Introduction
Anaplastic astrocytoma (AA) and glioblastoma (GBM)
represent ~20% of all central nervous system (CNS) tumors. These
two brain tumor subtypes respond poorly to therapy and they are the
most biologically aggressive malignancies and the principal cause
of cancer-related mortality and morbidity during childhood
(1). Although the outcome of
pediatric high-grade gliomas (HGGs) is marginally superior to that
of their adult counterparts, it remains poor, despite aggressive
treatment (2).
The two main prognostic factors are the primary
location and resectability. Currently, up to 85% of patients with
hemispheric GBM succumb to the disease within 2 years of diagnosis
(3). The genetic alterations
involved in the pathogenesis of pediatric HGGs, particularly the
role of somatic driver mutations (K27M) in the histone H3 genes
(H3F3 and H1ST1H3B), were previously investigated. Another mutation
in H3F3A, G34R/V, has also been described, and appears to be more
frequent among young adults (4).
The histone methyltransferases are responsible for
the methylation of histone H3 at Lys27 and the K27M mutation
replaces lysine with methionine, while the G34V/R mutation in amino
acid 34 initiates the replacement of glycine with either valine or
arginine (5). Histone proteins
undergo modifications, such as acetylation, methylation and
phosphorylation, which are mediated by writers
(methyltransferases), erasers (demethylases) and readers (which
recognize specific histone modifications). These mechanisms on
histone tails are important in facilitating gene transcription
(6). It is well-established that
the misregulation of histone lysine methylation is implicated in
the occurrence and development of numerous types of cancer
(4); however, the role of the K27M
mutation in the development of glioma remains unclear.
Considering the clinical implications, H3.3K27M
mutations have been found to be associated with poor prognosis of
pediatric HGGs, which appear to have diffuse growth patterns and a
midline localization. This group is classified as diffuse midline
glioma, H3K27M-mutant, and includes previously defined entities,
such as diffuse intrinsic pontine glioma (DIPG) (7).
Furthermore, the two main histone H3 variants have
been associated with distinct biological characteristics, thereby
driving different oncogenic programs. Tumors with mutated H1ST1H3B
were found to be less aggressive and more responsive to
radiotherapy and standard treatment. Histone H3F3A and HIST1H3B
K27M mutations in HGGs help define the two subgroups of HGG, which
exhibit different prognoses and phenotypes (8).
The BRAF V600E mutation is commonly present in
xanthoastrocytomas and gangliogliomas, but it has also been
described in diffuse astrocytoma and low-grade glioma (LGG). It was
previously demonstrated that the presence of mutations has
diagnostic, prognostic and therapeutic potential. In fact, BRAF
mutations may help differentiate benign from aggressive cancers;
tumors with a BRAF V600E mutation were strongly associated with an
increased risk of progression and development of a secondary cancer
with anaplastic evolution. Moreover, BRAF mutations were suggested
as possible targets for the improved adjuvant treatment of residual
or recurrent tumors (9); no
patients with double-mutant tumors (H3 and BRAF) were
identified.
In the present study, the presence of mutations and
their associations with clinical characteristics were compared in
pediatric HGGs, with the aim to define the prognostic role of these
mutations by comparing the genetic and clinical data with those
reported in the existing literature.
Materials and methods
Patient studies
A total of 42 patients with HGGs recruited between
January 2003 and January 2016 at the Meyer Children's University
Hospital (Florence, Italy) were considered to be eligible for
inclusion in the present study. Histological diagnosis and tumor
grading were performed based on the 2016 World Health Organization
criteria (10). The diagnoses were
confirmed following review by the CNS national panel of
pathologists. The study protocol was approved by the institutional
ethics board and informed consent was obtained from the parents or
legal guardians of all the patients.
Genetic analysis
The majority of the analyses were performed
following DNA extraction from fresh-frozen tissues using a specific
protocol (Qiagen GmbH). The sequence analysis of the coding region
of the gene was prepared using the BigDye Terminator v1.1 Cycle
Sequencing kit (Applied Biosystems; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol, and sequenced on a
310 Genetic analyzer (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The following primer sequences were used: BRAF-EX15 forward,
TCTAAGAGGAAAGATGAAGTACTATG and reverse, AGACCTTCAATGACTTTCTAGTAA;
and H3F3A forward, ATCGTGGCAGGAAAAGTT and reverse,
TTTAAGCAGTAGTTAAGTGTTCAAATG; and HIST1H3B forward,
GTCTCTGCAGGCAAGCTTTT and reverse, CAACTCGGTCGACTTTTGGT.
For the molecular analysis of the genes, the coding
portions and the respective flanking regions were amplified by PCR.
Once the systems were purified by capillary electrophoresis (CE),
software analysis was performed.
Statistical analysis
A Student's t-test was used to compare the levels
between the mutated and wild-type tumors. The log-rank (Mantel-Cox)
test was used to determine the association between the mutations
and overall survival (OS). The patient outcome (OS) was analyzed
using the following clinicopathological and treatment-related
variables: Age, sex, tumor size, stage at presentation,
histological subtype and response to chemotherapy. Survival was
measured from the time of diagnosis to the date of death or the
date of the last follow-up. Survival curves were drawn using the
Kaplan-Meier product limit method. P<0.05 was considered to
indicate a statistically significant difference.
Results
Patients and treatment
A total of 42 patients were enrolled in the present
study and their main clinicopathological characteristics are
summarized in Table I. The median
age at the time of diagnosis was 7 years (range, 0-32 years), while
10 patients were aged <3 years. All subjects were treated with
chemotherapy and/or radiotherapy according to the clinical studies
of Meyer Children's University Hospital. The tumors comprised 10
GBMs (23.8%) and 32 AAs (76.1%). In total, 21 patients underwent
partial resection of the lesion (R2), 11 underwent complete
resection (R0) and 10 patients had a biopsy. The second observation
was performed at the time of recurrence in 10 patients (1 R0, 9
R2).
 | Table IClinical characteristics of the
patients. |
Table I
Clinical characteristics of the
patients.
| Characteristics | No. (%) |
|---|
| Age, years | |
|
Median
(range) | 7 (0-32) |
|
<3 | 10 |
|
>3 | 32 |
| Sex | |
|
Female | 20 (47.6) |
|
Male | 22 (52.4) |
| Tumor histology | |
|
Anaplastic
astrocytoma | 32 (76.1) |
|
Glioblastoma
multiforme | 10 (23.8) |
| Mutations in patients
aged >3 years (n=24) | |
|
H3F3A
K27M | 7 (29.1) |
|
H1ST1H3B
K27M | 1 (4.1) |
|
H3F3A
G34R | 0 |
|
BRAF V600
E | 5 (20.8) |
|
IDH | 0 |
|
P53 | 0 |
The 32 patients aged >3 years were treated with
chemotherapy and radiotherapy. Patients aged <3 years were
treated with adjuvant chemotherapy [standard-dose chemotherapy
(SDCT) and high-dose chemotherapy (HDCT) with autologous stem cell
rescue]. Second-line chemotherapy at the time of recurrence was
administered in 23 patients, 10 of whom received two lines of
palliative chemotherapy.
Mutation analysis
K27M mutation analysis was performed in 24 of the
patients aged >3 years; the mutation in histone H3.3 leading to
the K27M amino acid substitution was identified in 7/24 (21%) HGGs.
However, the H1ST1H3B mutation was only recorded in 1 patient with
GBM. The subsequent analysis to determine the association between
OS and mutations was performed using the group of 24 patients.
The H3.3K27M mutation was present in 3 GBM and 3 AA
cases. No correlation was identified between the mutation and tumor
histology (P=0.61). Patients with the mutation appeared to have
progressive disease, but without a statistically significant
correlation (P=0.07). However, patients carrying this mutation had
a significantly worse prognosis compared with the wild-type
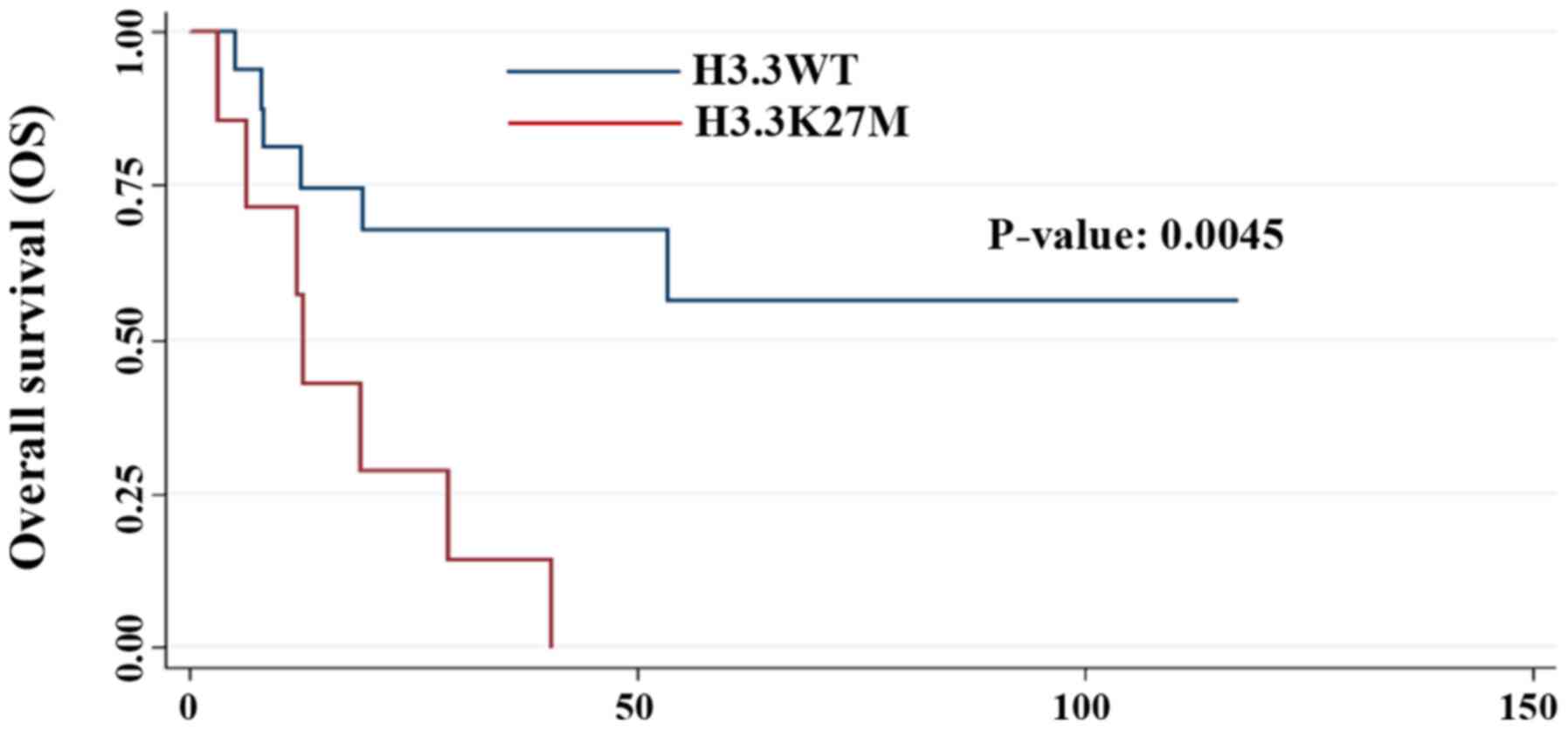
patients (P=0.0045; Fig. 1). As
regards localization, all the tumors with the H3.3K27M mutation
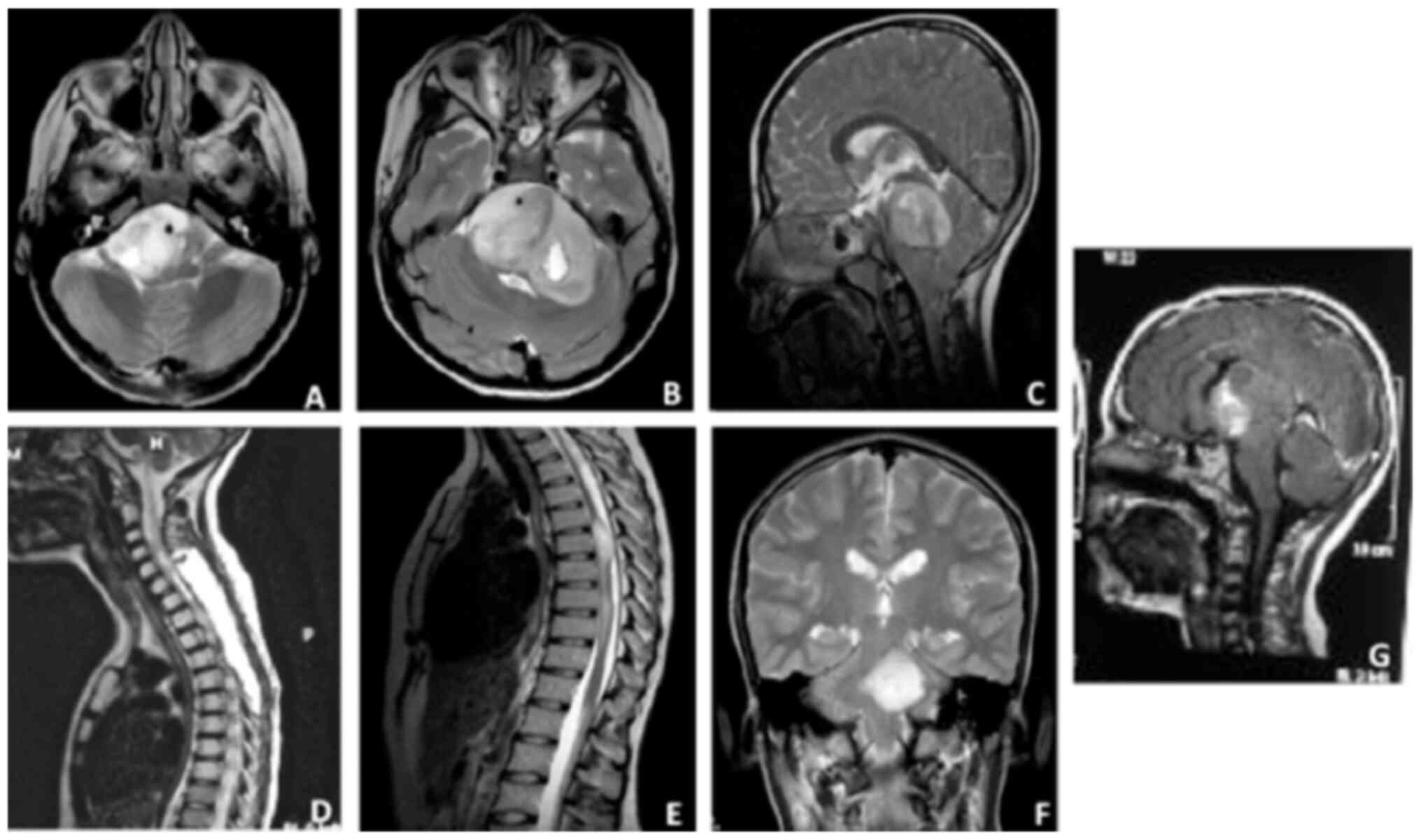
were midline tumors (2 in the spinal cord, 3 in the brainstem, 1 in
the cerebellopontine angle and 1 in the pineal gland; Fig. 2).
The BRAF V600E mutation was identified in 5 patients
(21%), in 3 of whom the HGG developed from a previous LGG
(P=0.001). The median age of the patients with this mutation was
6.8 years. In addition, 3 of these tumors were reported in
females.
Treatment and survival
The median OS the 42 patients was 77 months. The
median OS for patients aged >3 years was 38.5 months, while for
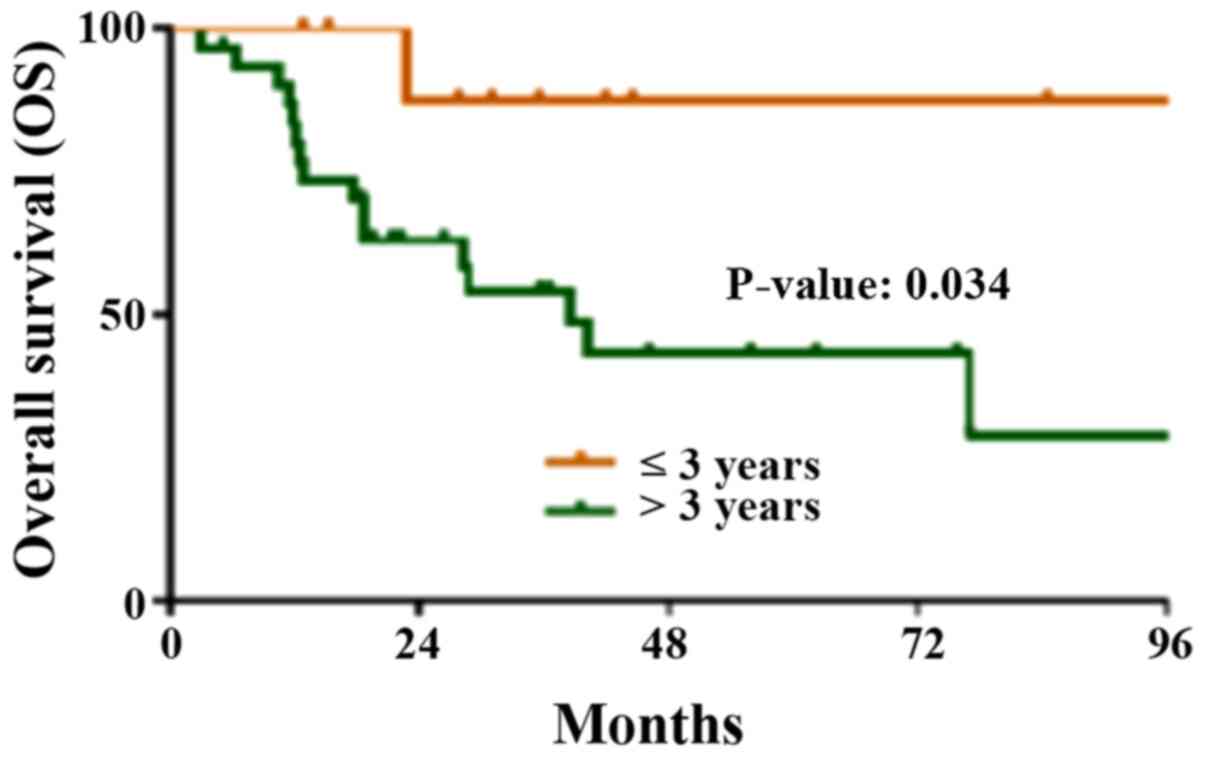
children aged <3 years the median OS was not reached (P=0.034;
Fig. 3). Considering the 42
patients with HGGs, the therapeutic goal was to achieve, where
possible, gross total resection following chemotherapy and
radiotherapy, with as less structural damage as possible. A total
of 11 patients underwent gross total resection (R0), while in
patients receiving non-curative resection (R2), consisting of
incomplete resection (n=21) and biopsy (n=10), the median OS were
not reached (40.1 and 1.5 months, respectively; P=0.18).
Subsequently, 10/42 patients (23.8%) aged <3
years were administered HDCT with autologous bone marrow stem cell
transplant. These patients did not reach median OS. Schedules with
exclusive SDCT were administered to 22/42 patients (53.6%), who
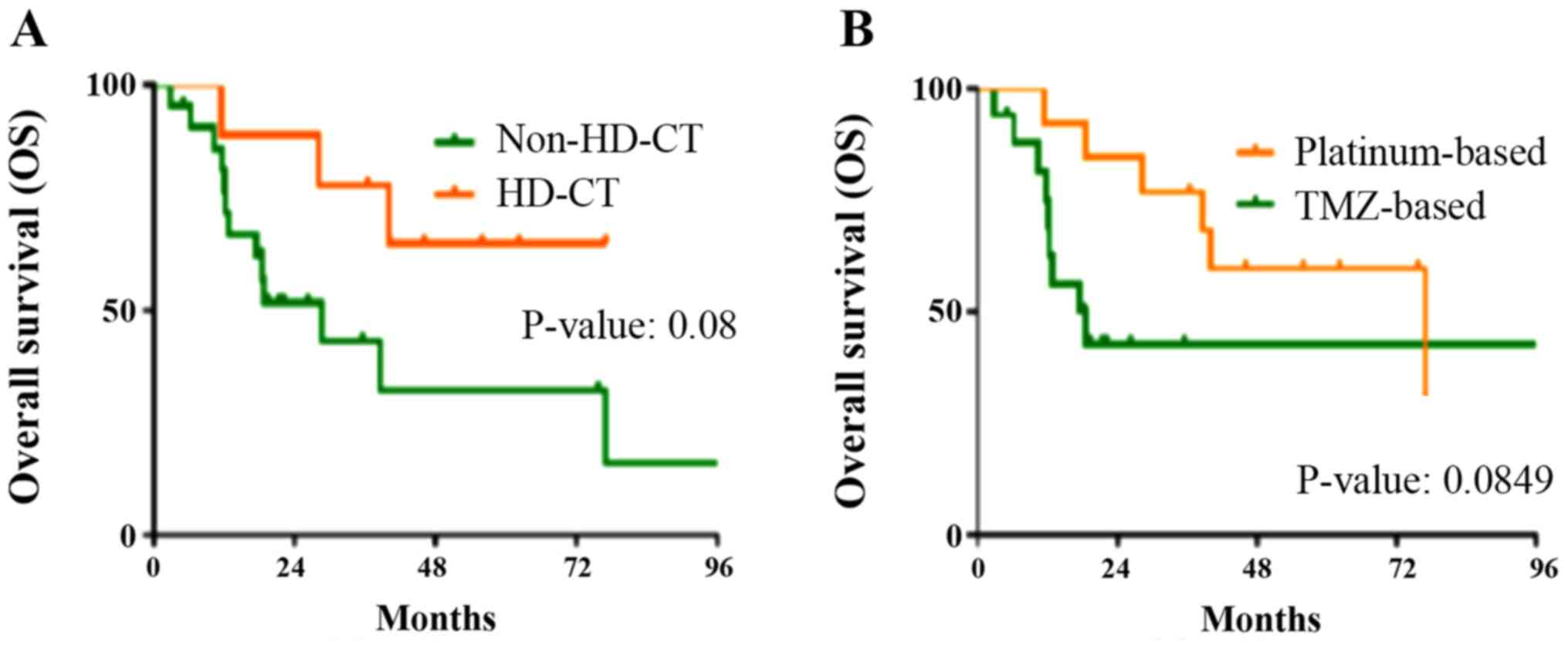
demonstrated a median OS of 28.6 months, without a statistically
significant trend (P=0.08; Fig.
4A). As regards SDCT, a temozolomide (TMZ)-based regimen was
used in 17 cases and a platinum (CDDP)-based regimen in the
remaining cases. The median OS of the patients who received
CDDP-based treatment was 77 months, while it was 18.5 months for
the patients who received TMZ-based treatment. No statistically
significant differences in survival were observed between the TMZ-
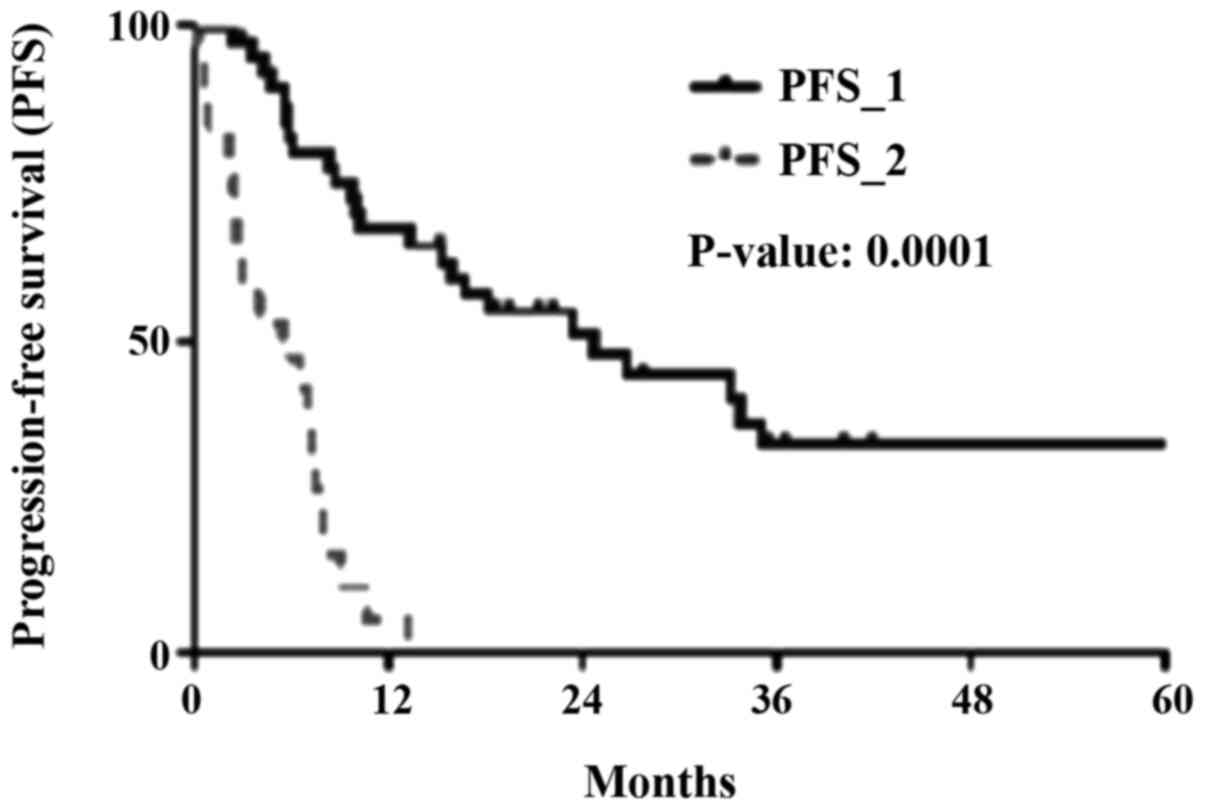
and CDDP-based chemotherapy regimens (P=0.0849; Fig. 4B). Regarding the palliative
treatments, the progression-free survival (PFS) of the secondary
and tertiary treatments was 6.5 and 2.1 months, respectively. The
univariate analysis revealed a significant difference in the PFS
curves between the primary (all patients or only patients with
progressive disease) and subsequent treatments (P=0.0001, Fig. 5).
In addition, 24 patients (57.1%) experienced disease
progression, 14 of whom were subjected to a salvage
second-observation surgery. The median OS of these patients was
28.7 months compared with 25.3 months when salvage surgery was not
performed (P=0.957). Furthermore, 22 patients (91.6%) received
palliative multimodal anticancer treatments, including chemotherapy
and radiotherapy, following disease progression.
Discussion
Pediatric HGGs are characterized by different
molecular drivers, including mutations in the H3 histone variants
K27M (HIST1H3B and H3F3A), H3F3A G34R, BRAF V660E, IDH and p53. The
H3.3K27M mutation has been reported in several high-risk CNS
malignancies, such as midline diffuse gliomas (5,11,12).
In particular, the K27M mutation is a well-established specific
marker of pediatric midline glioma. The confirmation of the
presence of this histone mutation is crucial for defining a new
histological category, namely diffuse midline glioma with histone
H3.3K27M mutation (10).
Previous studies have demonstrated that H3.3K27M
drives a distinct subset of glial neoplasms of the CNS, depending
not only on tumor type, but also on the site of origin and patient
age. Gielen et al (2)
analyzed 338 cases of pediatric brain tumors and revealed that the
H3.3K27M mutation was exclusively present in gliomas. In another
study, the mutation rate was 80% in thalamic pediatric GBMs with a
median age of 10.5 years (range, 5-23 years) (13,14).
Schwartzentruber et al reported the presence of the H3.3
variant in 31% of all pediatric GBMs; the H3.3K27M mutation was
more frequent compared with the H3.3G34R/V mutation in pediatric
diffuse high-grade astrocytomas, and the H3.3K27M mutation
primarily occurred in younger patients, whereas the H3.3G34R/V
mutation was found in older patients and young adults (5).
Fontebasso et al suggested that every typical
mutation in pediatric HGGs has a specific neuroanatomical position
and a correlation with age; H3.3K27M occurs in children (1-10
years) and presents with a midline localization; and the IDH1
mutation occurs in adolescents (11-20 years), while the H3.3G34V/R
occurs mutation in young adults (20-45 years), both of which occur
in the cerebral hemispheres (15).
Subsequent analysis demonstrated that the histone H3.3K27M mutation
was present in the majority of high-grade infiltrative astrocytomas
arising within the midline structures (thalamus, brainstem and
spinal cord) of pediatric and young adult patients. A high
prevalence was particularly recorded in cases of thalamic GBM in
young adults between 20 and 46 years of age, accounting for 90%
(9/10 cases) of thalamic HGGs (16,17).
In the present study, 24/42 patients were eligible
for mutational analysis. The H3.3K27M mutation was identified in 7
patients (29.1%). As regards tumor histology in patients with the
H3.3K27M mutation, 3 were diagnosed with AAs and 4 with GBMs. No
significant correlation was observed between tumor histology and
the H3.3K27M mutation (P=0.6126). These findings were consistent
with the findings of Karremann et al who also did not report
any prognostic impact on the survival rates related to tumor
histology (18).
It has been demonstrated that the presence of the
H3.3K27M mutation in pediatric HGG was associated with an
aggressive clinical behavior and a poor prognosis, including tumors
that are histologically low-grade (P=0.025) (4,13,19).
The multivariate analysis of the present study confirmed that
H3.3K27M positivity was a significant prognostic factor
(P=0.0045).
In a study published in 2018 involving 474 patients
with pediatric HGG, including 258 (54%) patients with the H3K27M
mutation and 216 (46%) without the mutation, the presence of the
mutation was independently and significantly associated with a
worse prognosis (HR=3.630; P<0.001). The patients with the
mutation had a significantly shorter OS (by 2.3 years; P=0.008) and
they were >3 times more likely to succumb to the disease
compared with H3 wild-type patients (20). However, a recent study concluded
that the poor prognosis was due to the diffuse or infiltrative
nature of the tumor; in fact, the prognosis of H3 wild-type diffuse
gliomas remained significantly worse compared with that of the
H3.3K27M-mutated gliomas (21).
The results of the present study indicated the
important diagnostic and prognostic role of the H3.3K27M mutation
in pediatric gliomas. In addition, the role of the H3.3K27M
mutation may not be limited to the prognostic aspect, but it may
also have therapeutic implications. Recently, retrospective studies
analyzed the efficacy of histone deacetylase (HDAC) inhibitor drugs
in patients with H3-mutant tumors. Felix and Fontenele (22) revealed a possible effect of valproic
acid on the survival of patients with GBM through a HDAC-inhibiting
effect. Thus, although this mechanism remains to be elucidated,
H3.3K27M mutations may make tumor cells more sensitive to HDAC
inhibitors. Moreover, it has been hypothesized that the small
molecule GSKJ4 may increase H3K27M methylation through inhibiting
JMJD3 H3K27 histone demethylase; several in vitro studies
have reported that patient-derived H3.3K27M cell lines treated with
GSKJ4 demonstrated decreased proliferation rates and cell viability
(23). In addition, an ongoing
phase 1 study at the University of California is currently
evaluating the efficacy of a H3.3K27M-specific vaccine in
HLA-A2+ children and young adults with H3.3K27M DIPGs
and other HGGs. In that study, patients with high-risk glioma
positive for HLA-A2 and the H3.3K27M mutation received the specific
H3.3K27M peptide vaccine following radiotherapy (ClinicalTrials.gov Identifier: NCT02960230).
Interestingly, advanced studies have emphasized the
discrepancies in H3.1 and H3.3 mutations, which have different
clinical and prognostic characteristics. In the present analysis,
only 1 patient with GBM carried a H1ST1H3B mutation, and the
patient succumbed to the disease after the first round of therapy.
Castel et al (8) reported
that patients with H3.1 mutations exhibited an improved clinical
response to radiotherapy with a more favorable OS compared with
patients with H3.3 mutations. Thus, a more in-depth genetic
characterization of the genetic signature of pediatric HGGs may
help guide clinical trials with drugs that inhibit histone
modifications. In addition to the effect of the treatment, it has
been suggested that a more appropriate predictor of survival may be
the type of histone H3 mutation rather than the
clinical-radiological risk score. A multi-agent approach,
inhibiting multiple targets, may increase treatment efficacy,
exhibit synergy and possibly avoid the development of resistance
(7).
The results obtained in the present study with
regards to the presence of the BRAF V600E mutation, which,
according to the literature, is associated with a poor prognosis,
suggested its association with progressive disease with a poor
response to conventional chemotherapy. It is therefore necessary to
screen for BRAF V600E in LGGs, so that they may be re-evaluated
over time, which may necessitate a different short-term and
long-term therapeutic approach and eventually define early targeted
therapies (9).
In conclusion, the results of the present study
underlined the role of H3.3K27M and BRAF V600E mutations in
pediatric gliomas, similar to previously reported studies. The
present study validated the important diagnostic and prognostic
role of these mutations in HGG, highlighting the worse prognosis
that accompanies H3K27M mutations and the high risk of progression
in patients harboring BRAF V600E. Thus, the presence of histone
mutations and all other mutations that characterize pediatric HGG
is crucial and should be further investigated.
Acknowledgements
The authors would like to thank Dr Maurizio Lucchesi
for his valuable and constructive support during the development of
this research. We also thank Dr Veronica De Gregorio for her
technical assistance.
Funding
The present study was funded by ‘La Forza di Giò-ovd’ and
Fondazione Anna Meyer- Firenze.
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article or are available from
the corresponding author on reasonable request.
Authors' contributions
MG carried out literature search, study design, data
collection, analysis and interpretation, figure preparation and
manuscript writing. LGi carried out genetic analysis. AMB and CC
carried out data collection and pathological analysis. MLC carried
out statistical and data analysis. LGa and LGe carried out data
collection and wrote the manuscript. IS conceved the study,
analysed and interpreted data, and wrote the manuscript. All the
authors have contributed to the critical revision of the
manuscript. All the authors have read and approved the final
version.
Ethics approval and consent to
participate
The study protocol was approved by the Institutional
Ethics Review Board and all patient data were derived from publicly
available datasets.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Merchant TE, Pollack IF and Loeffler JS:
Brain tumors across the age spectrum: Biology, therapy, and late
effects. Semin Radiat Oncol. 20:58–66. 2010.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Gielen GH, Gessi M, Hammes J, Kramm CM,
Waha A and Pietsch T: H3F3A K27M mutation in pediatric CNS tumors:
A marker for diffuse high-grade astrocytomas. Am J Clin Pathol.
139:345–349. 2013.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bouffet E, Tabori U, Huang A and Bartels
U: Possibilities of new therapeutic strategies in brain tumors.
Cancer Treat Rev. 36:335–341. 2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Venneti S, Santi M, Felicella MM, Yarilin
D, Phillips JJ, Sullivan LM, Martinez D, Perry A, Lewis PW,
Thompson CB and Judkins AR: A sensitive and specific
histopathologic prognostic marker for H3F3A K27M mutant pediatric
glioblastomas. Acta Neuropathol. 128:743–753. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Schwartzentruber J, Korshunov A, Liu XY,
Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA,
Tönjes M, et al: Driver mutations in histone H3.3 and chromatin
remodelling genes in paediatric glioblastoma. Nature. 482:226–231.
2012.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Bernstein BE, Mikkelsen TS, Xie X, Kamal
M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al:
A bivalent chromatin structure marks key developmental genes in
embryonic stem cells. Cell. 125:315–326. 2006.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Vanan MI, Underhill DA and Eisenstat DD:
Targeting epigenetic pathway in the treatment of pediatric diffuse
(high grade) gliomas. Neurotherapeutics. 14:274–283.
2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Castel D, Philippe C, Calmon R, Le Dret L,
Truffaux N, Boddaert N, Pagès M, Taylor KR, Saulnier P, Lacroix L,
et al: Histone H3F3A and HIST1H3B K27M mutations define two
subgroups of diffuse intrinsic pontine gliomas with different
prognosis and phenotypes. Acta Neuropathol. 130:815–827.
2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Lassaletta A, Zapotocky M, Mistry M,
Ramaswamy V, Honnorat M, Krishnatry R, Guerreiro Stucklin A,
Zhukova N, Arnoldo A, Ryall S, et al: Therapeutic and prognostic
implications of BRAF V600E in pediatric low-grade gliomas. J Clin
Oncol. 35:2934–2941. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Louis DN, Perry A, Reifenberger G, von
Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD,
Kleihues P and Ellison DW: The 2016 world health organization
classification of tumors of the central nervous system: A summary.
Acta Neuropathol. 131:803–820. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Rheinbay E, Louis DN, Bernstein BE and
Suvà ML: A tell-tail sign of chromatin: Histone mutations drive
pediatric glioblastoma. Cancer Cell. 21:329–331. 2012.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wu G, Broniscer A, McEachron TA, Lu C,
Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al:
Somatic histone H3 alterations in pediatric diffuse intrinsic
pontine gliomas and non-brainstem glioblastomas. Nat Genet.
44:251–253. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Khuong-Quang DA, Buczkowicz P, Rakopoulos
P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S,
Schwartzentruber J, Letourneau L, et al: K27M mutation in histone
H3.3 defines clinically and biologically distinct subgroups of
pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol.
124:439–447. 2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Sturm D, Witt H, Hovestadt V, Khuong-Quang
DA, Jones DT, Konermann C, Pfaff E, Tönjes M, Sill M, Bender S, et
al: Hotspot mutations in H3F3A and IDH1 define distinct epigenetic
and biological subgroups of glioblastoma. Cancer Cell. 22:425–437.
2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Fontebasso AM, Liu XY, Sturm D and Jabado
N: Chromatin remodeling defects in pediatric and young adult
glioblastoma: A tale of a variant histone 3 tail. Brain Pathol.
23:210–216. 2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Aihara K, Mukasa A, Gotoh K, Saito K,
Nagae G, Tsuji S, Tatsuno K, Yamamoto S, Takayanagi S, Narita Y, et
al: H3F3A K27M mutations in thalamic gliomas from young adult
patients. Neuro Oncol. 16:140–146. 2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Buczkowicz P, Bartels U, Bouffet E, Becher
O and Hawkins C: Histopathological spectrum of paediatric diffuse
intrinsic pontine glioma: Diagnostic and therapeutic implications.
Acta Neuropathol. 128:573–581. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Karremann M, Gielen GH, Hoffmann M, Wiese
M, Colditz N, Warmuth-Metz M, Bison B, Claviez A, van Vuurden DG,
von Bueren AO, et al: Diffuse high-grade gliomas with H3 K27M
mutations carry a dismal prognosis independent of tumor location.
Neuro Oncol. 20:123–131. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Korshunov A, Ryzhova M, Hovestadt V,
Bender S, Sturm D, Capper D, Meyer J, Schrimpf D, Kool M, Northcott
PA, et al: Integrated analysis of pediatric glioblastoma reveals a
subset of biologically favorable tumors with associated molecular
prognostic markers. Acta Neuropathol. 129:669–678. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Lu VM, Alvi MA, McDonald KL and Daniels
DJ: Impact of the H3K27M mutation on survival in pediatric
high-grade glioma: A systematic review and meta-analysis. J
Neurosurg Pediatr. 23:308–316. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Pratt D, Natarajan SK, Banda A, Giannini
C, Vats P, Koschmann C, Mody R, Chinnaiyan A and Venneti S:
Circumscribed/non-diffuse histology confers a better prognosis in
H3K27M-mutant gliomas. Acta Neuropathol. 135:299–301.
2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Felix F and Fontenele J: Valproic acid may
be tested in patients With H3F3A-mutated high-grade gliomas. J Clin
Oncol. 34:3104–3105. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Hashizume R, Andor N, Ihara Y, Lerner R,
Gan H, Chen X, Fang D, Huang X, Tom MW, Ngo V, et al: Pharmacologic
inhibition of histone demethylation as a therapy for pediatric
brainstem glioma. Nat Med. 20:1394–1396. 2014.PubMed/NCBI View
Article : Google Scholar
|