Introduction
Anorexia secondary to brain tumors has been
well-documented (1). Although
hypothalamic lesions cause anorexia more frequently, tumors located
in other brain regions are rarely associated with anorexia.
However, there is evidence suggesting that lesions of the posterior
fossa may also cause appetite loss and emaciation, indicating the
role of this region in food intake, satiety and body weight control
(2).
Hemangioblastoma (HB) is a typically benign vascular
tumor. The most frequent location of HB in the central nervous
system (CNS) is the cerebellum. Approximately 25% of these tumors
are encountered as part of the von Hippel-Lindau syndrome (VHLs)
(3,4). VHLs is caused by allelic variants in
the tumor suppressor gene VHL (NC_000003.12). Patients with
this genetic disease may develop tumors of the viscera and CNS
(5,6). When anorexia presents without other
symptoms, anorexia nervosa (AN) emerges as the most likely
diagnosis, particularly in young and previously healthy women
(7,8). This may lead to a misdiagnosis of AN
and delay the correct diagnosis, compromising prognosis and
appropriate treatment.
We herein report the case of a female patient
initially suspected to have AN. Due to lack of evidence supporting
AN on psychiatric evaluation, differential diagnosis was deemed
mandatory. In order to establish the cause of anorexia, magnetic
resonance imaging (MRI) of the CNS was performed, which detected a
tumor in the posterior fossa displaying the typical characteristics
of HB. Genetic sequencing of the VHL gene was further
performed, which confirmed VHLs type 1, presenting only with
anorexia and weight loss.
Case report
A 19-year-old woman was referred to our psychiatric
inpatient service in June 2017, with the suspicion of AN. At the
time of hospital admission, the patient's weight was 28 kg, her
height was 1.55 m and her body mass index (BMI) was 11.6
kg/m2. The patient had had no menstrual cycle for at
least 1 year. Over the previous 2 years, her food intake had
progressively diminished, without an immediately apparent cause.
The patient was hungry but felt satiated after ingesting only a
small amount of food, and had difficulty swallowing.
The patient is the second child of a
non-consanguineous relationship. The pregnancy was uneventful and
full-term; the patient was delivered vaginally, with normal weight
and height at birth. There were no abnormalities in developmental
milestones, but her weight never exceeded 35 kg. No other diseases
were reported.
On admission, the patient exhibited marked weight
loss and weakness, but her physical and neurological examinations
showed no alterations. The criteria of the Diagnostic and
Statistical Manual of Mental Disorders (DSM-5) or ICD-10
(International Statistical Classification of Diseases and Related
Health Problems, 10th revision), which are necessary for diagnosing
AN or another psychiatric disorder, were not met.
The Eating Attitude Test (EAT-26) and the Bulimic
Investigatory Test, Edinburgh (BITE) were performed on admission
(9,10) and the results (EAT: 11 and BITE: 7)
indicated no tendency towards an eating disorder. The patient did
not have an abnormal body image or fear of being overweight, but
rather felt ashamed of being this underweight. Purging, excessive
exercise or any other compensatory weight loss method were not
reported or observed.
An extensive clinical investigation was performed.
The findings of all blood tests, urinalysis and chest X-ray were
unremarkable. Esophagogastroduodenography and upper digestive
endoscopy revealed delay in the passage of food and esophageal
reflux, with mild local tissue inflammation. An abdominal CT scan
revealed cystic lesions throughout the parenchymatous portion of
the pancreas, which were suspected to be related to VHLs.
There were episodes of nausea during fasting after
waking up, which is highly uncommon in AN, followed by unprovoked
vomiting of gastric secretions. The patient frequently complained
of epigastric burning sensation, nocturnal headaches, postural
hypotension and difficulty in swallowing. Spontaneous vomiting also
occurred with slightly larger quantities of food. The patient was
able to eat palatable foods without nausea or abdominal
discomfort.
After 3 weeks of hospitalization, the patient
developed progressive gait abnormality, along with bilateral
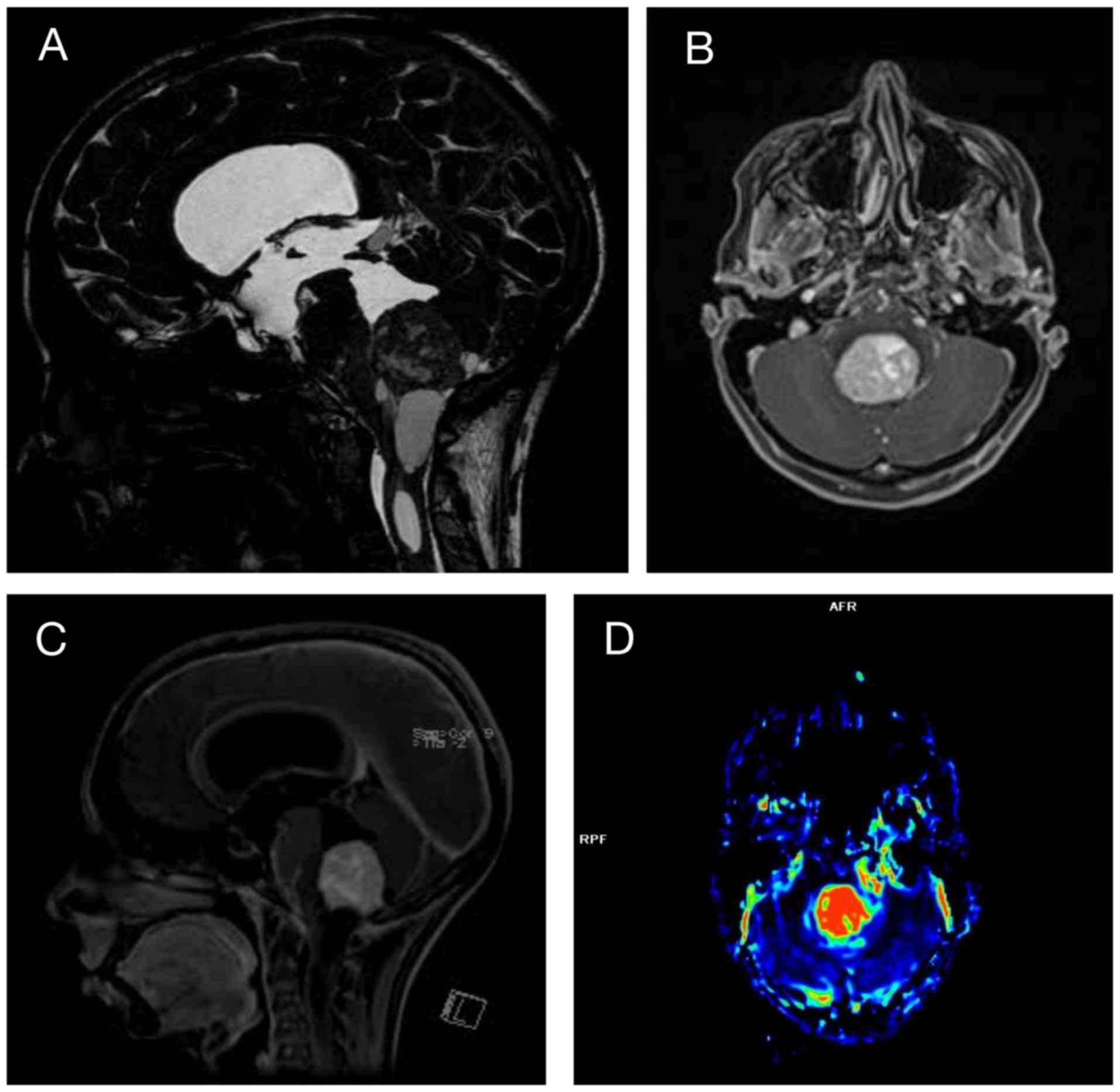
dysmetria, which was worse on the left side. A brain magnetic
resonance imaging (MRI) examination revealed a massive lesion with
vascular characteristics suggestive of a HB in the fourth ventricle
causing hypertensive hydrocephalus (Fig. 1). This lesion, together with the
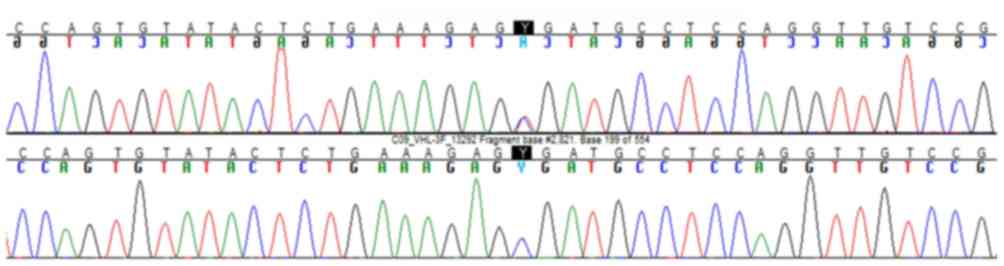
pancreatic cysts, supported the diagnosis of VHLs. Whole blood was
collected for genomic DNA analysis and VHL gene sequencing,
which confirmed VHLs (Fig. 2). The
VHL gene nucleotide sequence was obtained by EMBL Nucleotide
Sequence Database [BBC+00] (http://www.ebi.ac.uk/embl/index.html; ENST00000256196,
sequence: JQ821733). Sequencing was performed by polymerase chain
reaction using the following primers: F: TACTACAGAGGCATGAACACC and
R: CCCCTAAACATCACAATGC. In silico analyses by the mutation
analyzer 2.0.28 (https://mutalyzer.nl) suggested the
pathogenic potential of this variant. Pheochromocytoma was excluded
by 24-h metanephrine and serum catecholamine measurement.
The patient underwent surgery with insertion of a
ventriculoperitoneal shunt to relieve the hypertensive
hydrocephalus. On the first postoperative day, she reported
improvement of her appetite and felt hungrier than usual. For the
first 3 days, she was able to ingest larger quantities of food.
Over the next few days she progressively returned to the previous
state, tolerating small quantities of food at a time, with
unprovoked vomiting if she ate forcibly and nausea several times
during the day. However, there was no longer fasting nausea and
vomiting, or nocturnal headaches. Radiotherapy was considered as
the first choice of treatment due to the difficult surgical access
and risk of profuse bleeding. The patient underwent fractionated
radiotherapy to the posterior fossa (5,400 cGy in 30 fractions, 5
days per week in September and October 2017). During this period,
she still reported nausea throughout the day, difficulty in eating
large quantities of food, and her weight was maintained at ~31 kg,
with a BMI of 12.9 kg/m2. The patient was discharged for
outpatient treatment, as no psychiatric disorder was found on
investigation. The nausea and burning sensation progressively
diminished over the following months, but were not completely
eliminated. Six months post-radiotherapy, an MRI examination
revealed no significant alterations in the size of the lesion.
Radiotherapy was successful in controlling tumor growth. In
December 2018, the patient's weight remained ~31 kg and her BMI
12.9 kg/m2. She still reported difficulty in ingesting
larger quantities of food and nausea sporadically.
Discussion
VHLs is caused by germline mutations of the
VHL gene, which is located on chromosome 3 (3p25-26)
(11). Mutations in this gene have
been reported in all three of its exons and may affect several
organs, including the retina, kidneys, adrenal glands, nervous
system and pancreas (12,13). Haploinsufficiency of this gene may
promote the development of benign cystic lesions, but tumor
development depends on allele wild-type inactivation. Clinically,
VHLs may be divided into types 1 (without pheochromocytoma) and 2
(with pheochromocytoma) (6).
In VHLs type 1, most VHL mutations are
nonsense, including stop codons, deletions and insertions. HBs are
the most frequent tumors in VHLs, being present in ~60-80% of the
cases (6,14). HBs most frequently occur in the
cerebellum (45-50%) followed by the spinal cord (40-45%). The
clinical signs/symptoms of cerebellar HBs usually include headache
(75%), gait ataxia (55%), hydrocephalus (55%), dysmetria (29%) and
nausea/vomiting (28%). The growth of cerebellar HBs is usually
slow, and clinical series with short-term follow up report tumor
stability (3,14).
Our patient harbored a nonsense VHL mutation
(single-nucleotide change causing a premature stop codon in exon 3:
c.481C>T; p.Arg161*) without pheochromocytoma (type 1). What is
interesting in this clinical case is the presence of anorexia and
very low body weight, which are rarely reported in similar clinical
cases. Moreover, either HB or VHLs are not listed among the
differential diagnoses of patients with anorexia. A total of 8
cases of posterior fossa HBs causing anorexia were found in the
English literature, of which 3 were associated with VHLs (7,8,15-18).
Of the 8 cases, 6 had an excellent recovery from anorexia within a
few months after surgery.
In the previously reported cases, the mean time from
the earliest signs of anorexia until correct diagnosis and
treatment was ~4.5 years, indicating a long delay. Most of the
cases were first treated as AN for a long period of time until the
diagnosis of HB. In the present case, the patient was misdiagnosed
with AN for at least 6 months prior to admission to our
institution.
The possible explanations for that cerebral area
affecting satiety is that the fourth ventricle receives direct
vagal and sensory stimuli via afferent fibers and chemical
signaling through its loose blood-brain barrier, thus integrating
the perception of satiety, taste and visceral sensation (17); it also modulates the food reward
system through its projections to the thalamus and insular cortex
(19).
Considering all symptoms exhibited by the patient,
including inability to ingest large quantities of food at once,
tolerance of particularly palatable foods, sensation of being
‘full’, quick satiation and spontaneous vomiting, it was
hypothesized that the nucleus tractus solitarius (NTS) was
affected, possibly through compression, as suggested previously.
After radiotherapy, tumor growth ceased, but no significant
alterations in size were observed on further MRI scans. This may
explain the persistent anorexia, sporadic nausea and small meal
size, in contrast with previously described cases in which anorexia
completely resolved within a short time after tumor resection.
Therefore, in cases of HB with anorexia, neurosurgery may be more
effective for weight recovery and resolution of the eating
disorder.
In conclusion, a psychiatric diagnosis of AN can
only be confirmed after excluding all possible clinical conditions;
otherwise, it may delay correct diagnosis and lead to a poor
prognosis. Atypical symptoms, such as fasting nausea and vomiting,
difficulty swallowing and frequent, albeit mild headaches,
associated with no distortion of the body image and no fear of
being overweight, should prompt the search for an organic cause
rather than be considered an atypical case of AN. HBs, associated
or not with VHLs, should be included in the differential diagnosis
of AN, as the diagnosis directly affects treatment. In cases with
marked weight loss, resection of the tumor may be preferred over
radiotherapy, as total resection appears to be more effective for
treating anorexia. Moreover, the findings of this case support
previous evidence on the role of the brainstem and NTS in satiety
and meal size control.
Acknowledgements
Not applicable.
Funding
The present study was supported by FAPESP (Fundação
de Amparo à Pesquisa do Estado de São Paulo, grant no.
2014/50137-5) to SELA (Laboratório de Sequenciamento em Larga
Escala).
Availability of data and materials
All the data analyzed in the present study are
available from the corresponding author.
Authors' contributions
JHM and RLB were the major contributors to the
writing of the manuscript, and reviewed the clinical data, images
and follow-up records. MTDM and BAM contributed to the review of
the manuscript and clinical data records; FCGP and MJT discussed
the neurosurgical approach and the neurosurgical aspects of the
present study; MQA and DDCA sequenced the VHL gene; BBM and
TAC made substantial contributions regarding manuscript concept and
review. All authors have read and approved the final version of
this manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
The patient signed an informed consent authorizing
publication of case details, images and molecular
investigation.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lustig RH: Hypothalamic obesity after
craniopharyngioma: Mechanisms, diagnosis, and treatment. Front
Endocrinol (Lausanne). 2(60)2011.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Farr OM, Li CS and Mantzoros CS: Central
nervous system regulation of eating: Insights from human brain
imaging. Metabolism. 65:699–713. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bamps S, Calenbergh FV, Vleeschouwer SD,
Loon JV, Sciot R, Legius E and Goffin J: What the neurosurgeon
should know about hemangioblastoma, both sporadic and in Von
Hippel-Lindau disease: A literature review. Surg Neurol Int.
4(145)2013.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Neumann HP, Eggert HR, Weigel K, Friedburg
H, Wiestler OD and Schollmeyer P: Hemangioblastomas of the central
nervous system. A 10-year study with special reference to von
Hippel-Lindau syndrome. J Neurosurg. 70:24–30. 1989.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Decker J, Neuhaus C, Macdonald F, Brauch H
and Maher ER: Clinical utility gene card for: Von Hippel-Lindau
(VHL). Eur J Hum Genet 22, 2014.
|
|
6
|
Varshney N, Kebede AA, Owusu-Dapaah H,
Lather J, Kaushik M and Bhullar JS: A review of Von hippel-lindau
syndrome. J Kidney Cancer VHL. 4:20–29. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Oya S, Nejo T, Indo M and Matsui T: Pearls
& Oy-sters: Anorexia and emaciation in patients with cerebellar
hemangioblastoma. Neurology. 83:1298–1300. 2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Pavesi G, Berlucchi S, Feletti A, Opocher
G and Scienza R: Hemangioblastoma of the obex mimicking anorexia
nervosa. Neurology. 67:178–179. 2006.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Garner DM, Olmsted MP, Bohr Y and
Garfinkel PE: The eating attitudes test: Psychometric features and
clinical correlates. Psychol Med. 12:871–878. 1982.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Henderson M and Freeman CP: A self-rating
scale for bulimia The ‘BITE’. Br J Psychiatry. 150:18–24.
1987.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Latif F, Tory K, Gnarra J, Yao M, Duh FM,
Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L, et al:
Identification of the von Hippel-Lindau disease tumor suppressor
gene. Science. 260:1317–1320. 1993.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Rajpert-De Meyts E, Nielsen JE, Skakkebaek
NE and Almstrup K: Diagnostic markers for germ cell neoplasms: From
placental-like alkaline phosphatase to micro-RNAs. Folia Histochem
Cytobiol. 53:177–188. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Dornbos D III, Kim HJ, Butman JA and
Lonser RR: Review of the neurological implications of von
Hippel-Lindau Disease. JAMA Neurol. 75:620–627. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Rocha L, Noronha C, Taipa R, Reis J, Gomes
M and Carvalho E: Supratentorial hemangioblastomas in von
Hippel-Lindau wild-type patients-case series and literature review.
Int J Neurosci. 128:295–303. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Liebner EJ: A case of Lindau's disease
simulating anorexia nervosa; a roentgenologic report. Am J
Roentgenol Radium Ther Nucl Med. 78:283–288. 1957.PubMed/NCBI
|
|
16
|
Abecassis IJ, Smith T and Chandler JP:
Brain tumors and the area postrema. J Clin Neurosci. 20:1795–1797.
2013.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Song DK and Lonser RR: Pathological
satiety caused by brainstem hemangioblastoma. J Neurosurg Pediatr.
2:397–401. 2008.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Sutton LN, Lasner T, Hunter J, Rorke LB
and Sanford RA: Thirteen-year-old female with hemangioblastoma.
Pediatr Neurosurg. 27:50–55. 1997.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Morton GJ, Cummings DE, Baskin DG, Barsh
GS and Schwartz MW: Central nervous system control of food intake
and body weight. Nature. 443:289–295. 2006.PubMed/NCBI View Article : Google Scholar
|