Introduction
Lung cancer, common worldwide, is the leading cause
of cancer deaths in China. Effective and efficient measures to
target therapies are needed to reduce disease burden and improve
patient prognoses and outcomes. Precision medicine uses genomic
analysis, including next generation sequencing, to identify the
genetic profile of individual patients and lung cancer cells to
identify specific individual receptivity to available therapies.
The 2015 launch of the Precision Medicine Initiative by US
President Obama accelerated the integration of next generation
sequencing (NGS) methods in genomic medicine, especially in
oncology care (1,2). NGS technology is becoming more widely
employed as a novel genetic screening, prognostic and diagnostic
technique for clinical disease management (3-5)
and is becoming an effective and acceptable method for clinical
gene detection (6,7). Although it is still in the early stage
of clinical application for the diagnosis and treatment of tumors
(8-10),
its continuous innovation has generated increased awareness and
interest in the role of genetic markers and the molecular
mechanisms of diseases. Genome alterations play a significant role
in disease recurrence for lung cancer (11). Large-scale genomic sequencing
studies have revealed the complex genomic landscape of lung cancer,
with tumor heterogeneity (12).
NGS shows promise in the treatment of lung cancer to
identify candidate biomarkers for early diagnosis, identify
prognostic factors, and detect actionable mutations to guide
targeted therapy decisions (13).
However, NGS generates massive volumes of data. Analyzing and
interpreting this data creates challenges for analysts and
clinicians alike. As a result, the analysis of data and annotation
of variation have become a major bottleneck that inhibits wider
clinical adoption and usefulness of NGS technology.
There are many tools available to analyze NGS data
for variants (14). The standard
method for annotation of NGS mutations at cancer care centers like
Guangdong Lung Cancer Institute (GLCI) includes retrieval, analysis
and comparison of the annotation results with databases like
DrugBank, COSMIC, dbSNP, OMIM, ClinVar, 5000 Exomes and 1000
Genomes. Genes are further analyzed through screening under a
series of conditions examining zygosity, variant type, variant
effect, location, filtered coverage and minor allele frequency, on
the basis of preliminary analysis of NGS data.
This study compared the annotation and
interpretation of NGS results at a large volume cancer center in
Guangdong, China using standard methods and using IBM Watson for
Genomics (WfG), a cloud-based cognitive computing system. WfG is
trained to analyze molecular data at a massive scale to provide
clinically actionable insights that are supported by all available
relevant evidence. The tool is built on several different
predictive models that can perform analysis across the whole-genome
and accesses a comprehensive database of structured and
unstructured data sources using Natural Language Processing (NLP).
(Over 200 sources include DrugBank, NCI, COSMIC, ClinVar, and 1000
Genomes, as well as evidence extracted from the universe of
biological and medical literature.) WfG is in use in selected
markets (13,15,16),
however, its application in the analysis and annotation of results
of NGS data in Chinese patients has not been reported. This study
examined and compared the NGS data annotation process by comparing
the results of the gene mutation annotation for Chinese patients
with lung cancer generated by Guangdong Lung Cancer Institute
(GLCI) bioinformaticians and WfG. The ultimate goal of this study
was to leverage insights from the analysis to inform individual
treatment decisions to benefit future Chinese patients with lung
cancer.
Materials and methods
Materials
In terms of patient specimen collection and
analysis, we do have approval from our institutional ethics board
and informed consent from each patient. Actually, we have
established a tissue repository center (Tumor Sample Bank) in our
cancer center which was approved by Human Biomaterial and Genetic
Resource Office of China. Researchers at Guangdong Lung Cancer
Institute (GLCI) obtained a variety of tissue samples from the
Tumor Sample Bank in GLCI of Guangdong Provincial People's
Hospital, Guangzhou, China. The samples, from 115 randomly selected
patients diagnosed with lung cancer at Guangdong General Hospital
between 2014 and 2016, included 10 formalin fixed paraffin-embedded
(FFPE) samples, 12 small samples collected after puncture, 4 plasma
samples and 89 large tumor samples collected during surgery. All
patients were well informed and signed the informed consent.
DNA extraction
Researchers used QIAGEN QlAampDNA Mini Kit and
QIAGEN QlAampBlood Mini Kit for the gDNA extraction process from
each tissue sample (i.e., the FFPE samples, small samples collected
after puncture and large samples collected in surgery) and DNA from
1-4 ml plasma samples, respectively. DNA quantitative analysis was
completed using Qubit analyzer.
Design and synthesis of target capture
probe
Hybrid capture in target areas was implemented using
SureSelectXT Custom library. This probe library was
designed through SureDesign software based on genome hg19/GRCh37;
target areas were lung cancer-related high-frequency mutation gene
exon areas.
Establishment of NGS library
The NGS library was established using free DNA in
plasma and gDNA in tissue samples. Free DNA was extracted from 1-2
ml plasma to establish the library, without requirement of
fragmentation. Approximately, 50-1,000 ng of gDNA was extracted
from each tissue sample and cut into 100-200 bp segments through
enzyme digestion to establish the library. The NGS library was
established using Ion Xpress™ Plus Fragment Library Kit and Ion
Xpress™ Barcode Adapters 1-16 Kit, and the selection, purification
and recovery of DNA was completed via Agencourt AMPure XP beads.
Target areas were captured after hybridization through SureSelectXT
Custom library at 65˚C for 16-24 h following 11 cycles of
polymerase chain reaction (PCR) amplification in pre-library after
segment selection. The target area sequencing library was obtained
from the captured library after purification and 9 cycles of PCR
amplification. Finally, QIAxcel and Qubit were used to detect the
library segment length and library concentration, respectively.
Sequencing and data analysis
The library was diluted into 12 pM using water
according to its concentration and connected with microballoon
using Ion PI™ Template OT2 200 Kit v2, after which the samples were
spotted on P1 chips for sequencing. The sequencing data were
compared with human genome Hg 19 using Suite software (Life
Technologies, Version 5.0.2), and mutations were detected using
Variant Caller software (Life Technologies, Version 5.0.2.1), so as
to form corresponding variant calling files (VCF).
Target sequencing data interpretation
by GLCI using standard methods
Integrative Genomics Viewer (IGV) software was
applied by GLCI bioinformaticians to annotate the gene mutation
information by comparing with the databases DrugBank, COSMIC,
dbSNP, OMIM, ClinVar, 5000 Exomes and 1000 Genomes.
Gene variant interpretation executed
by Watson for Genomics
Upon sequence completion by GLCI lab, the research
team accessed the cloud-based cognitive computing tool, Watson for
Genomics (WfG). The following information was uploaded to WfG: (a)
tumor type, (b) a list of variants as a variant calling file (.vcf).
Uploading this data required approximately 1 min for each sample,
by a data technician. After these data were uploaded, WfG performed
the Molecular Profile Analysis (MPA) for each gene with a variant.
A subpart of the WfG cognitive tool, the MPA reviews evidence from
functional studies and protein structure and applies programming
logic to classify variants into five categories: Pathogenic, likely
pathogenic, benign, likely benign, and variables of unknown
significance (VUS). Alterations categorized as benign or likely
benign were removed from the report. Next, WfG identified a gene as
actionable if: a) the variant was pathogenic or likely pathogenic;
b) the variant was directly targetable or part of a pathway that
was targetable based on evidence from the literature; and c) a U.S.
Food and Drug Administration-approved or investigational target
therapy was available.
Statistical analysis
Statistical analysis of the difference between tumor
mutation burden obtained by WFG and GLCI was performed by the
paired t-test analysis. Counting data was expressed by the number
of cases/percentage n(%), and the measurement data were expressed
by mean number (mean ± SD). t-test analysis was performed using
SPSS v22.0 software (IBM Corp.). P<0.05 was considered to
indicate a statistically significant difference.
Results
Congruence rate of gene mutation
interpretation results of GLCI and WfG analyses/between two
methods
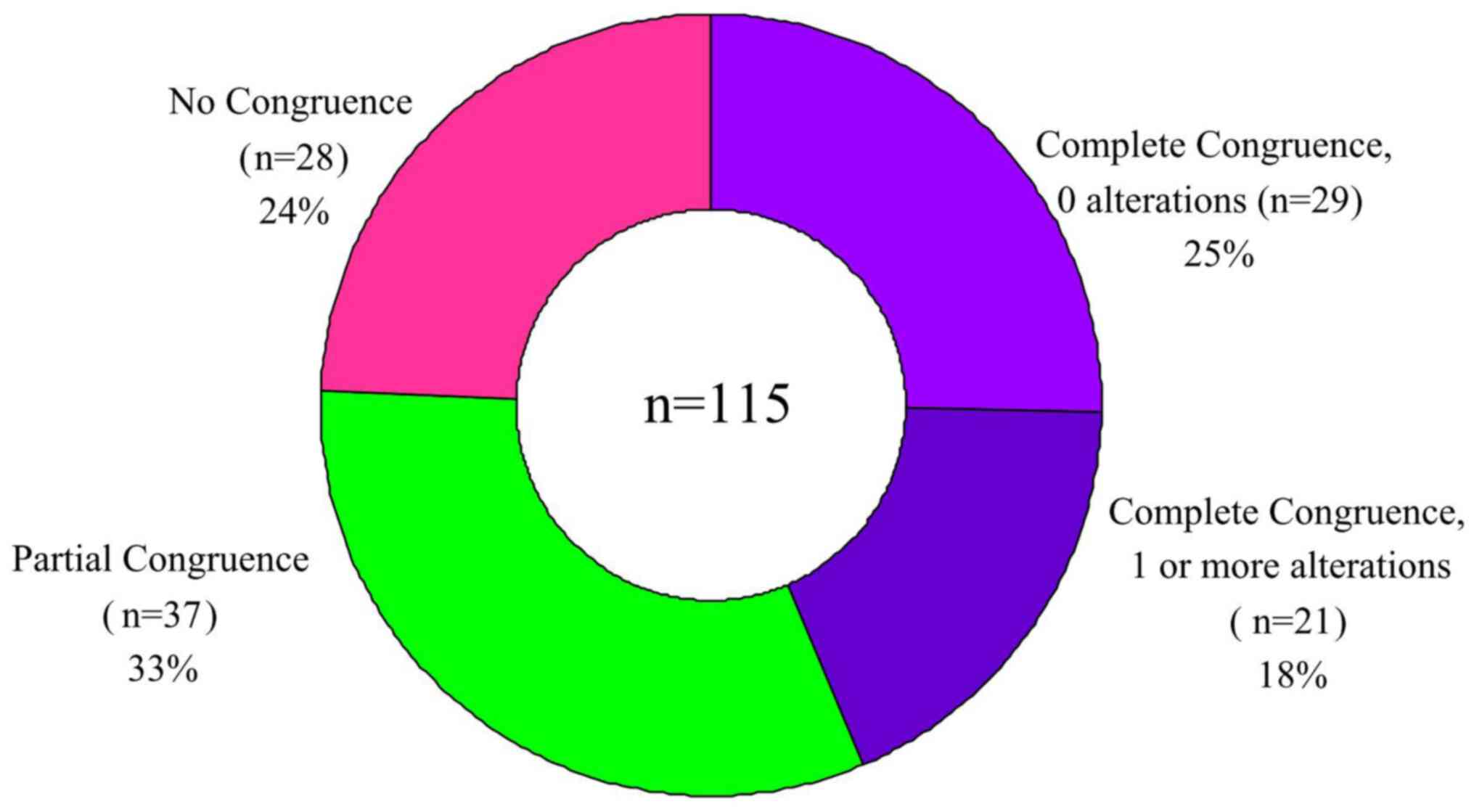
‘Complete congruence’ was defined as having
completely consistent interpretation results from the two methods,
which included the following two scenarios: In the first scenario,
no mutation sites were identified by either method, i.e., Complete
Congruence, 0 alterations reported. In the second scenario, the
same single or multiple mutation sites were interpreted by both
methods, i.e., Complete Congruence, 1 or more identical alterations
reported by both methods.
‘Partial congruence’ was defined as the partially
consistent interpretation results from the two methods, i.e.,
mutation sites interpreted by WfG analysis contained those by GLCI
bioinformaticians. ‘No congruence’ was defined as totally
inconsistent interpretation results from the two methods, i.e., no
mutation sites were interpreted by GLCI bioinformaticians while 1
or more mutation site were interpreted by WfG analysis, or vice
versa.
After annotation of the sequencing results of all
115 samples, across all samples WfG identified 180 alterations
whereas GLCI identified 80 mutation sites. The congruence rate in
detecting mutation sites across the entire sample was 44.44%.
Complete congruence was found in the analyses of 50 samples
(congruence rate 43.48%.) Of these completely congruent samples, 29
had no reported mutation sites (Complete Congruence, 0
alterations), and 21 had 29 mutation sites (Complete Congruence, 1
or more alterations).
In the remaining 65 samples (56.52%), a total of 180
mutation sites were found after analysis by both methods. Compared
with GLCI bioinformaticians, WfG analysis interpreted more mutation
sites (100), with an average of 1.54 more mutation sites in each
sample. (In one sample, WfG interpreted 11 more mutation sites than
were interpreted by the GLCI bioinformatician). In 37 samples
(32.17%) Partial Congruence was found in the analysis: GLCI
interpreted 51 mutation sites and WfG interpreted 103 mutation
sites. (WfG interpreted an average of 1.41 more mutation sites in
each sample). Finally, in the 28 samples with no congruence, WfG
reported 48 mutation sites and GLCI reported zero mutation sites.
In these samples, WfG analysis interpreted 1.71 more mutation sites
in each sample when compared to GLCI analysis WfG (Fig. 1 and Table I).
 | Table INumber of mutation sites identified by
WfG and GLCI. |
Table I
Number of mutation sites identified by
WfG and GLCI.
| Type of
uniformity | No. | Mutation sites
identified by WfG, n | Mutation sites
identified by GLCI, n |
|---|
| Complete congruence,
0 alterations | 29 | 0 | 0 |
| Complete congruence,
1 or more alterations | 21 | 29 | 29 |
| Partial
congruence | 37 | 103 | 51 |
| No congruence | 28 | 48 | 0 |
| Total | 115 | 180 | 80 |
Differential genes and signal pathways
in data interpretation results between two methods
NGS data from 115 samples were annotated by WfG and
GLCI bioinformaticians respectively. In 50 samples (43.5%), no
mutations were found by either GLCI or WfG. However, in the
remaining 65 samples (56.5%), WfG analysis interpreted more
mutation sites (100), whereas in 54 (47.0%) samples, WfG analysis
interpreted 1-2 more mutation sites when compared with GLCI
bioinformaticians' results (Table
II).
| Table IIVariability in mutation sites
interpreted by Watson for Genomics and Guangdong Lung Cancer
Institute. |
Table II
Variability in mutation sites
interpreted by Watson for Genomics and Guangdong Lung Cancer
Institute.
| Variability (number
of different mutation sites) | Incidence, no. of
samples | Share of samples,
% | Cumulative
percentage, % |
|---|
| 0 | 50 | 43.5 | 43.5 |
| 1 | 41 | 35.7 | 79.1 |
| 2 | 13 | 11.3 | 90.4 |
| 3 | 3 | 2.6 | 93.0 |
| 4 | 4 | 3.5 | 96.5 |
| 5 | 2 | 1.7 | 98.3 |
| 6 | 1 | 0.9 | 99.1 |
| 11 | 1 | 0.9 | 100.0 |
| Total | 115 | 100.0 | |
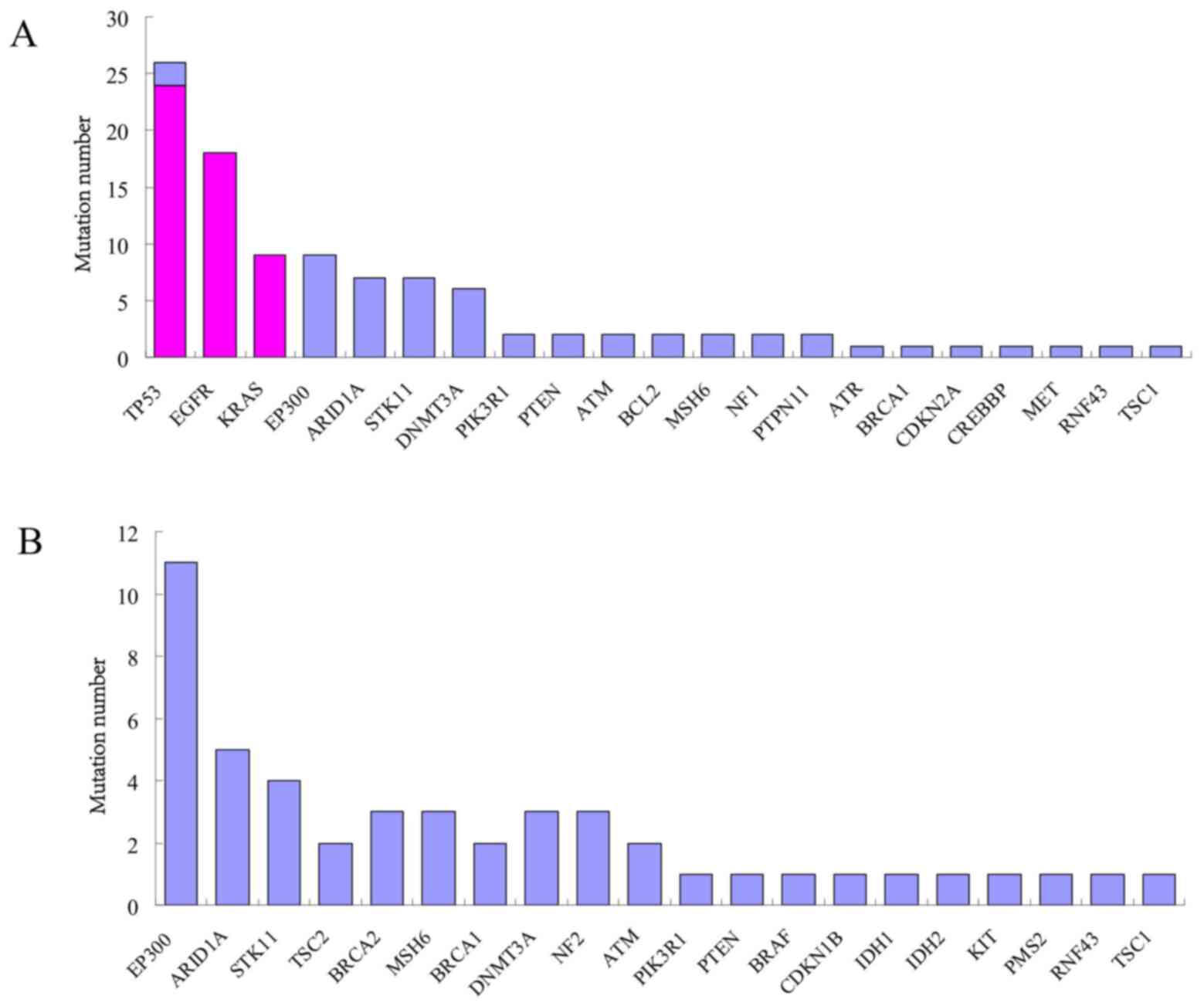
The 100 mutation sites that were additionally
identified by WfG analysis were located on 27 genes, including
EP300, ARID1A, STK11 and DNMT3A. The
mutation rate was 30.77% (20/65) in EP300 gene; 18.46%
(12/65), 16.92% (11/65) and 13.85% (9/65) in ARID1A,
STK11 and DNMT3A genes, respectively. In the 37
(32.17%) samples out of 65, 51 mutation sites were interpreted by
GLCI bioinformaticians while 103 by WfG analysis, with 51
co-interpreted mutation sites located on EGFR, KRAS
and TP53 genes. WfG interpreted 52 more mutation sites
located on EP300, ARID1A, STK11,
DNMT3A, PIK3R1, PTEN, ATM, BCL2,
MSH6, NF1, PTPN11, ATR, BRCA1,
CDKN2A, CREBBP, MET, RNF43 and
TSC1 genes. In another 28 (24.35%) samples out of 65, no
mutation sites were interpreted by GLCI bioinformaticians while 48
mutation sites were interpreted by WfG analysis and were located on
EP300, ARID1A, STK11, TSC2,
BRCA2, MSH6, BRCA1, DNMT3A, NF2,
ATM, PIK3R1, PTEN, BRAF, CDKN1B,
IDH1, IDH2, KIT, PMS2, RNF43 and
TSC1 genes (Fig. 2).
Analysis of congruence rate of common
driver gene mutation annotation
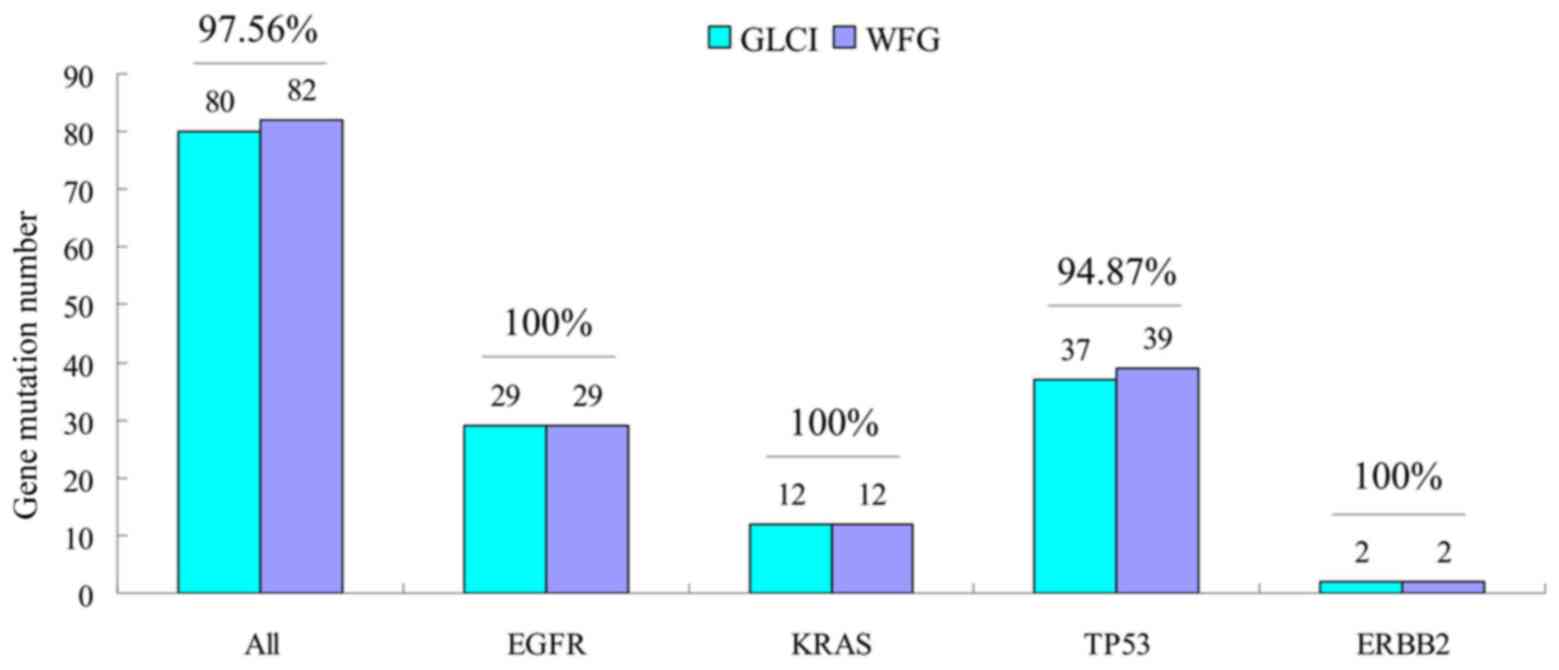
The two methods reported similar annotation results
in the mutations analysis of 4 common driver genes of EGFR,
KRAS, TP53 and ERBB2. Of the 115 samples, the
total number of mutation sites for these 4 genes was 82, 80 of
which were identically reported by both methods (congruence rate
97.56%). The congruence rates of EGFR, KRAS,
TP53 and ERBB2 were 100% (29/29), 100% (12/12),
94.87% (37/39) and 100% (2/2) respectively (Fig. 3).
Analysis of tumor mutation burden
(TMB) interpretation results between two methods
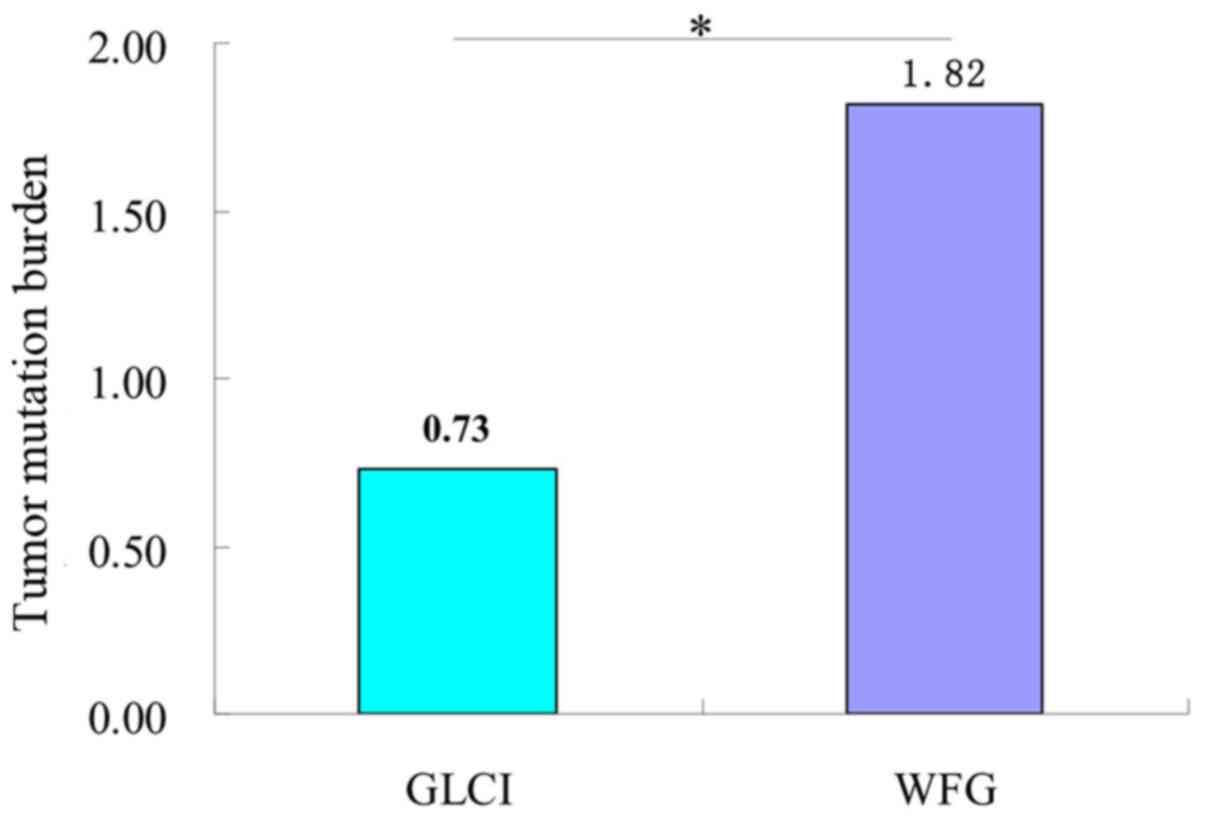
The average number of missense mutations obtained
before preliminary analysis and filtration of NGS data of target
0.947 kb genes in the 115 samples was 130.43, with a TMB of 137.78.
After WfG interpretation and filtration, the average number of
missense mutations and TMB obtained was 1.72 and 1.82,
significantly higher than the 0.69 and 0.73, respectively, obtained
by GLCI bioinformaticians (P<0.05) (Fig. 4).
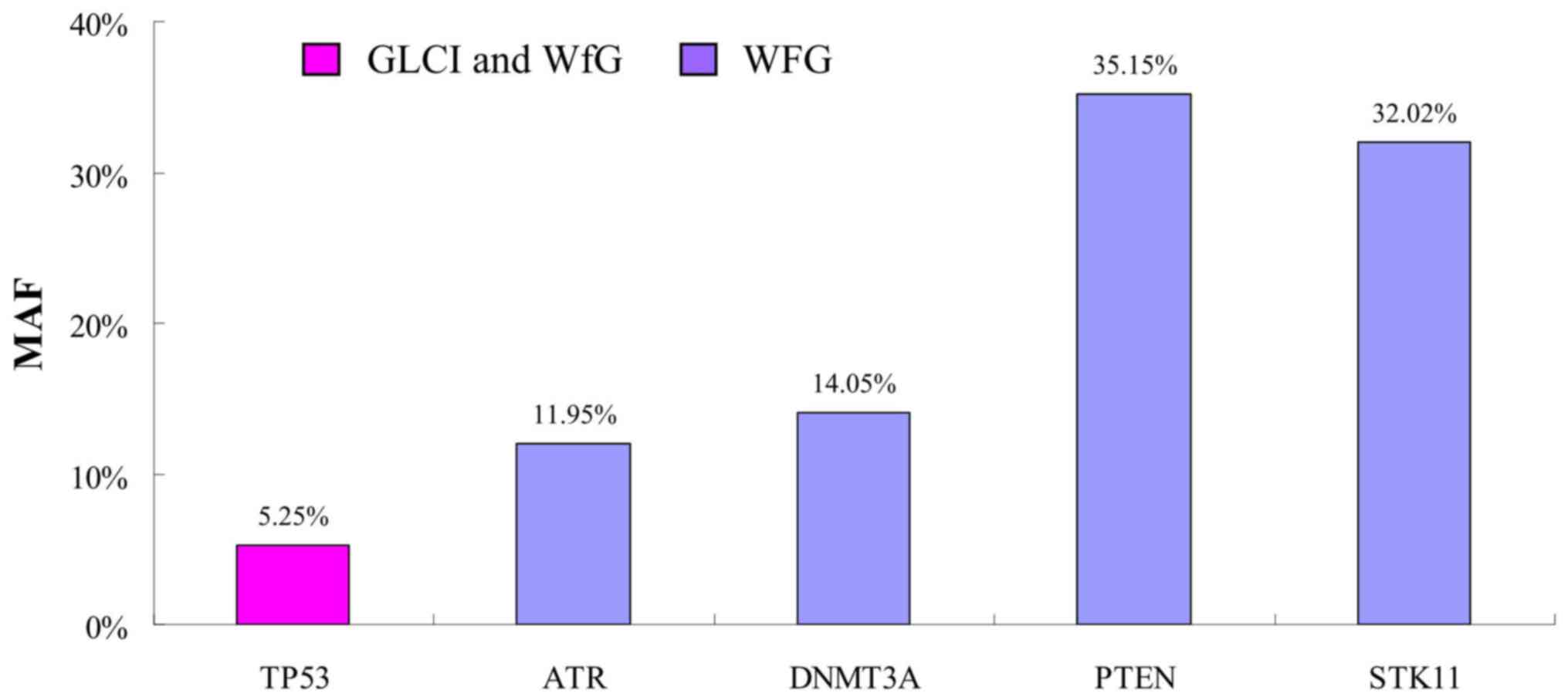
New high-Minor Allele Frequency (MAF)
mutations indicated by WfG
In one of the 37 samples with Partial Congruence,
(sample no. 29002) WfG analysis identified 4 new mutation sites
with high MAF. The MAF of TP53 G245V mutation found by both
methods was 5.25%, while the MAF was 11.95, 14.05, 35.15 and 32.02%
in new mutation sites ATR G492fs, DNMT3A A222fs,
PTEN R130Q and STK11 G257fs interpreted by WfG only,
respectively (Fig. 5).
Discussion
Several cancer centers are beginning to use
artificial intelligence (AI) computing systems to analyze NGS data
(14). The speed and volume of
research, discovery and reporting on new genes and mutations, and
their relationships to tumorigenesis is accelerating. Monitoring
and integrating this knowledge for use in clinical decision-making
is a task well-suited to cognitive computing technologies. WfG is a
cognitive computing technology which can learn new information and
analyze data at a rate that far exceeds manual curation and
analysis (14). During this study,
the number of cases analyzed and annotated by one GLCI
bioinformatician working full time was approximately 10 cases per
one week. In contrast, WfG completed analysis and annotation of
each sample in ~3 min, evidence that WfG was able to perform this
analysis at a much faster rate than even a highly trained human
analyst.
To compare the effectiveness of the WfG cognitive
computing tool with human-only targeted panels at identifying
potentially actionable gene mutations for Chinese patients with
lung cancer, we retrospectively analyzed 115 cases from GLCI that
had undergone targeted DNA sequencing of 285 genes and subsequent
analysis by the GLCI bioinformaticians. We conducted an independent
analysis of these 115 samples using WfG. WfG was provided the full
list of variants on each sequenced sample and identified 180
mutation sites in the 115 samples vs. only 80 mutation sites
identified by GLCI bioinformaticians. This indicated that WfG could
interpret more mutation sites than GLCI bioinformaticians using
standard methods. This is particularly valuable as it is well known
that target therapy or individualized therapy focuses on specific
variant genes or specified gene mutation sites. WfG's ability to
interpret more gene mutation sites from 56.52% samples demonstrates
that it can provide more opportunities for targeted therapy for
patients with lung cancer and additional useful information for
clinical doctors developing therapeutic strategies.
In this study, the congruence rate of the analyses
of NGS results was 43.48% and congruence rate of mutations was
44.44% between WfG analysis and GLCI bioinformaticians', and is
similar to the results of other studies which have compared WfG and
manual mutation analysis (13,16).
There are several possible reasons that fewer mutation sites were
interpreted by GLCI bioinformaticians than by WfG. For example, the
GLCI biological analysis was completed by one individual
bioinformatician and was relatively conservative, emphasizing
mutation sites that were common driver gene mutation sites that had
been studied thoroughly, as demonstrated by the 97.56% congruence
rate of significant driver gene mutation sites. This is because
GLCI bioinformaticians mainly focus on driving genes, while the new
analysis methods are more comprehensive, which not only does not
lose the driving gene targets, but also increases the discovery of
many new rare mutation targets. A defining feature of the WfG
cognitive tool and its analysis is its ability to retrieve almost
all the research published worldwide, and to extract actionable
information to analyze and annotate gene mutation sites. In
contrast, the bioinformatician relied on extensive individual
experience in reading the literature and analyzing results. As to
most uncommon variant genes, WfG annotation was superior to the
GLCI bioinformatician's. In this study results, the genes in the
list interpreted by WfG analysis only, such as MSH6,
DNMT3A, NF2, ATM, PIK3R1,
CDKN1B, IDH1, IDH2, KIT, PMS2,
RNF43 and TSC1, reflect WfG's ability to access and
integrate more recent research consistently during analysis. This
may also explain discrepancies between the annotation results of
bioinformaticians and WfG analysis in samples with mutation sites
occurring in uncommon genes. This may also suggest that uncommon
variant genes should be of increased concern to bioinformaticians,
especially in this rapidly evolving arena.
This retrospective analysis of individual tissue
samples representing 115 cases offers an opportunity to review
treatment therapies with the benefit of new information gleaned
from WfG. In 24.35% samples, targeted therapy was not performed for
patients as no clinically significant mutation sites were found by
GLCI bioinformaticians. In contrast, mutation sites were found in
uncommon genes by WfG analysis in 56.52% of the samples. Patients
with these mutations may have been eligible to participate in
clinical trials with corresponding agents, had this information
been available to their clinicians at the point of care. Going
forward, it is therefore possible that cognitive technologies such
as WfG will be able to assist clinicians by providing the
comprehensive and timely analysis needed to help them guide
patients to appropriate therapies. This is consistent with the view
of Itahashi et al (17).
They believe that WFG is useful for a clinician at a general
hospital additional survey of evidence by a clinician is required
when evaluating functions (17). In
our sample of Chinese patients with lung cancer, opportunities for
targeted therapy might have been available for 24.35% of patients,
informed by the WfG analysis. Of particular interest, in one sample
in which there was no congruence between GLCI and WfG analysis, and
in which there was no common mutation gene variation, this analysis
found that the MAF value of multiple new gene mutation sites
interpreted by WfG analysis was evidently higher than that of
discovered insignificant gene mutation sites. This finding suggests
that the therapeutic strategies for patients with findings like
these might be changed accordingly.
This report is a preliminary study to compare
AI-aided analysis using the cognitive computing technology WfG to
the standard manual method. Limitations of the study included
relatively small sample size, absence of in-association analysis
with clinical therapy and, as a result, no specific information
about how the results of this analysis might have affected clinical
outcomes. Our study did not systematically measure the time
required to annotate, interpret and report NGS results by either
the standard method used by GLCI, or by WfG. Future studies will be
necessary to address these limitations.
In conclusion, this study provides evidence that
analysis of NGS results by the cognitive computing technology WfG
can provide an accurate and comprehensive interpretation of more
gene mutation sites through a more rapid process than routine
manual analysis, generating potential opportunities of targeted
therapy for cancer patients. The basis of targeted therapy is to
obtain the effective gene mutation information of patients in time.
For patients diagnosed with lung cancer in China and elsewhere, the
timely provision of actionable information that may affect
treatment options can be critical to individual therapy and
research. The abundance of mutation sites interpreted uniquely by
WfG analysis in partial samples was relatively high, further
suggesting opportunities to optimize clinical decision making for a
greater number of affected patients in the future.
Acknowledgements
This abstract was presented at the 2018 ASCO Annual
Meeting (June 1, 2018; Chicago, USA) and was published as Abstract
no. e24254.
Funding
The present study was supported by the following
grants: Guangdong Provincial Natural Science Program (grant no.
2019A1515010900; to XZ); GDPH Dengfeng Program (grant nos.
DFJH201903, KJ012019444 and 8197103306; to XZ); Guangdong
Provincial Applied S&T R&D Program [grant no.
2016B020237006; to Professor Peng Li (Guangzhou Institutes of
Biomedicine and Health, Chinese Academy of Sciences, Guangzhou,
China) and XZ].
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YC and XZ designed the study, YC, WY, ZX, WG, DL and
ZL performed the experiments. YC interpreted the data and drafted
the initial manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The protocol of the present study was approved by
the Ethics Committee of Guangdong Provincial People's Hospital and
written informed consent was obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
The White House Office of the Press
Secretary. Remarks by the President in State of the Union.
(2015-01-20) [2015-03-20]. urihttps://www.whitehouse.gov/the-press-office/2015/01/20/remarks-president-state-union-address-january-20-2015simplehttps://www.whitehouse.gov/the-press-office/2015/01/20/remarks-president-state-union-address-january-20-2015.
|
|
2
|
The White House Office of the Press
Secretary. FACT SHEET: President Obama's Precision Medicine
Initiative. (2015-01-30) [2015-03-20]. urihttps://www.whitehouse.gov/the-press-office/2015/01/30/fact-sheet-president-obama's-precision-medicine-initiativesimplehttps://www.whitehouse.gov/the-press-office/2015/01/30/fact-sheet-president-obama's-precision-medicine-initiative.
|
|
3
|
Yang Y, Muzny DM, Reid JG, Bainbridge MN,
Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al:
Clinical whole-exome sequencing for the diagnosis of mendelian
disorders. N Engl J Med. 369:1502–1511. 2013.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Lee H, Deignan JL, Dorrani N, Strom SP,
Kantarci S, Quintero-Rivera F, Das K, Toy T, Harry B, Yourshaw M,
et al: Clinical exome sequencing for genetic identification of rare
Mendelian disorders. JAMA. 312:1880–1887. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Dewey FE, Grove ME, Pan C, Goldstein BA,
Bernstein JA, Chaib H, Merker JD, Goldfeder RL, Enns GM, David SP,
et al: Clinical interpretation and implications of whole-genome
sequencing. JAMA. 311:1035–1045. 2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Hayes DN and Kim WY: The next steps in
next-gen sequencing of cancer genomes. J Clin Invest. 125:462–468.
2015.PubMed/NCBI View
Article : Google Scholar
|
|
7
|
Garraway LA: Genomics-driven oncology:
Framework for an emerging paradigm. J Clin Oncol. 31:1806–1814.
2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Good BM, Ainscough BJ, McMichael JF, Su AI
and Griffith OL: Organizing knowledge to enable personalization of
medicine in cancer. Genome Biol. 15(438)2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Griffith M, Miller CA, Griffith OL,
Krysiak K, Skidmore ZL, Ramu A, Walker JR, Dang HX, Trani L, Larson
DE, et al: Optimizing cancer genome sequencing and analysis. Cell
Syst. 1:210–223. 2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Hyman DM, Solit DB, Arcila ME, Cheng DT,
Sabbatini P, Baselga J, Berger MF and Ladanyi M: Precision medicine
at memorial sloan kettering cancer center: Clinical next generation
sequencing enabling next-generation targeted therapy trials. Drug
Discov Today. 20:1422–1428. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Hu Z, Sun R and Curtis C: A population
genetics perspective on the determinants of intra-tumor
heterogeneity. Biochim Biophys Acta Rev Cancer. 1867:109–126.
2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Jamal-Hanjani M, Hackshaw A, Ngai Y, Shaw
J, Dive C, Quezada S, Middleton G, de Bruin E, Le Quesne J, Shafi
S, et al: Tracking genomic cancer evolution for precision medicine:
The lung TRACERx Study. PLoS Biol. 12(e1001906)2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Patel NM, Michelini VV, Snell JM, Balu S,
Hoyle AP, Parker JS, Hayward MC, Eberhard DA, Salazar AH, McNeillie
P, et al: Enhancing next-generation sequencing-guided cancer care
through cognitive computing. Oncologist. 23:179–185.
2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Pabinger S, Dander A, Fischer M, Snajder
R, Sperk M, Efremova M, Krabichler B, Speicher MR, Zschocke J and
Trajanoski Z: A survey of tools for variant analysis of
next-generation genome sequencing data. Brief Bioinform.
15:256–278. 2014.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Oncologists partner with Watson on
genomics. Cancer Discov. 5(788)2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wrzeszczynski KO, Frank MO, Koyama T,
Rhrissorrakrai K, Robine N, Utro F, Emde AK, Chen BJ, Arora K, Shah
M, et al: Comparing sequencing assays and human-machine analyses in
actionable genomics for glioblastoma. Neurol Genet.
3(e164)2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Itahashi K, Kondo S, Kubo T, Fujiwara Y,
Kato M, Ichikawa H, Koyama T, Tokumasu R, Xu J, Huettner CS, et al:
Evaluating clinical genome sequence analysis by Watson for
genomics. Front Med (Lausanne). 5(305)2018.PubMed/NCBI View Article : Google Scholar
|