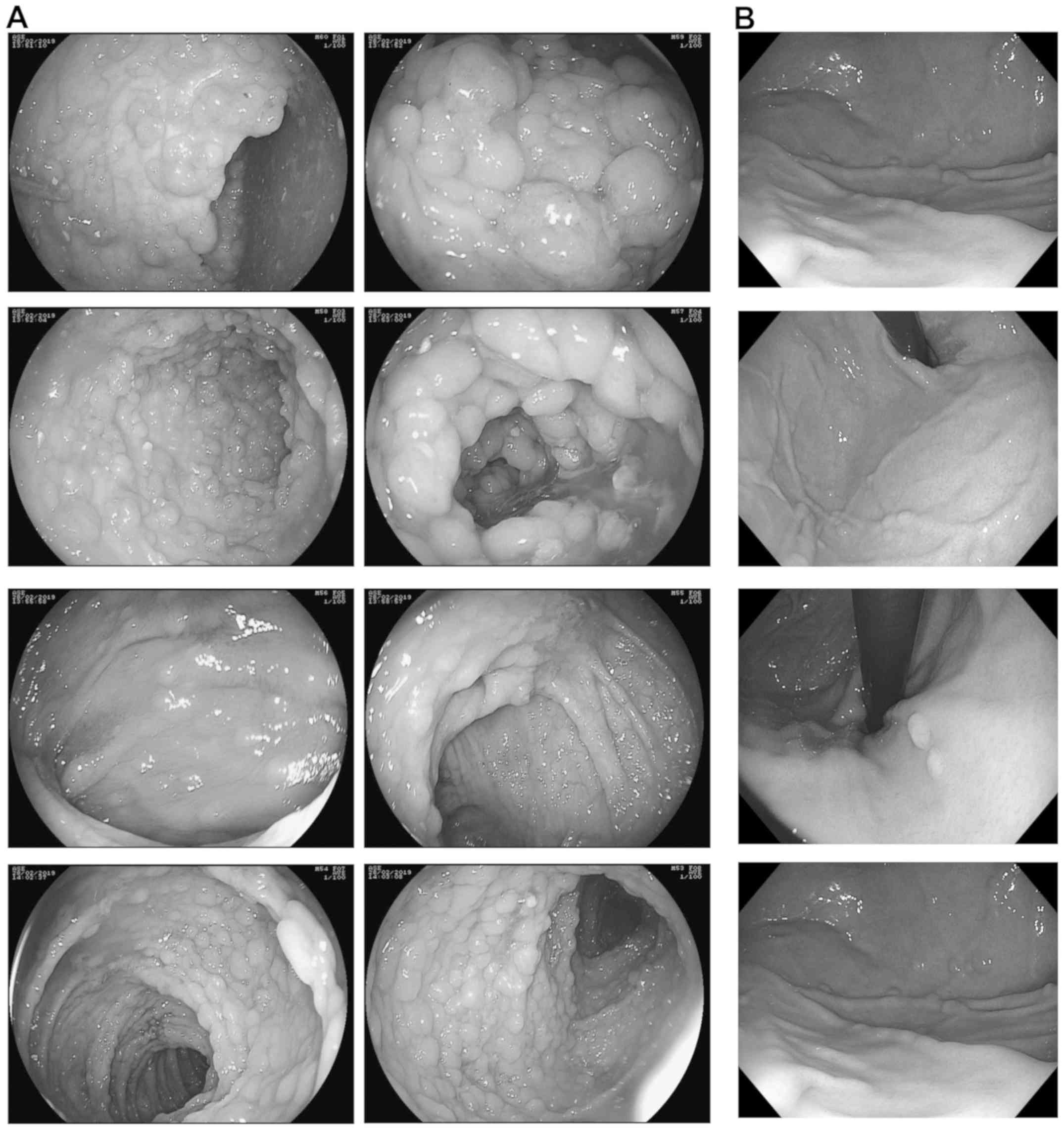
more frequent during late childhood. Colonoscopies and
esophagogastroduodenoscopy performed in December 2015, December
2016, March 2018 and February 2019 showed hundreds of subcentimetre
polyps throughout the colon (Fig.
1A) as well as polyps in the gastric fundus (>10 mm) and
body (<5 mm) (Fig. 1B),
respectively, thereby confirming the diagnosis of FAP. Histological
analysis revealed a Helicobacter pylori infection-negative
chronic gastritis-like inflammation of the stomach, inflammatory
infiltrate in the ileum and low grade dysplasia of the glandular
epithelium of the colon. Haematochemical analysis revealed
microcytic anaemia with a haemoglobin level of 10.3 g/dl and a mean
cellular volume of 66.5fl. The results of physical analysis were
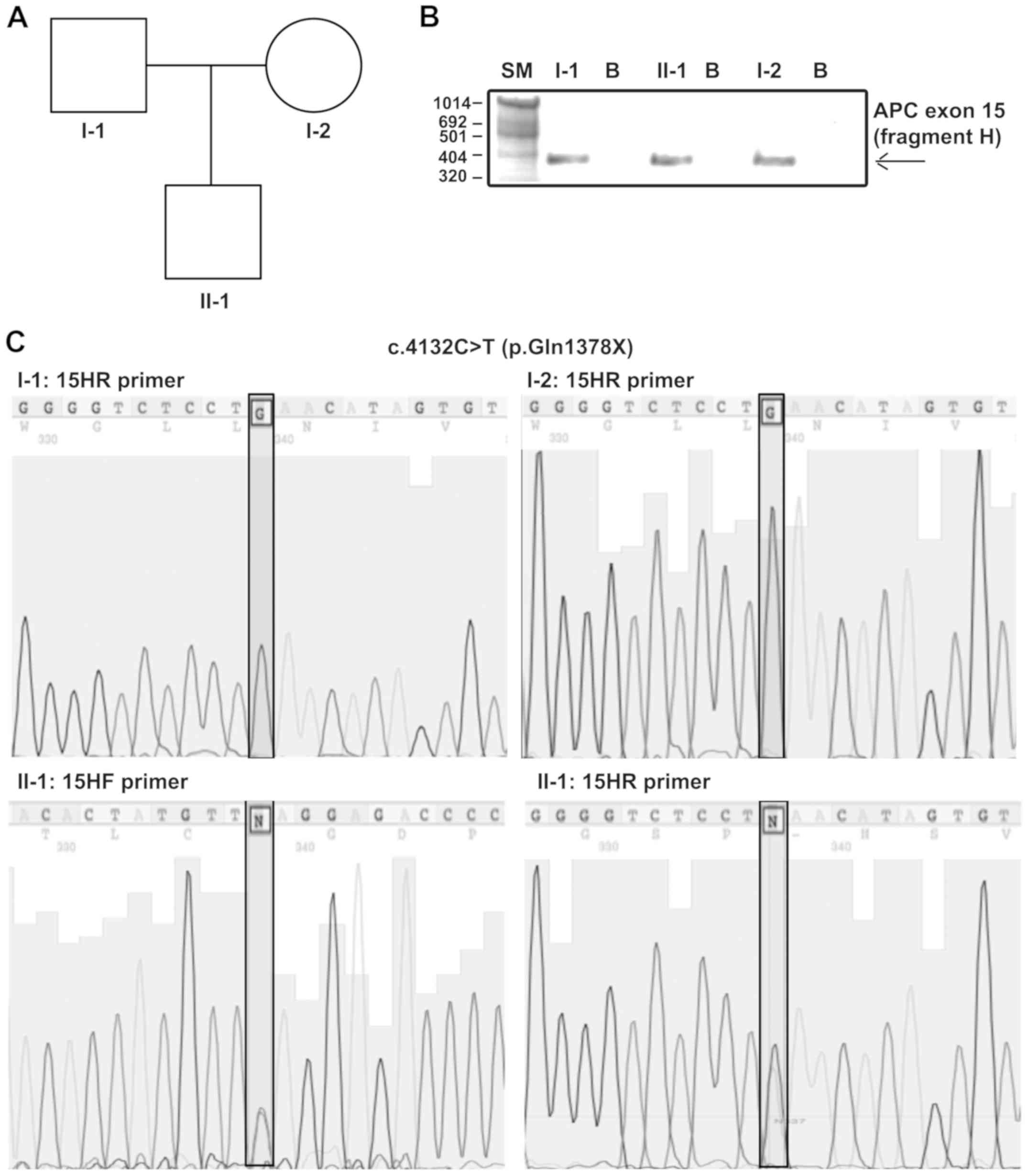
unremarkable. As shown in Fig. 2A,
there was no family history of FAP, other polyposis syndromes or
colon cancer. The patient's parents received clinical and genetic
counselling and provided informed consent to molecular
screening.
Genomic DNA was extracted from peripheral blood
lymphocytes of the proband and his parents as previously described
(13). Briefly, 2 ml of the
patient's blood were incubated at 37˚ in a red blood lysis buffer
(0.15 M NH4Cl2 and 0.17 M Tris-HCl, pH 7.65)
for 15 min and centrifuged. The lymphocyte pellet was resuspended
in a DNA extraction buffer (1 M Tris.HCl, 0.5 M EDTA and 5 M NaCl)
and digested with Proteinase K and 10% SDS at 60˚C for ten minutes.
After adding 6M NaCl, the sample was centrifuged, the DNA in the
supernatant was precipitated with absolute ethanol and resuspended
in an appropriate volume of deionized sterile water. The quality
and the quantity of the DNA was spectrophotometrically assessed
with the NanoDrop™ 2000 spectrophotometer (Thermo Fisher
Scientific, Inc.). An absorbance ratio at 260 and 280 nm in the
range of 1.8-2.0 was considered good for further analysis.
APC exons 1 to 15 were amplified by
polymerase chain reaction (PCR) using 100 ng of the proband genomic
DNA and primer pairs described by Groden et al (7) in a 50 µl reaction mixture (Table I). The amplification protocol was as
follows: 5 min at 95˚C, then 35 cycles of 20 sec at 95˚C, 30 sec at
60˚C and 45 sec at 72˚C, and a final extension of 5 min at 72˚C.
All reactions were run in the MyCycler thermal cycler (Bio-Rad).
Amplified fragments were run on a 1X agarose gel and visualized
with ethidium bromide, then purified using the QIAquick PCR
Purification Kit (Qiagen) according to the manufacturer's
recommendations and subjected to automated Sanger sequencing
(Fig. 2B and C).
 | Table IPrimer pairs used for the genetic
analysis of adenomatous poliposis coli exons 1 to 15. |
Table I
Primer pairs used for the genetic
analysis of adenomatous poliposis coli exons 1 to 15.
| Primers | Forward sequence
(5'-3') | Reverse sequence
(5'-3') |
|---|
| 1 FP/RP |
AGGTCCAAGGGTAGCCAAGG |
TAAAAATGGATAAACTACAATTAAAAG |
| 2 FP/RP |
AAATACAGAATCATGTCTTGAAGT |
ACACCTAAAGATGACAATTTGAG |
| 3 FP/RP |
TAACTTAGATAGCAGTAATTTCCC |
ACAATAAACTGGAGTACACAAGG |
| 4 FP/RP |
ATAGGTCATTGCTTCTTGCTGAT |
TGAATTTTAATGGATTACCTAGGT |
| 5 FP/RP |
CTTTTTTTGCTTTTACTATTAACG |
TGTAATTCATTTTATTCCTAATAGCTC |
| 6 FP/RP |
GGTAGCCATAGTATGATTATTTCT |
CTACCTATTTTTATACCCACAAAC |
| 7 FP/RP |
AAGAAAGCCTACACCATTTTTGC |
GATCATTCTTAGAACCATCTTGC |
| 8 FP/RP |
ACCTATAGTCTAAATTATACCATC |
GTCATGGCATTAGTGACCAG |
| 9 FP/RP |
AGTCGTAATTTTGTTTCTAAACTC |
TGAAGGACTCGGATTTCACGC |
| 9a FP/RP |
TCATTCACTCACAGCCTGATGAC |
GCTTTGAAACATGCACTACGAT |
| 10 FP/RP |
AAACATCATTGCTCTTCAAATAAC |
TACCATGATTTAAAAATCCACCAG |
| 10a FP/RP |
AGACTAGGACTGAGACATTAATCATC |
GGTGAGGAGTGAGAAGAAGGTAATC |
| 11 FP/RP |
GATGATTGTCTTTTTCCTCTTGC |
CTGAGCTATCTTAAGAAATACATG |
| 12 FP/RP |
TTTTAAATGATCCTCTATTCTGTAT |
ACAGAGTCAGACCCTGCCTCAAAG |
| 13 FP/RP |
TTTCTATTCTTACTGCCTAGCATT |
ATACACAGGTAAGAAATTAGGA |
| 14 FP/RP |
TAGATGACCCATATTCTGTTTC |
CAATTAGGTCTTTTTGAGAGTA |
| 15A FP/RP |
GTTACTGCATACACATTGTGAC |
GCTTTTTGTTTCCTAACATGAAG |
| 15B FP/RP |
AGTACAAGGATGCCAATATTATG |
ACTTCTATCTTTTTCAGAACGAG |
| 15C FP/RP |
ATTTGAATACTACAGTGTTACCC |
CTTGTATTCTAATTTGGCATAAGG |
| 15D FP/RP |
CTGCCCATACACATTCAAACAC |
TGTTTGGGTCTTGCCATCTT |
| 15E FP/RP |
AGTCTTAAATATTCAGATGAGCAG |
GTTTCTCTTCATTATATTTTATGCTA |
| 15F FP/RP |
AAGCCTACCAATTATAGTGAACG |
AGCTGATGACAAGATGATAATG |
| 15G FP/RP |
AAGAAACAATACAGACTTATTGTG |
ATGAGTGGGGTCTCCTGAAT |
| 15H FP/RP |
ATCTCCCTCCAAAAGTGGTGC |
TCCATCTGGAGTACTTTCTGTG |
| 15I FP/RP |
AGTAAATGCTGCAGTTCAGAGG |
CCGTGGCATATCATCCCCC |
| 15J FP/RP |
CCCAGACTGCTTCAAAATTACC |
GAGCCTCATCTGTACTTCTGA |
| 15K FP/RP |
CCCTCCAAATGAGTTACGTGA |
TTGTGGTATAGGTTTTACTGGTG |
| 15L FP/RP |
ACCCAACAAAAATCAGTTAGATG |
GTGGCTGGTAACTTTAGCCTC |
| 15M FP/RP |
ATGATGTTGACCTTTCCAGGG |
ATTGTGTAACTTTTCATCAGTTGC |
| 15N FP/RP |
AAAGACATACCAGACAGAGGG |
CTTTTTTGGCATTGCGGAGCT |
| 15O FP/RP |
AAGATGACCTGTTGCAGGAATG |
GAATCAGACGAAGCTTGTCTAGAT |
| 15P FP/RP |
CCATAGTAAGTAGTTTACATCAAG |
AAACAGGACTTGTACTGTAGGA |
| 15Q FP/RP |
CAGCCCCTTCAAGCAAACATG |
GAGGACTTATTCCATTTCTACC |
| 15R FP/RP |
CAGTCTCCTGGCCGAAACTC |
GTTGACTGGCGTACTAATACAG |
| 15S FP/RP |
TGGTAATGGAGCCAATAAAAAGG |
TGGGAGTTTTCGCCATCCAC |
| 15T FP/RP |
TGTCTCTATCCACACATTCGTC |
ATGTTTTTCATCCTCACTTTTTGC |
| 15U FP/RP |
GGAGAAGAACTGGAAGTTCATA |
TTGAATCTTTAATGTTTGGATTTGC |
| 15V FP/RP |
TCTCCCACAGGTAATACTCCC |
GCTAGAACTGAATGGGGTACG |
| 15W FP/RP |
CAGGACAAAATAATCCTGTCCC |
ATTTTCTTAGTTTCATTCTTCCTC |
The sequences analysis was performed by alignment
with those present in the GenBank database using the BLASTn
software (http://www.ncbi.nlm.nih.gov/blast/html). The accession
number of the used reference sequence was NM_000038.4. The same
procedure was followed for the genetic analysis of APC exon
15 (fragment H) on the DNA of both the patient's parents (Fig. 2B and C).
Genetic counselling excluded a family history of
adenomatous polyposis syndrome. Indeed, the patient's parents
reported that none of his first grade relatives, including
themselves, had symptoms correlated to FAP or to those developed by
the proband, such as chronic gastritis-like inflammation of the
stomach in the absence of Helicobacter pylori infection.
However, none of them underwent colonoscopy.
Sequence analysis of the APC gene revealed
the causative pathogenetic variant in the proband: A heterozygous C
to T transition in the exon 15H called c.4132 C>T (p.Gln1378X),
which changes the glutamine at codon 1378 in a premature stop codon
(Fig. 2C). The proband was also
carrier of the rs459552, [c.5465 T>A (p.Asp1822Val)]
polymorphism, in fragment L of exon 15 of the APC gene (data not
shown). Such polymorphism causes the substitution of a valine with
an aspartate at codon 1822 and is reported as benign in the ClinVar
database (https://www.ncbi.nlm.nih.gov). Furthermore, the
APC genetic carrier test for c.4132 C>T (p.Gln1378X)
mutation was negative in the proband's parents (Fig. 2C), whereas both carried the rs459552
polymorphism.
Discussion
Approximately 70-80% of FAP cases are caused by
inherited pathogenetic variants of the APC gene, whereas up
to 25% of cases are attributed to de novo germline
pathogenetic variants (3). Herein,
we report the case of a ten-year old boy, clinically diagnosed with
severe FAP, in the absence of a family history of polyposis.
Genetic analysis of APC confirmed the clinical diagnosis. In
fact, a stop codon variant, namely c.4132 C>T (p.Gln1378X) in
fragment H of exon 15, was identified as the cause of FAP.
Although pathogenetic APC germline variants
have a penetrance of ~100%, very close genotype-phenotype
correlations and marked heterogeneity in the phenotypic expression
of FAP are well known. Severe phenotypes, characterized by more
than 5,000 polyps and early onset of the disease are associated
with pathogenetic variants between codons 1250 and 1464, whereas
patients with classical FAP develop hundreds to thousands of
adenomatous polyps in the colorectum during the second and third
decades of life (14,15). Germline pathogenetic variants
between codons 168 and 1680 and deletion of entire APC locus
are responsible for classical FAP (4,16).
Attenuated phenotypes, characterized by a few polyps (~10-100) are
associated with pathogenetic variants at the extreme 5' or 3' end
of the APC gene or in alternatively spliced exon 9.
Alterations in the region between codons 1286 and 1513, i.e., the
mutation cluster region, probably provide a strong selective
advantage to tumour cells. Indeed, the majority of somatic variants
and germline APC variants are clustered in this region and
cause aggressive phenotype (4,16).
Thus, in accordance with previous studies, our patient, who carried
a pathogenetic truncating variant at codon 1378, had a very early
onset of the disease and severe FAP phenotype (17,18).
The c.4132 C>T variant is reported as somatic by Liu et
al (19) and in the
‘Catalogue of Somatic Mutations in Cancer (COSMIC)’
(http://cancer.sanger.ac.uk/cosmic/mutation/overview?id=18862).
However, Friedl and Aretz (20)
found this variant as germlinal in a FAP patient, but no
information about the patient's history was provided (20). Moreover, to date, no
genotype/phenotype correlations have been reported.
The patient also carried a second DNA variant,
namely, c.5465 T>A (Asp1822Val). This polymorphism, which was
also identified in the patient's parents, causes a valine/aspartate
substitution at codon 1822. Several studies have investigated the
role of polymorphisms in determining the risk of developing FAP or
CRC, hypothesizing that the common variants in CRC genes now
considered benign variants are, rather, low-risk alleles.
De Rosa et al (21) were the first to identify the
Asp1822Val polymorphism. They analysed the genotype of two
generations of individuals in ten families and concluded that it
was not a disease-causing variant since it was found almost with
the same frequency in normal and affected individuals, and it
didn't modify the phenotype in any of the FAP patients.
Furthermore, according to Fernández-Rozadilla et al
(22), Asp1822Val is unrelated to
CRC development and alleles T and A are equally distributed among
cases and controls (P=0.2197) (22). In contrast, in a case-control study
in which 1,785 CRC patients and 1,306 controls were analyzed,
Picelli et al (23) found
that the Asp1822Val polymorphism showed an odds ratio slightly
lower in subjects carrying CRC than in controls (OR=0.75,
CI=0.59-0.94), which suggested that the Asp1822Val substitution
could protect from CRC (23).
Finally, Wong et al (24)
evaluated the association between eight APC single
nucleotide polymorphisms and the risk of developing colorectal
adenoma and concluded that none of the single nucleotide
polymorphisms were associated with an increased risk. However, p.
Asp1822Val was reported to influence the risk of colorectal adenoma
in individuals with higher fat intake.
In conclusion, we found that the germline Gln1378X
APC variant is the cause of a very severe sporadic FAP in the
proband analyzed in the present study. Notably, the patient had the
initial typical symptoms of the disease at the age of two years,
although the clinical diagnosis was made when he was nine years
old. Young symptomatic FAP patients have been described previously,
but rarely in families with negative family history (25). We reported a severe case of FAP with
onset in the toddler years, which represents a de novo
pathogenetic variant (9). In both
cases, the very young age at initial presentation (~2 years), as
well as the absence of a family history of FAP, delayed the
diagnosis for many years. Therefore, we reiterate the importance of
endoscopic investigation in a child with iron-deficiency anaemia
and a history of rectal bleeding should undergo endoscopic
investigation notwithstanding no having a family history of
FAP.
Further studies are required to determine whether
Gln1378X and p.Val1822Asp play an additive role in his FAP
phenotypic manifestations or not.
Acknowledgements
The authors would like to thank Dr Jaen Ann Gilder
for the text editing.
Funding
The present study was supported by Università di
Napoli Federico II (CTB_ATENEO_2017_DR_3577/2017).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
MDR and PI designed the study. MDR, AC, AA and FC
performed genetic analysis. EM and MR provided sample collection
and clinical support. MDR and PI contributed to data
interpretation. MDR and AC wrote the manuscript, and FD and RL
critically revised the manuscript and participated in the analysis
and interpretation of the data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Written informed consent has been provided by the
proband's parents. All methods are part of the clinical practice
necessary to carry out the molecular analysis of the APC gene,
requested by the proband's parents. Furthermore, the proband's
parents agree that the DNA sample no longer needed for the study is
used for medical research purposes in an anonymous form and/or in
epidemiological cases. The procedures reported in this study were
performed in accordance with the rules of the Good Clinical
Practice Guidelines (GCP) and the ethical principles set out in the
Declaration of Helsinki. The study was also authorized by ‘Comitato
etico per le attività Biomediche-Carlo Romano of the University of
Naples Federico II’ (protocol no. 35/17).
Patient consent for publication
Not applicable. All identifying information, case
details, personal information or images that may enable an
individual to be identified, are not included in the text.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386.
2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
De Rosa M, Rega D, Costabile V, Duraturo
F, Niglio A, Izzo P, Pace U and Delrio P: The biological complexity
of colorectal cancer: Insights into biomarkers for early detection
and personalized care. Therap Adv Gastroenterol. 9:861–886.
2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ma H, Brosens LA, Offerhaus GJ, Giardiello
FM, de Leng WW and Montgomery EA: Pathology and genetics of
hereditary colorectal cancer. Pathology. 50:49–59. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Fearnhead NS, Britton MP and Bodmer WF:
The ABC of APC. Hum Mol Genet. 10:721–733. 2001.PubMed/NCBI View Article : Google Scholar
|
|
5
|
De Rosa M, Galatola M, Borriello S,
Duraturo F, Masone S and Izzo P: Implication of adenomatous
polyposis coli and MUTYH mutations in familial colorectal
polyposis. Dis Colon Rectum. 52:268–274. 2009.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Half E, Bercovich D and Rozen P: Familial
adenomatous polyposis. Orphanet J Rare Dis. 4(22)2009.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Groden J, Thliveris A, Samowitz W, Carlson
M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson
M, et al: Identification and characterization of the familial
adenomatous polyposis coli gene. Cell. 66:589–600. 1991.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Leoz ML, Carballal S, Moreira L, Ocaña T
and Balaguer F: The genetic basis of familial adenomatous polyposis
and its implications for clinical practice and risk management.
Appl Clin Genet. 8:95–107. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Auricchio R, De Rosa M, Quaglietta L,
Miele E, Boccia G, Staiano A and Izzo P: A dramatic case of
early-onset familial adenomatous polyposis. Clin Genet. 67:104–106.
2005.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Aoki K and Taketo MM: Adenomatous
polyposis coli (APC): A multi-functional tumor suppressor gene. J
Cell Sci. 120:3327–3335. 2007.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Polakis P: The adenomatous polyposis coli
(APC. tumor suppressor. Biochim Biophys Acta. 1332:F127–F147.
1997.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Dodaro C, Grifasi C, Florio J, Santangelo
ML, Duraturo F, De Rosa M, Izzo P and Renda A: The role of mutation
analysis of the APC gene in the management of FAP patients. A
controversial issue. Ann Ital Chir. 87:321–325. 2016.PubMed/NCBI
|
|
13
|
Galatola M, Paparo L, Duraturo F, Turano
M, Rossi GB, Izzo P and De Rosa M: Beta catenin and cytokine
pathway dysregulation in patients with manifestations of the ‘PTEN
hamartoma tumor syndrome’. BMC Med Genet. 13(28)2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Nagase H and Nakamura Y: Mutations of the
APC (adenomatous polyposis coli) gene. Hum Mutat. 2:425–434.
1993.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Gayther SA, Wells D, SenGupta SB, Chapman
P, Neale K, Tsioupra K and Delhanty JD: Regionally clustered APC
mutations are associated with a severe phenotype and occur at a
high frequency in new mutation cases of adenomatous polyposis coli.
Hum Mol Genet. 3:53–56. 1994.PubMed/NCBI View Article : Google Scholar
|
|
16
|
De Rosa M, Scarano MI, Panariello L,
Carlomagno N, Rossi GB, Tempesta A, Borgheresi P, Renda A and Izzo
P: Three submicroscopic deletions at the APC locus and their rapid
detection by quantitative-PCR analysis. Eur J Hum Genet. 7:695–703.
1999.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Newton KF, Mallinson EK, Bowen J, Lalloo
F, Clancy T, Hill J and Evans DG: Genotype-phenotype correlation in
colorectal polyposis. Clin Genet. 81:521–531. 2012.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Fostira F, Thodi G, Sandaltzopoulos R,
Fountzilas G and Yannoukakos D: Mutational spectrum of APC and
genotype-phenotype correlations in Greek FAP patients. BMC Cancer.
10(389)2010.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Liu X, Shan X, Friedl W, Uhlhaas S,
Propping P, Li J and Wang Y: May the APC gene somatic mutations in
tumor tissues influence the clinical features of Chinese sporadic
colorectal cancers? Acta Oncol. 46:757–762. 2007.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Friedl W and Aretz S: Familial adenomatous
polyposis: Experience from a study of 1164 unrelated german
polyposis patients. Hered Cancer Clin Pract. 3:95–114.
2005.PubMed/NCBI View Article : Google Scholar
|
|
21
|
De Rosa M, Scarano MI, Panariello L,
Salvatore F and Izzo P: A novel Mbo II polymorphism in exon 15 of
the human adenomatous polyposis coli gene. Clin Genet. 53:315–316.
1998.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Fernández-Rozadilla C, de Castro L,
Clofent J, Brea-Fernández A, Bessa X, Abulí A, Andreu M, Jover R,
Xicola R, Llor X, et al: Single nucleotide polymorphisms in the Wnt
and BMP pathways and colorectal cancer risk in a spanish cohort.
PLoS One. 5(e12673)2010.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Picelli S, Zajac P, Zhou XL, Edler D,
Lenander C, Dalén J, Hjern F, Lundqvist N, Lindforss U, Påhlman L,
et al: Common variants in human CRC genes as low-risk alleles. Eur
J Cancer. 46:1041–1048. 2010.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wong HL, Peters U, Hayes RB, Huang WY,
Schatzkin A, Bresalier RS, Velie EM and Brody LC: Polymorphisms in
the adenomatous polyposis coli (APC) gene and advanced colorectal
adenoma risk. Eur J Cancer. 46:2457–2466. 2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Septer S, Lawson CE, Anant S and Attard T:
Familial adenomatous polyposis in pediatrics: Natural history,
emerging surveillance and management protocols, chemopreventive
strategies, and areas of ongoing debate. Fam Cancer. 15:477–485.
2016.PubMed/NCBI View Article : Google Scholar
|