Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of
the most aggressive epithelial tumors with a 5-year survival rate
of <10% (1), which dismal
prognosis is greatly related to a diagnosis at late stages and few
effective treatment options. The prognosis of patients with PDAC
has barely changed over the past two decades, as there are no
reliable biomarkers for early detection (2). Although modest advances have been made
in treatment options with combination therapies (3-5),
recurrence rates remain high (~80%), with patients relapsing within
2 years (6). Hence, implementation
of new diagnostic methods, such as liquid biopsy, may help enhance
detection accuracy and monitoring tumor progression in real
time.
PDAC occurs due to the accumulation of multiple
genetic alterations, including activation of oncogenes or loss of
tumor-suppressors, as well as aberrant function of signaling
pathways (7). Acquisition of
mutations in KRAS (KRAS proto-oncogene, GTPase) is regarded
as a driver event in PDAC. However, several in vivo studies
showed that mutated KRAS alone is insufficient to trigger
metastatic transformation (8).
Combination with other frequently found inactivating mutations in
genes such as CDKN2A (cyclin-dependent kinase inhibitor 2A),
SMAD4 (SMAD family member 4), and TP53 (tumor protein
p53), or epigenetic changes in key genes, are recognized to further
enhance tumorigenesis and metastasis. In fact, 70-90% of PDAC cases
harbor co-occurring KRAS and TP53 mutations (9), constituting the most common genetic
alterations in PDAC. Although overexpression of wild-type and
mutated KRAS is well recognized in colorectal and non-small
cell lung cancers (10,11), it remains poorly known in PDAC.
Here, we report the case of a patient with advanced
PDAC with multiple liver metastases found to bear marked
amplification of the oncogenic KRASG12D allele as
detected by liquid biopsy. Mutant KRAS amplification may
have important clinical implications, including increased risk for
resistance to treatment.
Case report
A 52-years-old Japanese man with no relevant medical
history visited our hospital in early July 2019 with chief
complaints of persistent upper abdominal pain for 2 months. The
patient had a 36-year history of smoking and daily alcohol
consumption. No family history of cancer was reported. Physical
examination showed high fever (>38.0˚C) and tenderness in the
upper abdomen. Laboratory data revealed mild liver dysfunction and
normal levels of carbohydrate antigen 19-9 (CA19-9; 31.9 U/ml),
with duke pancreatic monoclonal antigen type 2 (DUPAN-2) >1,600
U/ml. Abdominal contrast-enhanced computed tomography (CT) revealed
a hypovascular tumor mass of 25 mm in the head of the pancreas.
Multiple liver metastases with different masses were detected and
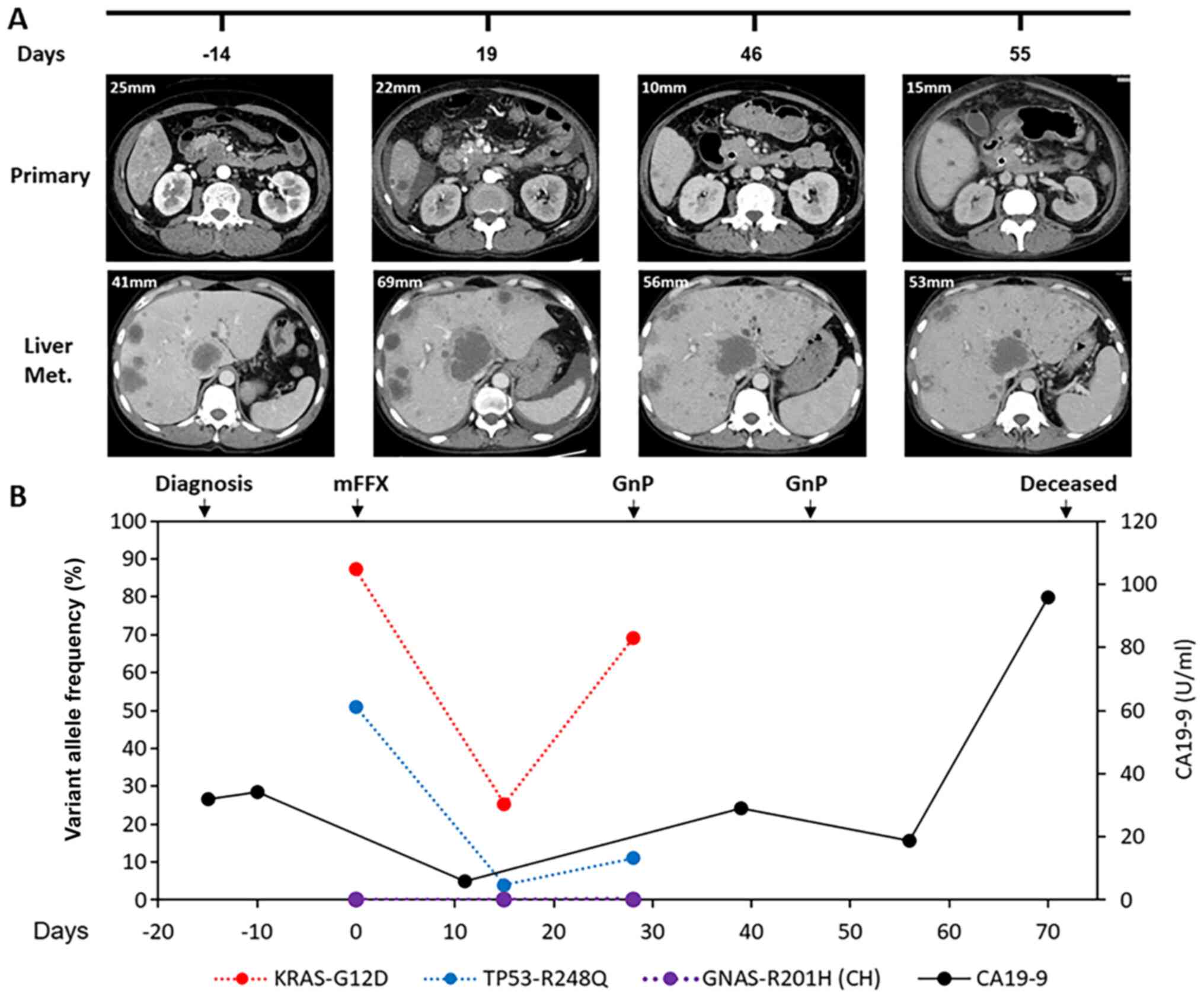
no metastases at other sites were evident on CT (Fig. 1A). The patient was immediately
admitted, and endoscopic ultrasound-guided fine needle aspiration
was performed on the primary tumor and metastases. Histological
analysis confirmed that it was an adenocarcinoma, classified as cT3
cN0 cM1(Hep) and cStage IV according to the Union for International
Cancer Control criteria and Tumor-Node-Metastasis classification
(12). The patient started
FOLFIRINOX (mFFX; folinic acid, fluorouracil, irinotecan, and
oxaliplatin combo) therapy 2 weeks after the diagnosis in July
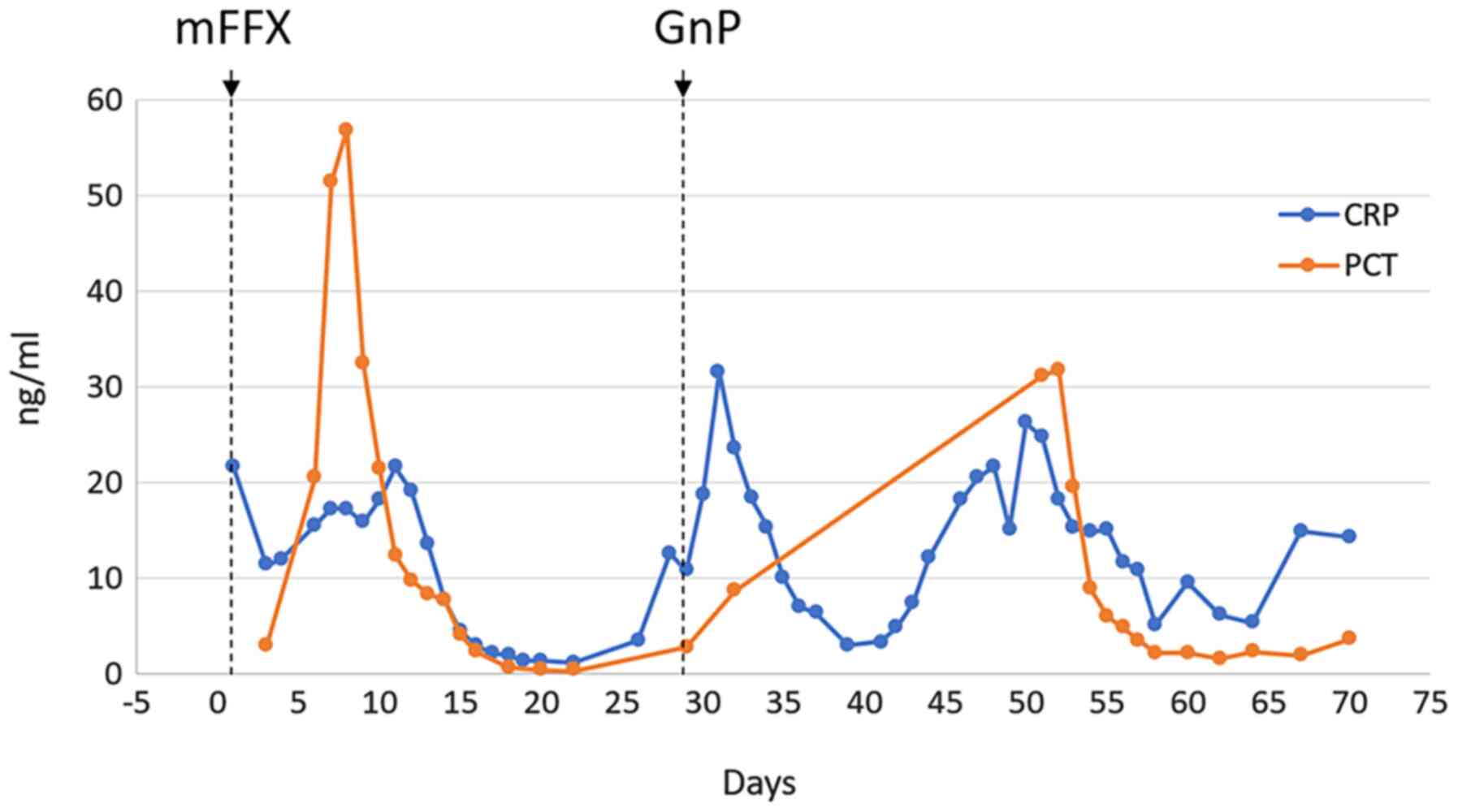
2019. On day 3 of treatment, the patient experienced liver
dysfunction with increased levels of uric acid and creatinine. On
day 6, a strong myelosuppressive effect [white blood cell (WBC):
600/µl, neutrophils: 256/µl, and platelets: 40,000/µl] was observed
along with disseminated intravascular coagulation and acute renal
failure. On day 8, the patient experienced encephalopathy and a
marked increase in the levels of procalcitonin (PCT; 56.9 ng/ml)
and C-reactive protein (CRP; 21.62 mg/dl) were observed, most
likely as a result of tumor tissue damage. On day 19, a CT
assessment revealed a reduction in the primary lesion; thus, the
mFFX treatment was initially considered to be effective. However,
soon after, a strong fever recurred with increased CRP levels.
Therefore, mFFX re-administration was considered severely adverse
and intolerable. With the patient informed consent, the regimen was
changed and treatment continued with an intravenously administrated
second-line therapy, named GnP, comprising gemcitabine (1,000
mg/m2) and nab-paclitaxel (125 mg/m2)
(13). GnP did not induce adverse
reactions as potent as mFFX, but still resulted in an inflammatory
response and elevated levels of procalcitonin (Fig. 2). Despite a slight recovery after
chemotherapy, the patient general condition continued to
deteriorated and a myriad of new metastatic liver tumors emerged
with uncontrollable growth patterns. The patient died of
gastrointestinal bleeding associated with disseminated
intravascular coagulation 70 days after treatment initiation.
Genetic analysis was performed in both tumor tissue
and liver biopsies by amplicon-based next-generation sequencing
(NGS) with the Ion AmpliSeq Comprehensive Cancer Panel (Thermo
Fisher Scientific, Inc.) of 509 genes. Activating
KRASG12D and TP53R248Q
mutations, along with increased copy number variations of the
proto-oncogenes MYC (MYC proto-oncogene, bHLH transcription
factor) and MAF (MAF bZIP transcription factor) were
detected in both primary tumor and metastasis samples (Tables I and II). Plasma samples were collected just
before the first-line chemotherapy and in weeks 2 and 4 after
treatment initiation, and circulating tumor DNA (ctDNA) was
analyzed (Tables III and IV). Ultradeep targeted NGS with the
Oncomine pan-cancer cell-free assay (Thermo Fisher Scientific) was
used to investigate genetic alterations in 52 genes. ctDNA from
before the first line of treatment confirmed the genetic
alterations found in the primary tumor and revealed an
amplification in the mutant KRASG12D allele
[variant allele frequency (VAF) = 87.2%). Mutant allele
amplification was detected based on the capped molecular depth
(19,999x] obtained in the ctDNA sequence, which surpassed by at
least 4-fold the maximum expected molecular depth in unamplified
regions (4,980x) based on the sample input (16.6 ng). A 5-fold
amplification of KRASG12D in the metastasis
samples (VAF=91.5%) was subsequently confirmed by digital
polymerase chain reaction and NGS sequencing. A
GNASR201H mutation (VAF=0.05%) was also detected
in the plasma liquid biopsy. Since this mutation was not present in
the primary tumor tissue, genomic DNA from the WBCs was also
analyzed. The GNASR201H mutation was confirmed in
the WBCs (VAF=0.1%), indicating its association with clonal
hematopoiesis rather than with the pancreatic tumor (Table I). Analysis of ctDNA 2 weeks after
the first mFFX cycle showed an initial decrease in
KRASG12D and TP53R248Q
frequency (VAF = 25.3 and 3.9%, respectively). However, ctDNA
analysis at week 4 of mFFX indicated an upregulation of the mutant
VAF levels and in KRASG12D amplification, close
to the levels prior to treatment (Fig.
1B; Table III).
 | Table IMutations detected in tissue
samples. |
Table I
Mutations detected in tissue
samples.
| | Primary tissue | Liver metastasis | White blood
cells |
|---|
| Mutation | Mol depth, x | Counts, n | VAF, % | Mol depth, x | Counts, n | VAF, % | Mol depth, x | Counts, n | VAF, % |
|---|
| KRAS-G12D | 3,831 | 1,766 | 46.10 | 18,844a | 17,245 | 91.51 | 3,529 | 0 | 0.00 |
| TP53-R248Q | 2,194 | 1,129 | 51.46 | 1,975 | 1,209 | 61.22 | 9,544 | 0 | 0.00 |
| GNAS-R201H | 4,073 | 0 | 0.00 | 4,949 | 0 | 0.00 | 6,812 | 6 | 0.10 |
| Table IICNVs detected in tissue samples. |
Table II
CNVs detected in tissue samples.
| | Primary tissue | Liver metastasis | White blood
cells |
|---|
| Gene <CNV> | Copy no. | CNV ratio | Copy no. | CNV ratio | Copy no. | CNV ratio |
|---|
| KRAS | 4 | 2.0 | 10 | 5.0 | 0 | 0 |
| MYC | 6 | 3.1 | 6 | 3.0 | 0 | 0 |
| MAF | 7 | 3.5 | 8 | 4.0 | 0 | 0 |
| Table IIIMutations detected in ctDNA
samples. |
Table III
Mutations detected in ctDNA
samples.
| | Before treatment | 2 weeks after
treatment | 4 weeks after
treatment |
|---|
| Mutation | Mol depth, x | Counts, n | VAF, % | Mol depth, x | Counts, n | VAF, % | Mol depth, x | Counts, n | VAF, % |
|---|
| KRAS-G12D | 19,999a | 17,449 | 87.20 | 7,111 | 1,798 | 25.30 | 13,736a | 9,493 | 69.10 |
| TP53-R248Q | 3,144 | 1,602 | 51.00 | 4,207 | 165 | 3.92 | 3,794 | 418 | 11.00 |
| GNAS-R201H | 3,787 | 4 | 0.05 | 6,175 | 5 | 0.08 | 6,606 | 8 | 0.12 |
| Table IVCNVs detected in ctDNA samples. |
Table IV
CNVs detected in ctDNA samples.
| | Before
treatment | 2 weeks after
treatment | 4 weeks after
treatment |
|---|
| Gene
<CNV> | Copy no. | CNV ratio | Copy no. | CNV ratio | Copy no. | CNV ratio |
|---|
| MYC | 2.8 | 1.4 | 2.1 | 1.0 | 2.4 | 1.2 |
Discussion
KRAS activating mutations and TP53,
CDKN2A, and SMAD4 loss-of-function alterations are
the most common genetic alterations found in PDAC. Nevertheless, a
large number of infrequent mutations and copy number variations in
multiple genes are also detected, resulting in significant
interindividual heterogeneity (7,14). In
addition, the oncogenic effect of MYC is well established as a
critical effector of activated RAS in several cancer types,
including PDAC (15).
Allelic imbalance caused by amplification of mutant
KRAS is more frequently reported in high-grade tumors of
NSCLC and can affect its response to therapy (10,11).
Amplification of KRASmut in PDAC, although less
documented, confers an increased metastatic potential by inducing
robust epithelial-mesenchymal transition signatures, being
associated to worse prognosis (16). However, one of the challenges in
accurately detecting gene amplification in PDAC is the presence of
high stromal cell content within the tumor tissue (17), with non-neoplastic stroma
confounding precise gene dosage and comprehensive interpretation of
copy number alterations. Although tissue biopsies are the gold
standard for diagnosis and molecular characterization of tumors,
the analysis of ctDNA from liquid biopsies can avoid the
interference of non-neoplastic stromal cells and capture the
intrinsic influence of tumor heterogeneity during the course of the
disease. In this case, we detected KRASG12D
amplification in the ctDNA but not in the primary tumor.
Amplification was later confirmed in the metastatic tissue,
reflecting the heterogeneous evolution of the tumor.
KRASG12D amplification was associated with rapid
tumor growth, suggesting that it may play an important role in
promoting the metastatic spread of PDAC cells. In addition, the
poor response of the patient to both lines of treatment suggests
that the presence of amplified KRASG12D may also
impair the tumor sensitivity to chemotherapy. Recognition of this
information in advance may help predict treatment-related
deterioration of the patient general condition. This is of
particular importance for selecting treatment approaches that
consider the rate of tumor collapse to help control poor prognosis
PDAC cases. Hence, albeit underrated in the clinical setting,
amplification of oncogenic KRASG12D, which
constitutes a key driving force that adds to an aggressive PDAC
phenotype, can be detected in ctDNA through routine liquid
biopsy.
This case highlights the importance of accurate
detection of gene-dosage gains in oncogenic KRAS mutations
in PDAC. KRASG12D amplification in combination
with TP53 mutation and deregulated MYC expression may be
associated an aggressive PDAC phenotype. Hence, in light of the
heterogeneous characteristics of aggressive pancreatic cancers,
monitoring tumor evolution through liquid biopsies can help
identify such cases at earlier stages. Importantly, the
amplification of oncogenic KRASG12D can be
successfully detected through liquid biopsy and is feasible for
implementation in the clinical setting.
Acknowledgements
The authors would like to thank Rie Hayashi, Dr Hiu
Ting Chan and Dr Yoon Ming Chin for technical support.
Funding
The present study was supported by the Council for Science,
Technology and Innovation (CSTI), cross-ministerial Strategic
Innovation Promotion Program (SIP), ‘Innovative AI Hospital System’
[Funding Agency: National Institute of Biomedical Innovation,
Health and Nutrition (NIBIOHN); grant no. SIPAIH18C03].
Availability of data and materials
All data generated or analysed during this study are
included in this published article.
Authors' contributions
MM, YN, SKL, YK and FPS were involved in the
conception, design and execution of the study and manuscript
writing. MM, SKL and FPS collected and analyzed the data. MM and YK
provided study material. MM and FPS confirm the authenticity of all
the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The patient provided informed consent in writing for
participation in the study approved by the Japanese Foundation for
Cancer Research Review Board (IRB 2018-1016).
Patient consent for publication
The patient provided informed consent in writing
regarding the publication of the study approved by the Japanese
Foundation for Cancer Research Review Board (IRB 2018-1016).
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rahib L, Smith BD, Aizenberg R, Rosenzweig
AB, Fleshman JM and Matrisian LM: Projecting cancer incidence and
deaths to 2030: The unexpected burden of thyroid, liver, and
pancreas cancers in the United States. Cancer Res. 74:2913–2921.
2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ducreux M, Cuhna AS, Caramella C,
Hollebecque A, Burtin P, Goéré D, Seufferlein T, Haustermans K, Van
Laethem JL, Conroy T, et al: Cancer of the pancreas: ESMO clinical
practice guidelines for diagnosis, treatment and follow-up. Ann
Oncol. 26 (Suppl 5):v56–v68. 2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Neoptolemos JP, Stocken DD, Bassi C,
Ghaneh P, Cunningham D, Goldstein D, Padbury R, Moore MJ, Gallinger
S, Mariette C, et al: Adjuvant chemotherapy with fluorouracil plus
folinic acid vs. gemcitabine following pancreatic cancer resection:
A randomized controlled trial. JAMA. 304:1073–1081. 2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Uesaka K, Boku N, Fukutomi A, Okamura Y,
Konishi M, Matsumoto I, Kaneoka Y, Shimizu Y, Nakamori S, Sakamoto
H, et al: Adjuvant chemotherapy of S-1 versus gemcitabine for
resected pancreatic cancer: A phase 3, open-label, randomised,
non-inferiority trial (JASPAC 01). Lancet. 388:248–257.
2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sinn M, Bahra M, Liersch T, Gellert K,
Messmann H, Bechstein W, Waldschmidt D, Jacobasch L, Wilhelm M, Rau
BM, et al: CONKO-005: Adjuvant chemotherapy with gemcitabine plus
erlotinib versus gemcitabine alone in patients after R0 resection
of pancreatic cancer: A multicenter randomized phase III trial. J
Clin Oncol. 35:3330–3337. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Gbolahan OB, Tong Y, Sehdev A, O'Neil B
and Shahda S: Overall survival of patients with recurrent
pancreatic cancer treated with systemic therapy: A retrospective
study. BMC Cancer. 19(468)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Waddell N, Pajic M, Patch AM, Chang DK,
Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al:
Whole genomes redefine the mutational landscape of pancreatic
cancer. Nature. 518:495–501. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Tuveson DA, Shaw AT, Willis NA, Silver DP,
Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D, et
al: Endogenous oncogenic K-ras(G12D) stimulates proliferation and
widespread neoplastic and developmental defects. Cancer Cell.
5:375–387. 2004.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ying H, Kimmelman AC, Lyssiotis CA, Hua S,
Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff
JL, et al: Oncogenic Kras maintains pancreatic tumors through
regulation of anabolic glucose metabolism. Cell. 149:656–670.
2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Junttila MR, Karnezis AN, Garcia D,
Madriles F, Kortlever RM, Rostker F, Brown Swigart LB, Pham DM, Seo
Y, Evan GI and Martins CP: Selective activation of p53-mediated
tumour suppression in high-grade tumours. Nature. 468:567–571.
2010.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Burgess MR, Hwang E, Mroue R, Bielski CM,
Wandler AM, Huang BJ, Firestone AJ, Young A, Lacap JA, Crocker L,
et al: KRAS Allelic imbalance enhances fitness and modulates MAP
kinase dependence in cancer. Cell. 168:817–829.e15. 2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Sobin LH, Gospodarowicz MK and Wittekind C
(eds): International Union Against Cancer (UICC): TNM
Classification of Malignant Tumours. 7th edition, Wiley-Blackwell,
Oxford, 2009.
|
|
13
|
Okusaka T, Nakamura M, Yoshida M, Kitano
M, Uesaka K, Ito Y, Furuse J, Hanada K and Okazaki K: Committee for
Revision of Clinical Guidelines for Pancreatic Cancer of the Japan
Pancreas Society. Clinical practice guidelines for pancreatic
cancer 2019 from the Japan pancreas society: A synopsis. Pancreas.
49:326–335. 2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Jones S, Zhang X, Parsons DW, Lin JC,
Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et
al: Core signaling pathways in human pancreatic cancers revealed by
global genomic analyses. Science. 321:1801–1806. 2008.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Sodir NM, Kortlever RM, Barthet VJA,
Campos T, Pellegrinet L, Kupczak S, Anastasiou P, Swigart LB,
Soucek L, Arends MJ, et al: MYC instructs and maintains pancreatic
adenocarcinoma phenotype. Cancer Discov. 10:588–607.
2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Mueller S, Engleitner T, Maresch R,
Zukowska M, Lange S, Kaltenbacher T, Konukiewitz B, Öllinger R,
Zwiebel M, Strong A, et al: Evolutionary routes and KRAS dosage
define pancreatic cancer phenotypes. Nature. 554:62–68.
2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Moffitt RA, Marayati R, Flate EL, Volmar
KE, Loeza SG, Hoadley KA, Rashid NU, Williams LA, Eaton SC, Chung
AH, et al: Virtual microdissection identifies distinct tumor- and
stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat
Genet. 47:1168–1178. 2015.PubMed/NCBI View
Article : Google Scholar
|