Introduction
Retinoblastoma (RB) is the most common intraocular
malignancy, characterized by high mortality if not detected early
and treated promptly. Early diagnosis and intervention play a key
role in the successful treatment of RB (1). Delayed diagnosis of RB for >6
months from the first clinical sign has been reported to be
associated with a mortality rate of ~70% (2). Therefore, although in patients with RB
diagnosed at stage A of the disease the eyes or vision can be
salvaged, currently, there is not an effective treatment approach
for those diagnosed at stage E (3,4).
RB is considered as a monogenetic hereditary type of
cancer since 97% of RB cases are caused by the disruption of the RB
transcriptional corepressor 1 (RB1) tumor suppressor gene
(5). The RB1-encoded protein
(pRB) acts as a scaffold protein, which interacts with other
proteins to regulate multiple cellular processes essential for cell
fate and function. Consequently, RB1 deficiency may
predispose cells to tumorigenesis. In fact, it has been reported
that RB1 inactivation is detected in several types of
cancer. Since pRB interacts with other proteins through cyclin
folds in the N-terminus and pocket domain, and intrinsically
disordered structures in the C-terminus, a wide spectrum of
mutations dispersed throughout the RB1 gene has been
identified in patients with cancer (6).
It has been also reported that 45% of patients with
RB are suffering from the inherited type of the disease (RB1
carriers), where the first allele of RB1 is mutated during
preconception or shortly after conception, predisposing the child
to retinal tumorigenesis (7). The
remaining RB1 allele is functionally lost during retinal
development, promoting the initiation of RB in either both eyes
(bilateral RB) or one eye (unilateral RB) with multifocal tumors.
‘Non-heritable or sporadic RB’ (non-RB1 carriers) is
commonly referred to as patients without germline RB1
mutations, who usually present with unifocal tumors in one eye
(8). Patients with hereditary RB
exhibit a worse prognosis, are sensitive to certain treatments,
have a high risk of developing second primary malignancies and can
pass the mutations on to their offspring (3,9).
Therefore, identifying germline RB1 mutations is of great
importance for implementing the appropriate treatment approach, and
assessing the risk of developing second primary malignancies, both
secondary RB and other primary malignancies, in patients with RB,
and the risk of RB onset in the patient's relatives.
Genetic testing and counseling (GTC) are recommended
for all patients with RB and are integrated in the management of RB
in developed countries (10), thus
resulting in high survival rate of patients with RB and
cost-effective medical treatments for RB (11,12).
GTC for patients with bilateral or familial RB is straightforward.
However, for unilateral, non-familial RB cases, GTC is not as easy
due to the low risk of RB1 mutation carriers, and the
inefficient detection of germline RB1 mosaicism (13). Genetic testing for RB1 is a
time-consuming and expensive procedure given the large size of the
RB1 gene, which can be inactivated by multiple mutations,
and the absence of mutational hotspots (14,15).
Direct sequencing is widely applied for detecting RB1
mutations, however, this method is not recommended for identifying
low allelic-fraction variants (16). For the detection of these types of
variants, PCR can be applied only when the mutations are already
known. In addition, Multiplex Ligation-dependent Probe
Amplification (MLPA), quantitative PCR (qPCR) or array Comparative
Genomic Hybridization (aCGH) can identify large RB1
rearrangements. The combination of the aforementioned methods is
essential for detecting all possible RB1 mutations (17-19).
Recently, Next Generation Sequencing (NGS) has been implemented as
rapid and effective strategy for identification of RB1
mutations since all variations can be detected in a single test,
thus providing a number of advantages, including high sensitivity
and cost-effectiveness (20-25).
In Vietnam, Sanger sequencing coupled with MLPA (SS-MLPA) could
detect germline RB1 mutations in 82-84% of bilateral cases
(26,27). Nevertheless, the sensitivity of NGS
in a routine clinical practice remains unknown.
The two-hit hypothesis suggests that patients with
bilateral or unilateral multifocal RB, and/or diagnosed at an early
age are more likely to carry germline RB1 mutations
(28). This hypothesis is supported
by clinical data. Therefore, a study demonstrated that up to 100%
of patients with bilateral carried germline RB1 mutations,
and their age at diagnosis was 10 months younger compared with that
of the unilateral cases (13).
Bilateral and unilateral eye diseases account for 40 and 60% of all
RB cases (7), respectively. All
bilateral RB cases are considered heritable, whereas ~15% of
unilateral cases carry constitutional RB1 mutations
(29,30). Since age at diagnosis is younger in
bilateral cases, previous studies have associated age at diagnosis
with germline RB1 status in order to predict age associated
with increased risk of patients with unilateral disease being
RB1 carriers. Unfortunately, these studies yielded
conflicting results (31-34).
The current study retrospectively analyzed the
clinicopathological and genetic data of patients with RB to
evaluate the sensitivity of NGS for detecting constitutional
RB1 variants, to detect novel germline RB1 mutations,
and to consider age at diagnosis as a risk factor for patients
being RB1 carriers.
Materials and methods
Patients and samples
In the current retrospective study, a total of 62
patients with RB were included, who were referred to Department of
Cancer Research, Vinmec Research Institute of Stem Cell and Gene
Technology and the Department of Medical Genetics, Vinmec Hi-Tech
Center for genetic testing between 2017 and 2019. Signed informed
consents were obtained from parents/caregivers of all subjects and
the study was approved by the Vinmec's Institutional Review Board.
All data, including age at diagnosis, sex, tumor stages, laterality
and family history were retrieved from the patients' medical
records. Genetic testing for germline RB1 variants was
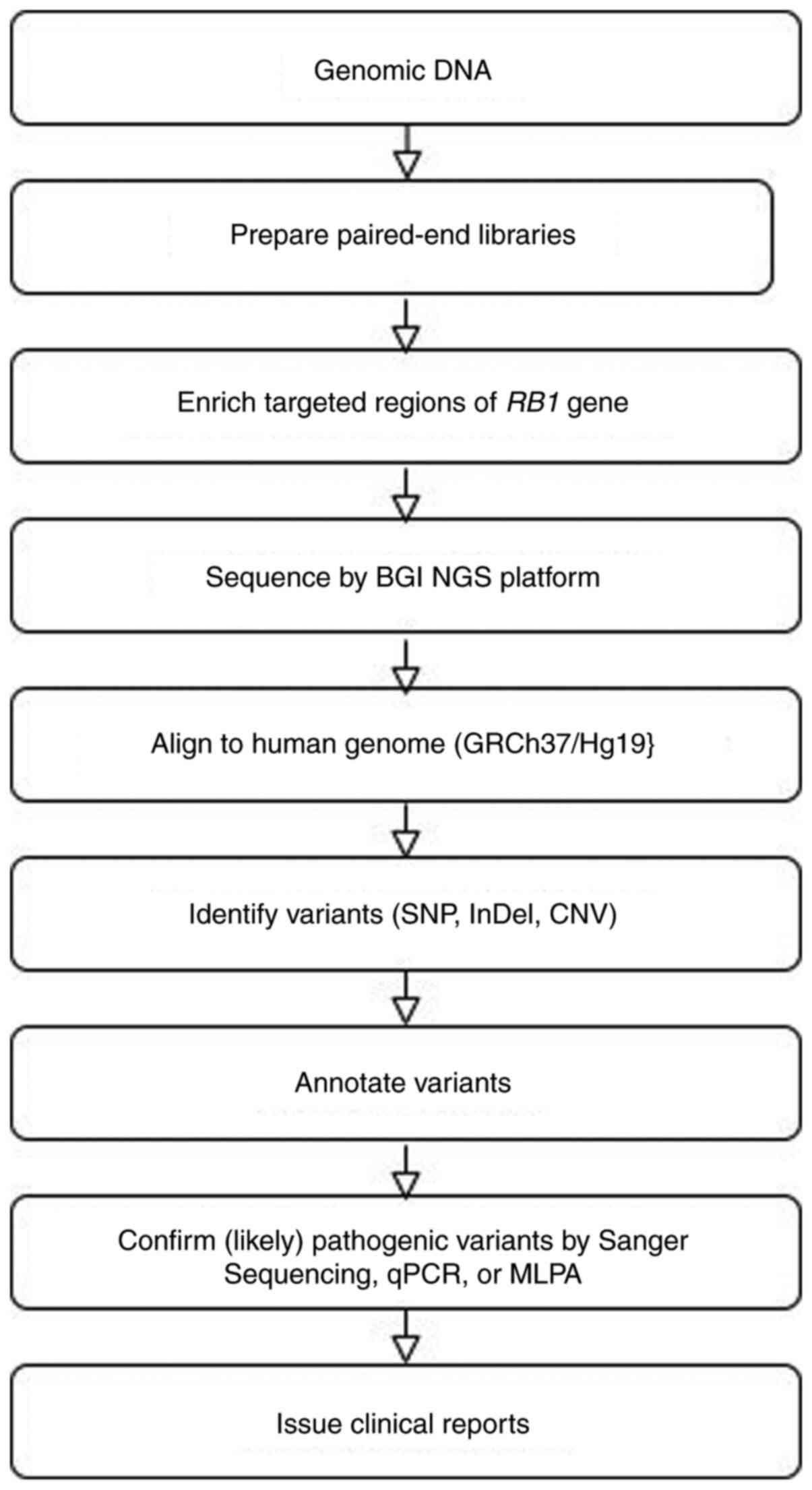
performed at the Beijing Genomics Institute (BGI), HongKong. The
workflow for analyzing germline RB1 variants is illustrated
in Fig. 1.
Preparation of tissue samples, NGS and
variant calling
Genomic DNA (gDNA) was extracted from peripheral
blood mononuclear cells and its concentration was quantified using
a Qubit Fluorometer (Thermo Fisher Scientific, Inc.). Subsequently,
gDNA was fragmented, indexed and amplified. The RB1 promoter
and all its exons, plus 20 nucleotides proximal to either 5' or 3'
of each exon, were captured by a BGI chip. Library size and
quantity were verified using Qubit® 2.0 (Thermo Fisher
Scientific, Inc.) and an Agilent Bioanalyzer 2100 (Agilent
Technologies, Inc.). Sequencing was performed on the BGIseq
platform. All samples identified with pathogenic or likely
pathogenic variants were confirmed by Sanger Sequencing, qPCR or
MLPA.
Bioinformatics analysis
The sequence reads were aligned to the human
reference genome (GRCh37/Hg19) using the Burrows-Wheeler Aligner.
Single nucleotide variants, insertion/deletion variants (InDel) and
copy number variations were identified using the BGI internal NGS
software (BGISEQ-500).
Variant annotation
Interpretation of germline variants followed the
American College of Medical Genetics and Genomics (ACMG) standards
and guidelines (35). The variants
were designated according to nomenclature and the recommendations
of the Human Genome Variation Society (36). Public databases, including Clinvar
(37), Universal Mutation Database
(38), Leiden Open Variation
Database (LOVD) (39) and ARUP
(40) were used for data analysis.
The SIFT (41), POLYPHEN2(42) and Mutation taster (43) tools were applied to predict
deleterious mutations, while variants were classified using the
Varsome (44) and InterVar
(45) classification tools.
Identification of novel variants
All variants identified by the BGI laboratory were
annotated with the Ensemble Variant Effect Predictor (EVEP) tool
(46). Variants with existing ID
were checked on corresponding databases. The unmatched variants and
those without existing ID were screened on PubMed. Variants not
previously reported in public databases and PubMed were considered
as novel ones. The allele frequency of the identified novel
variants was investigated in four databases, including the 1000
Genomes Project (browser.1000genomes.org), the Exome Sequencing Project
(esp.gs.washington.edu/drupal), the
Genome Aggregation Database and the Vietnamese Genetic Variation
Database (47). Deleterious novel
variants were evaluated by Combined Annotation Dependent Depletion
(CADD) (48) and EVEP, while their
pathogenicity was interpreted using VarSome.
Statistical analysis
The differences between the characteristics of
RB1 and non-RB1 carriers were compared using a
χ2 test, Fisher's exact test or Student's t-test.
Fisher's exact test and odds ratio (OR) were calculated using
calculators provided by the Social Science Statistics website
(49) and MedCalc statistical
software (50), respectively.
Results
Characteristics of RB1and non-RB1
carriers
A total of 25 bilateral and 37 unilateral RB cases
were included in the present study. There was no statistically
significant difference in the total number of RB1 and
non-RB1 carriers, as well as in sex distribution between the
two groups. However, the mean age at diagnosis of RB1
carriers was significantly younger compared with that of
non-RB1 carriers (22.14 vs. 32.66 months). Although 80% of
all patients were diagnosed with stage E RB, the distribution of
the RB stages was not statistically different between RB1
and non-RB1 carriers. Additionally, the proportion of
RB1 carriers was notably higher in patients with bilateral
RB and significantly decreased in the unilateral cases.
Constitutional RB1 mutations were detected in 100% (25/25)
and 27% (10/37) of patients with bilateral and unilateral RB,
respectively (Tables I and II), suggesting a sensitivity rate of 100%
for detecting germline RB1 variants using the NGS
technology.
 | Table ISummary of germline RB1
mutations identified in Vietnamese patients with
retinoblastoma. |
Table I
Summary of germline RB1
mutations identified in Vietnamese patients with
retinoblastoma.
| Case ID | Family history | Months at
diagnosis | Laterality | Sex | Staging | RB1 germline
mutations | Molecular
consequences | Interpretation | Exon/Intron | Previously reported
(Refs.) |
|---|
| VinRB01 | Yes | 48 | Bil | F | C/E | Ex1_27 Del | Large
rearrangement | Pathogenic | Exon 1-27 | (53,61-63) |
| VinRB02 | No | 38 | Bil | M | A/E | Ex1_27 Del | Large
rearrangement | Pathogenic | Exon 1-27 | (53,61-63) |
| VinRB03 | No | 25 | Bil | F | E/C | Ex1_27 Del | Large
rearrangement | Pathogenic | Exon 1-27 | (53,61-63) |
| VinRB04 | No | 10 | Bil | F | E/A | Ex18_23 Dup | Large
rearrangement | Pathogenic | Exon 18-23 | (64) |
| VinRB05 | No | 30 | Bil | F | D/E | Ex12_dup & c.83
C>G | Large
rearrangement | Pathogenic &
VUS | Exon 12 & Exon
1 | Novel &
rs776175164 |
| VinRB06 | No | 3 | Bil | M | E/B | c.210_211insAG | Frameshift | Pathogenic | Exon 2 | Novel |
| VinRB07 | No | 31 | Bil | M | A/E | c.515_516insA | Frameshift | Pathogenic | Exon 5 | Novel |
| VinRB08 | No | 6 | Bil | M | B/E | c.380G>A | Missense | Pathogenic | Exon 3 | COSV57302882 |
| VinRB09 | No | 18 | Bil | M | E/C | c.1072C>T | Nonsense | Pathogenic | Exon 11 | rs121913301 |
| VinRB10 | No | 36 | Bil | M | D/E |
c.1403_1404insA | Frameshift | Pathogenic | Exon 15 | Novel |
| VinRB11 | No | 7 | Bil | F | E/B | c.1735C>T | Nonsense | Pathogenic | Exon 18 | rs121913305 |
| VinRB12 | Yes | 6 | Bil | M | B/B | c.1735C>T | Nonsense | Pathogenic | Exon 18 | rs121913305 |
| VinRB13 | No | 5 | Bil | F | B/E | c.2359C>T | Nonsense | Pathogenic | Exon 23 | rs137853293 |
| VinRB14 | No | 1 | Bil | M | B/B | c.306T>A | Nonsense | Pathogenic | Exon 3 | Novel |
| VinRB15 | No | 26 | Bil | F | E/E | c.958C>T | Nonsense | Pathogenic | Exon 10 | rs121913300 |
| VinRB16 | No | 29 | Bil | F | E/E | c.958C>T | Nonsense | Pathogenic | Exon 10 | rs121913300 |
| VinRB17 | No | 31 | Bil | M | B/E |
c.1953_c.1960+1delTAAAAAAGG | Splice | Likely
pathogenic | Exon 19 | Novel |
| VinRB18 | No | 19 | Bil | F | B/E | c.1128-1G>A | Splice | Pathogenic | Intron 11 | COSV57294096 |
| VinRB19 | No | 20 | Bil | M | B/E | c.1215+1G>A | Splice | Pathogenic | Intron 12 | rs587776783 |
| VinRB20 | No | 2 | Bil | F | B/B | c.1696-1G>A | Splice | Pathogenic | Intron 17 | COSV57329272 |
| VinRB21 | No | 3 | Bil | F | B/B |
c.2520+1_2520+4delGTGA | Splice | Pathogenic | Intron 24 | rs1131690858 |
| VinRB22 | No | 22 | Bil | M | E/B | c.607+1G>A | Splice | Pathogenic | Intron 6 | COSV57310480 |
| VinRB23 | No | 14 | Bil | M | D/D | c.607+1G>T | Splice | Pathogenic | Intron 6 | COSV57310480 |
| VinRB24 | No | 54 | Uni | M | /E | c.2107-2A>C | Splice | Likely
pathogenic | Intron 20 | Novel |
| VinRB25 | No | 21 | Bil | M | E/B | c.719-6_719-2del
TTACA | Splice | Likely
pathogenic | Intron 7 | Novel |
| VinRB26 | No | 30 | Uni | F | E | c.1981C>T | Missense | Pathogenic | Exon 20 | rs137853294 |
| VinRB27 | No | 19 | Uni | F | E/ | Ex24 Del | Large
rearrangement | Pathogenic | Exon 24 | (65) |
| VinRB28 | No | 14 | Uni | M | E/ |
c.1953_1954insA | Frameshift | Pathogenic | Exon 19 | rs1566234123 |
| VinRB29 | Yes | 2 | Uni | M | /B | c.958C>T | Nonsense | Pathogenic | Exon 10 | rs121913300 |
| VinRB30 | No | 74 | Uni | F | /D | c.1072C>T | Nonsense | Pathogenic | Exon 11 | rs121913301 |
| VinRB31 | No | 17 | Uni | F | E/ | c.1303G>T | Nonsense | Likely
pathogenic | Exon 13 | COSV57313162 |
| VinRB32 | Yes | 34 | Uni | M | E/ | c.1735C>T | Nonsense | Pathogenic | Exon 18 | rs121913305 |
| VinRB33 | No | 21 | Bil | F | D/E | c.958C>T | Nonsense | Pathogenic | Exon 10 | rs121913300 |
| VinRB34 | No | 24 | Uni | F | /E | c.2106+1G>C | Splice | Likely
pathogenic | Intron 20 | Novel |
| VinRB35 | No | 35 | Uni | M | D/ | c.264+1G>C | Splice | Pathogenic | Intron 2 | COSV57317171 |
| VinRB36 | No | 14 | Uni | F | E/ | No | | | | |
| VinRB37 | No | 10 | Uni | F | /E | No | | | | |
| VinRB38 | No | 11 | Uni | M | E/ | No | | | | |
| VinRB39 | No | 22 | Uni | F | E/ | No | | | | |
| VinRB40 | No | 30 | Uni | M | E/ | No | | | | |
| VinRB41 | No | 24 | Uni | F | /E | No | | | | |
| VinRB42 | No | 25 | Uni | F | E/ | No | | | | |
| VinRB43 | No | 57 | Uni | M | /D | No | | | | |
| VinRB44 | No | 25 | Uni | M | E/ | No | | | | |
| VinRB45 | No | 17 | Uni | M | E | No | | | | |
| VinRB46 | No | 31 | Uni | M | /E | No | | | | |
| VinRB47 | No | 22 | Uni | M | E/ | No | | | | |
| VinRB48 | No | 108 | Uni | F | E/ | No | | | | |
| VinRB49 | No | 59 | Uni | F | E/ | No | | | | |
| VinRB50 | No | 84 | Uni | F | /E | No | | | | |
| VinRB51 | No | 8 | Uni | M | E/ | No | | | | |
| VinRB52 | No | 10 | Uni | M | D/ | No | | | | |
| VinRB53 | No | 8 | Uni | F | E/ | No | | | | |
| VinRB54 | No | 44 | Uni | M | E/ | No | | | | |
| VinRB55 | No | 23 | Uni | M | E/ | No | | | | |
| VinRB56 | No | 49 | Uni | F | D/ | No | | | | |
| VinRB57 | No | 21 | Uni | F | D/ | No | | | | |
| VinRB58 | No | 56 | Uni | M | E/ | No | | | | |
| VinRB59 | No | 41 | Uni | F | E/ | No | | | | |
| VinRB60 | No | 33 | Uni | M | E/ | No | | | | |
| VinRB61 | No | 42 | Uni | M | E/ | No | | | | |
| VinRB62 | No | 8 | Uni | F | /E | No | | | | |
| Table IIClinicopathological distribution of
RB1 (n=35) and non-RB1 (n=27) carriers by family
history, laterality, sex, age at diagnosis and stage of RB
tumorigenesis. |
Table II
Clinicopathological distribution of
RB1 (n=35) and non-RB1 (n=27) carriers by family
history, laterality, sex, age at diagnosis and stage of RB
tumorigenesis.
|
Characteristics | RB1 carriers
(n=35) | Non-RB1 carriers
(n=27) | Both (n=62) | P-value |
|---|
| Family history, n
(%) | 4 (100.0) | 0 (0.0) | 4 (100.0) | |
| Laterality, n
(%) | | | | 0.001a |
|
Bilateral
RB | 25 (100.0) | 0 (0.0) | 25 (100.0) | |
|
Unilateral
RB | 10 (27.0) | 27 (73.0) | 37 (100.0) | |
| Sex, n (%) | | | | 0.87b |
|
Female | 17 (57.0) | 13 (43.0) | 30 (100.0) | |
|
Male | 18 (56.0) | 14 (44.0) | 32 (100.0) | |
| Mean age,
months | 22.14 | 32.67 | | 0.04c |
| RB stage, n
(%) | | | | 0.80a |
|
A | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
|
B | 4 (11.4) | 1 (3.7) | 5 (8.0) | |
|
C | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
|
D | 3 (8.6) | 4 (14.8) | 7 (11.3) | |
|
E | 28 (80.0) | 22 (81.5) | 50 (80.6) | |
Novel RB1 germline mutations
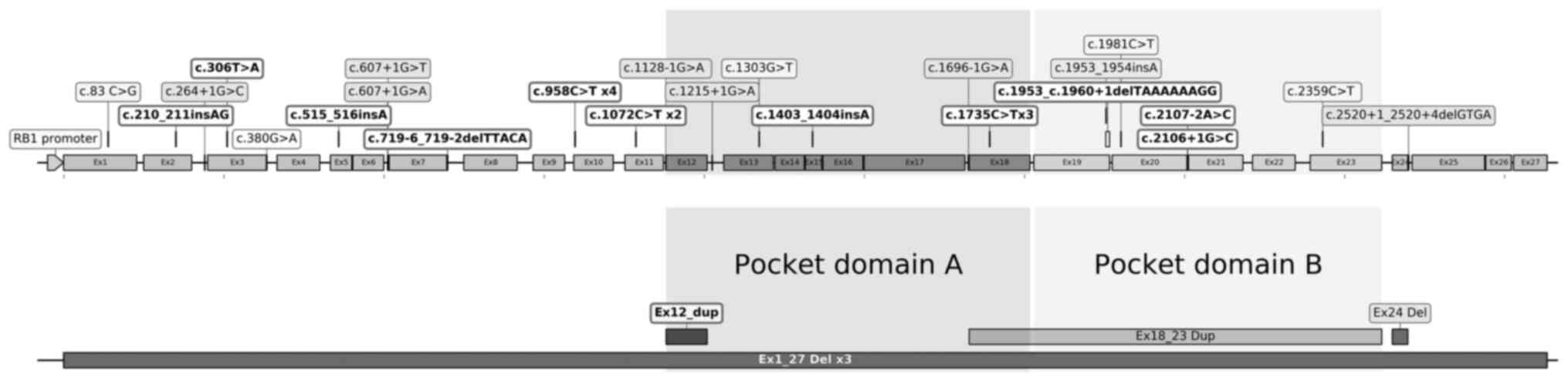
A total of 28 distinct variants, including four
recurrent and 24 non-recurrent ones, were identified in 56% (35/62)
of patients with. RB. In addition, the four recurrent mutations
were found in 33% (12/35) of RB1 carriers. Point and small
InDel mutations in RB1 were dispersed along the gene.
However, large rearrangements were only identified in or nearby the
pocket domain. The majority of mutations (26/28) were located in
the N-terminus or pocket domain of pRB (Fig. 2). The EVEP tool predicted that all
these variations, except one (c.83C>G), exerted a highly
disruptive effect in pRB and were classified as pathogenic variants
using the Clinvar, Cosmic or Varsome databases. The c.83C>G
mutation was concurrently found with an exon 12 duplication in one
patient with bilateral RB. Nonsense and slice mutations were the
two most frequent mutations, identified in 34 (12/35) and 31%
(11/35) of all RB1 carriers, respectively. Additionally,
large rearrangements, frameshift and missense mutations were
detected in 17 (6/35), 11.4 (4/35) and 8.6% (3/35) of RB1
carriers, respectively (Tables I
and III).
| Table IIIA total of 28 distinct RB1
germline alterations, including 9 novel ones, were identified in 35
patients with retinoblastoma. |
Table III
A total of 28 distinct RB1
germline alterations, including 9 novel ones, were identified in 35
patients with retinoblastoma.
| RB1 germline
variants | Molecular
consequences | Amino acid
alterations | Clinical
significance | Existing
variations | No. of
carriers | Subunits |
|---|
| c.210_211insAG | Frameshift |
p.(Ala74Glufs*4) | Pathogenic | Newly
identified | 1 | N-terminus |
| c.306T>A | Nonsense | p.(Cys102Ter) | Pathogenic | Newly
identified | 1 | N-terminus |
| c.515_516insA | Frameshift |
p.(Tyr173Ilefs*12) | Pathogenic | Newly
identified | 1 | N-terminus |
| c.719-6_7192
delTTACA | Splice | - | Pathogenic | Newly
identified | 1 | N-terminus |
| Ex12 Dup | Large
rearrangement | - | Pathogenic | Newly
identified | 1 | Pocket |
| c.1403_1404insA
c.1953_c.1960+1 | Frameshift |
p.(Ser469Ilefs*6) | Pathogenic | Newly
identified | 1 | Pocket |
| delTAAAAAAGG | Splice | - | Pathogenic | Newly
identified | 1 | Pocket |
| c.2106+1G>C | Splice | - | Pathogenic | Newly
identified | 1 | Pocket |
| c.2107-2A>C | Splice | - | Pathogenic | Newly
identified | 1 | Pocket |
| c.83C>G
(concurrent with Ex12dup) | Missense | p.(Pro28Arg) | VUS | rs776175164 | 1 | N-terminus |
| c.1215+1G>A | Splice | - | Pathogenic | rs587776783 | 1 | Pocket |
|
c.1953_1954insA | Frameshift | p.(V654Sfs*14) | Pathogenic | rs1566234123 | 1 | Pocket |
| c.1981C>T | Missense | p.(Arg661Trp) | Pathogenic | rs137853294 | 1 | Pocket |
| c.2359C>T | Nonsense | p.(Arg787Ter) | Pathogenic | rs137853293 | 1 | Pocket |
| c.1735C>T | Nonsense | p.(Arg579Ter) | Pathogenic | rs121913305 | 3 | Pocket |
| c.1072C>T | Nonsense | p.(Arg358Ter) | Pathogenic | rs121913301 | 2 | N-terminus |
| c.958C>T | Nonsense | p.(Arg320Ter) | Pathogenic | rs121913300 | 4 | N-terminus |
|
c.2520+1_2520+4delGTGA | Splice | - | Pathogenic | rs1131690858 | 1 | C-terminus |
| Ex1_27 Del | Large
rearrangement | - | Pathogenic | (53,61-63) | 3 | Whole gene |
| Ex18_23 Dup | Large
rearrangement | - | Pathogenic | (64) | 1 | Pocket |
| Ex24 Del | Large
rearrangement | - | Pathogenic | (65) | 1 | C-terminus |
| c.1696-1G>A | Splice | - | Pathogenic | COSV57329272 | 1 | Pocket |
| c.264+1G>C | Splice | - | Pathogenic | COSV57317171 | 1 | N-terminus |
| c.1303G>T | Nonsense | p.(Gly435Ter) | Pathogenic | COSV57313162 | 1 | Pocket |
| c.607+1G>A | Splice | - | Pathogenic | COSV57310480 | 1 | N-terminus |
| c.607+1G>T | Splice | - | Pathogenic | COSV57310480 | 1 | N-terminus |
| c.380G>A | Missense | p.(Ser127Asn) | Pathogenic | COSV57302882 | 1 | N-terminus |
| c.1128-1G>A | Splice | - | Pathogenic | COSV57294096 | 1 | Pocket |
The novel variants were defined by screening 28
RB1 mutations into the EVEP tool. Among them, 16 RB1
alterations exerted Variation ID on the Clinvar, Cosmic or LOVD
databases. For the remaining 12 unidentified variants, screening on
the PubMed platform was carried out. The analysis revealed three
large rearrangement, including Ex1_27 DEL, Ex24 DEL and Ex13_18
DUP. Therefore, nine variants were considered as novel. These nine
variations, including one large rearrangement, one nonsense, four
slice and three frameshift mutations, were all null mutations,
identified in seven patients with bilateral and two with unilateral
RB. These variants were located at either the pocket (5/9) or
N-terminus (4/9) domain. Furthermore, the EVEP tool predicted that
these variants could have a significant disruptive effect on pRB,
and all, except EX12 DUP, were also predicted to be among the top
1% of the most deleterious variants in the human genome by CADD.
Finally, these mutations were not found in polymorphism databases,
and were classified as pathogenic by VarSome (Table III; Fig. 2).
Association between age at diagnosis
and the genetic status of patients with RB
The proportion of RB1 and non-RB1
carriers with an age at diagnosis of 0-36 and >36 months is
presented in Table IV. It was
found that the proportion of RB1 carriers with age at
diagnosis of 0-36 months was notably higher compared with those of
>36 months. Consequently, the relative risk of children aged
between 0-36 months being RB1 carriers was significantly
higher than that of children >36 months (63.3 vs. 36%;
OR=3.88; 95% CI=1.04-14.4; Table
IV). In terms of unilateral cases only, although the number of
RB1 carriers was reduced compared with the non-RB1
ones, the difference between the two groups was not statistically
significant. Accordingly, the relative risk of children aged
between 0-36 months suffering from the inherited form of the
disease was not markedly different compared with children >36
months of age (30.8 vs. 18%; OR=2; 95% CI=0.35=11.4;
Table IV).
| Table IVDistribution of RB1 and
non-RB1 carriers diagnosed at 0-36 vs. >36 months. |
Table IV
Distribution of RB1 and
non-RB1 carriers diagnosed at 0-36 vs. >36 months.
| A, All patients
with RB |
|---|
| Characteristic | RB1
carriers, n=35 | Non-RB1
carriers, n=27 | Total | P-value |
|---|
| Age, n (%) | | | | 0.036a |
|
0-36
months | 31 (63.3) | 18 (36.7) | 49 (100.0) | |
|
>36
months | 4 (36.0) | 9 (64.0) | 13 (100.0) | |
| OR, 0-36 vs. >36
months | 3.88 (95% CI,
1.04-14.40) | 0.26 (95% CI,
0.07-0.96) | | 0.040 |
| B, Patients with
unilateral RB only |
| Characteristic | RB1
carriers, n=10 | Non-RB1
carriers, n=27 | Total | P-value |
| Age, n (%) | | | 0.62 | 0.043a |
|
0-36
months | 8 (30.8) | 18 (69.2) | 26 (100.0) | |
|
>36
months | 2 (18.0) | 9 (82.0) | 11 (100.0) | |
| OR, 0-36 vs. >36
months | 2 (95% CI,
0.35-11.40) | 0.5 (95% CI,
0.09-2.86) | | 0.043 |
Discussion
Integrated RB1 genetic testing for the
management of RB may reduce RB-associated mortality and treatment
costs. However, the test sensitivity rate should be ≥90% to ensure
a negative result would indicate a low risk of hereditary RB. In
the current study, patients who were diagnosed with bilateral RB
were used as a positive control group to evaluate the sensitivity
of NGS in routine clinical practice. The detection rate of NGS was
100% for patients with bilateral RB, while all common RB1
mutation types were detected. Additionally, nine novel pathogenic
mutations were identified. The present study also supported the
potential use of age at diagnosis as a risk factor for inherited
RB.
Recently, NGS is considered a highly sensitive and
efficient approach for the detection of RB1 mutations due to
increasing use of NGS in gene mutation analysis of RB; especially
screening and identification of RB1 mutations with NGS
substantially benefits the prepotency, early diagnosis and
treatment of retinoblastoma (20-25).
Furthermore, the sensitivity rate of NGS in the present study was
similar with that reported to previous studies. For example, Li
et al (51) detected
germline RB1 mutations in 100% (19/19) of patients with
bilateral RB, which was consistent with previously reported
laboratory data. Additionally, Singh et al (52) demonstrated a detection rate of 100%
(22/22) for bilateral cases, following validation of data using the
TruSight Cancer Sequencing Panel (Illumina, Inc.), with a
sensitivity rate of 98.2%, specificity of 100%, and reproducibility
of 99.5%. Furthermore, Grotta et al (53) showed a determination rate of 96%
(28/29) for patients with bilateral RB when combining NGS and aCGH.
Herein, the detected RB1 variants had common characteristics
with RB1 alterations commonly observed in patients with RB.
For instance, mutations were not identified in the exons 26 and 27
of the RB1 gene and were scattered along the RB1
gene. Another study revealed that null mutations, including large
rearrangements, nonsense, splicing and frameshift mutations were
detected at high frequency, whereas missense mutations exhibited a
low frequency (6). Overall, the
aforementioned data indicated that NGS could be considered as an
accurate method for detecting germline RB1 mutations.
The detection rate of germline RB1 mutations
in Vietnamese patients with RB using SS-MLPA has been previously
reported. Therefore, Kiet et al (26) and Nguyen et al (27) detected germline RB1 mutations
in 84 (21/25) and 82% (9/11) of patients with bilateral RB,
respectively. However, the detection rates were lower compared with
those reported in other Asian studies applying the same approach.
For example, Tomar et al (18) and He et al (54) revealed a detection rate of 94
(17/18) and 92% (36/39) in Singaporean and Chinese patients with
bilateral RB, respectively. In addition, Rojanaporn et al
(55) and Mohd Khalid et al
(56) also reported a detection
rate of germline RB1 mutations of 92 (25/27) and 100% (7/7)
in Thai and Malaysian patients with bilateral RB, respectively.
In the present study nine novel RB1 mutations
were identified in the cyclin fold-contained subunits of the pRB.
The allele frequencies of these null mutations were <0.0001% in
the public databases, and had not been recorded in the genetic
polymorphism database consisting of 400 healthy Vietnamese
individuals (47). The low
frequency of these variations in the general population indicated
that these mutations could be eliminated during evolution due to
their disadvantages. Additionally, the CADD tool predicted that all
these alterations, except one (EX12 DUP), were among the top 1% of
deleterious variants in the human genome. CADD integrates multiple
annotations, including genomic features, gene-annotation models,
evolutionary and epigenetic features, and gene functional
predictions to generate a single, quantitative scoring system.
These scores are then used to rank the deleterious effect of a
given variant (48,57). For example, a CADD score of 20 or 30
suggests that a variant is among the top 1% or 0.1% of deleterious
variants in the human genome, respectively. Consequently, VarSome
(44), a tool for implementing the
ACMG standards, is used to classify variants as pathogenic ones
when they meet very strong evidence of pathogenicity (PSV1). The
PSV1 criterion assumes that certain null mutations, such as
nonsense, frameshift, splice sites of +/-1 or 2, initiation codon
and exon deletions, can lead to a complete absence of the gene
product due to impaired transcription or nonsense-mediated decay of
an altered transcript (35). The
predicted null variants in a gene, whose loss of function is the
known mechanism underlying the development of a particular disease,
such as RB1 for RB, can be considered as the common cause of
the disease. Devarajan et al (58) also applied the stringent criteria
for defining pathogenic variants and reported no false-positive
results during the detection of constitutional RB1
variants.
The two-hit hypothesis suggests that in patients
with hereditary RB the tumors are formed at younger ages compared
with sporadic cases (28). In 1998,
Zajaczek et al (31)
reported four patients with unilateral RB, who were diagnosed with
germline RB1 mutations at the age of <19 months. This
initial evidence supported the hypothesis that age at diagnosis
could differentiate the hereditary from the sporadic form of
unilateral RB. However, further studies with larger sample sizes
did not reveal any association between age at diagnosis and
germline RB1 status in patients with unilateral RB (32-34).
In addition, Tomar et al (18) showed that Singaporean individuals
diagnosed with RB at 0-36 months of age had a 53% risk of being
RB1 carriers, whereas those >36 months had 8% risk of
suffering from hereditary RB. The results of the present study were
consistent with those of the previous one, suggesting that children
diagnosed with RB at 0-36 months of age were more likely to be
RB1 carriers compared with those aged >36 months.
Nevertheless, similar analyses in patients with unilateral RB did
not reveal significant difference in the risk of hereditary RB.
The present study revealed three clinical
implications. Firstly, the results supported that the NGS method
implemented at the Vinmec Hi-Tech Center was highly reliable in
detecting germline RB1 variants. Secondly, this study could
provide novel insights into the mechanisms underlying RB1
inactivation. Finally, the current findings suggested that age at
diagnosis could be considered as a risk factor for hereditary
RB.
Nevertheless, there are some limitations in the
present retrospective study, including the small sample sizes and
delay in diagnosis. Therefore, the results could not be
generalizable to all patients with RB. The number of patients with
bilateral RB, who underwent RB1 testing in each of the
aforementioned studies was <30. Furthermore, the small sample
size and different detection strategies used to identify germline
RB1 mutations could result in variations in the detection
rate between NGS and SS-MPLA. However, previous studies revealed
that NGS-based methods could detect germline RB1 mutations
in patients with RB, whose constitutional RB1 mutations
could not be detected by either Sanger sequencing nor MPLA
(59,60). In addition, the lack of statistical
significance in the relative risk of patients with unilateral RB
diagnosed at the age of 0-36 and those >36 months being carriers
of RB1 could be also due to the small number of unilateral
cases.
In conclusion, the results of the present study
indicated that NGS could be considered a reliable method for
screening for constitutional RB1 mutations, and age at
diagnosis could be used to assess the risk of hereditary RB.
Furthermore, the newly identified RB1 mutations could
provide useful information for an in-depth understanding of the
mechanisms underlying RB1 inactivation, and for the
development of rapid assays for detecting RB1 mutations.
Altogether, the current study suggested that NGS could be used for
detecting germline RB1 mutations in routine clinical
practice.
Acknowledgements
The authors would like to thank Professor Paolo
Boffetta (Icahn School of Medicine, Mount Sinai School of Medicine,
Tisch Cancer Institute, New York City, NY, USA) for the critical
review of this manuscript.
Funding
No funding was received.
Availability of data and materials
The FASTQ data that support the findings of this
study are available from the Beijing Genomics Institute (BGI)
laboratory (Hong Kong) but restrictions apply to the availability
of these data, which were used under license for the current study,
and so are not publicly available. Data are however available from
the authors upon reasonable request and with permission of the BGI
laboratory.
Authors' contributions
CQH and HQD conceived and designed the study. BDN,
LTP, DTN, CTMP and TLD recruited and referred patients to the study
and helped with their follow-up. HQD, CQH, NTN, SAHN, CN, BDN, LTP,
DTN, CTMP, TLD and MHT performed the experiments. HQD, CQH, NTN,
SAHN, CN and MHT analyzed and interpreted the results. CQH and HQD
wrote the manuscript. HQD and CQH are responsible for confirming
the authenticity of the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Signed informed consents were obtained from
parents/caregivers of all subjects, and the study was approved by
the Vinmec's Institutional Review Board (Vinmec Healthcare System,
Hanoi, Vietnam).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Canty CA: Retinoblastoma: An overview for
advanced practice nurses. J Am Acad Nurse Pract. 21:149–155.
2009.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Aerts I, Lumbroso-Le Rouic L,
Gauthier-Villars M, Brisse H, Doz F and Desjardis L:
Retinoblastoma. Orphanet J Rare Dis. 1(31)2006.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Kim JY and Park Y: Treatment of
retinoblastoma: The role of external beam radiotherapy. Yonsei Med
J. 56:1478–1491. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Abramson DH: Retinoblastoma: Saving life
with vision. Annu Rev Med. 65:171–184. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Soliman SE, Racher H, Zhang C, MacDonald H
and Gallie BL: Genetics and molecular diagnostics in
retinoblastoma-an update. Asia Pac J Ophthalmol (Phila). 6:197–207.
2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Valverde JR, Alonso J, Palacios I and
Pestaňa A: RB1 gene mutation up-date, a meta-analysis based on 932
reported mutations available in a searchable database. BMC Genet.
6(53)2005.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Draper GJ, Sanders BM, Brownbill PA and
Hawkins MH: Patterns of risk of hereditary retinoblastoma and
applications to genetic counselling. Br J Cancer. 66:211–219.
1992.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Rushlow D, Piovesan B, Zhang K,
Prigoda-Lee NL, Marchong MN, Clark RD and Gallie BL: Detection of
mosaic RB1 mutations in families with retinoblastoma. Hum Mutat.
30:842–851. 2009.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ali A, Kletke S, Gallie B and Lam WC:
Retinoblastoma for pediatric ophthalmologists. Asia Pac J
Ophthalmol (Phila). 7:160–168. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Canadian Retinoblastoma Society. National
retinoblastoma strategy Canadian guidelines for care. Can J
Ophthalmol. 44 (Suppl 2):S9–S47. 2009.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Dhar SU, Chintagumpala M, Noll C,
Chévez-Barrios P, Paysse EA and Plon SE: Outcomes of integrating
genetics in management of patients with retinoblastoma. Arch
Ophthalmol. 129:1428–1434. 2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Joseph B, Shanmugam MP, Srinivasan MK and
Kumaramanickavel G: Retinoblastoma: Genetic testing versus
conventional clinical screening in India. Mol Diagn. 8:237–243.
2004.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Berry JL, Kim JW, Damato BE and Singh AD
(eds): Clinical Ophthalmic Oncology: Retinoblastoma. Springer,
p303, 2019.
|
|
14
|
Ali MJ, Parsam VL, Honavar SG, Kannabiran
C, Vemuganti GK and Reddy VA: RB1 gene mutations in retinoblastoma
and its clinical correlation. Saudi J Ophthalmol. 24:119–123.
2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Lohmann DR: RB1 gene mutations in
retinoblastoma. Hum Mutat. 14:283–288. 1999.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ellison G, Donald E, McWalter G, Knight L,
Fletcher L, Sherwood J, Cantarini M, Orr M and Speake G: A
comparison of ARMS and DNA sequencing for mutation analysis in
clinical biopsy samples. J Exp Clin Cancer Res.
29(132)2010.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Richter S, Vandezande K, Chen N, Zhang K,
Sutherland J, Anderson J, Han L, Panton R, Branco P and Gallie B:
Sensitive and efficient detection of RB1 gene mutations enhances
care for families with retinoblastoma. Am J Hum Genet. 72:253–269.
2003.PubMed/NCBI View
Article : Google Scholar
|
|
18
|
Tomar S, Sethi R, Sundar G, Quah TC, Quah
BL and Lai PS: Mutation spectrum of RB1 mutations in retinoblastoma
cases from Singapore with implications for genetic management and
counselling. PLoS One. 6(e0178776)2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Thirumalairaj K, Abraham A, Devarajan B,
Gaikwad N, Kim U, Muthukkaruppan V and Vanniarajan A: A stepwise
strategy for rapid and cost-effective RB1 screening in Indian
retinoblastoma patients. J Hum Genet. 60:547–552. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Mehyar M, Mosallam M, Tbakhi A, Saab A,
Sultan I, Deebajah R, Jaradat I, AlJabari R, Mohammad M, AlNawaiseh
I, et al: Impact of RB1 gene mutation type in retinoblastoma
patients on clinical presentation and management outcome. Hematol
Oncol Stem Cell Ther. 13:152–159. 2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhang Z, Xiao YS, Shen R, Jiang HC, Tan L,
Li RQ, Yang XH, Gu HY, He WJ and Ma J: Next generation sequencing
of RB1 gene for the molecular diagnosis of ethnic minority with
retinoblastoma in Yunnan. BMC Med Genet. 21(230)2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Xu L, Shen L, Polski A, Prabakar RK, Shah
R, Jubran R, Kim JW, Biegel J, Kuhn P, Cobrinik D, et al:
Simultaneous identification of clinically relevant RB1 mutations
and copy number alterations in aqueous humor of retinoblastoma
eyes. Ophthalmic Genet. 41:526–532. 2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Chai P, Luo Y, Yu J, Li Y, Yang J, Zhuang
A, Fan J, Han M and Jia R: Clinical characteristics and germline
mutation spectrum of RB1 in Chinese patients with retinoblastoma: A
dual-center study of 145 patients. Exp Eye Res.
205(108456)2021.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zou Y, Li J, Hua P, Liang T, Ji X and Zhao
P: Spectrum of germline mutations in RB1 in Chinese patients with
retinoblastoma: Application of targeted next-generation sequencing.
Mol Vis. 27:1–16. 2021.PubMed/NCBI
|
|
25
|
Francis JH, Richards AL, Mandelker DL,
Berger MF, Walsh MF, Dunkel IJ, Donoghue MTA and Abramson DH:
Molecular changes in retinoblastoma beyond RB1: Findings from
next-generation sequencing. Cancers (Basel). 13(149)2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kiet NC, Khuong LT, Minh DD, Nguyen The
Vinh, Quan NHM, Xinh PT, Trang NNC, Luan NT, Khai NM and Vu HA:
Spectrum of mutations in the RB1 gene in Vietnamese patients with
retinoblastoma. Mol Vis. 25:215–221. 2019.PubMed/NCBI
|
|
27
|
Nguyen HH, Nguyen HTT, Vu NP, Le QT, Pham
CM, Huyen TT, Manh H, Pham HLB, Nguyen TD, Le HTT and Van Nong H:
Mutational screening of germline RB1 gene in Vietnamese patients
with retinoblastoma reveals three novel mutations. Mol Vis.
24:231–238. 2018.PubMed/NCBI
|
|
28
|
Knudson AG Jr: Mutation and cancer:
Statistical study of retinoblastoma. Proc Natl Acad Sci USA.
68:820–823. 1971.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Dimaras H, Kimani K, Dimba EA, Gronsdahl
P, White A, Chan HS and Ballie BL: Retinoblastoma. Lancet.
379:1436–1446. 2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Lohmann DL and Gallie BL: Retinoblastoma:
Revisiting the model prototype of inherited cancer. Am J Med Genet
C Semin Med Genet. 129C:23–28. 2004.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Zajaczek S, Jakubowska A, Kurzawski G,
Krzystolik Z and Lubiński J: Age at diagnosis to discriminate those
patients for whom constitutional DNA sequencing is appropriate in
sporadic unilateral retinoblastoma. Eur J Cancer. 34:1919–1921.
1998.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Schüler A, Weber S, Neuhäuser M, Jurklies
C, Lehnert T, Heimann H, Rudolph G, Jöckel KH, Bornfeld N and
Lohmann DR: Age at diagnosis of isolated unilateral retinoblastoma
does not distinguish patients with and without a constitutional RB1
gene mutation but is influenced by a parent-of-origin effect. Eur J
Cancer. 41:735–740. 2005.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Lohmann DR, Gerick M, Brandt B,
Oelschläger U, Lorenz B, Passarge E and Horsthemke B:
Constitutional RB1-gene mutations in patients with isolated
unilateral retinoblastoma. Am J Hum Genet. 61:282–294.
1997.PubMed/NCBI View
Article : Google Scholar
|
|
34
|
Berry JL, Lewis L, Zolfaghari E, Green S,
Le BHA, Lee TC, Murphree AL, Kim JW and Jubran R: Lack of
correlation between age at diagnosis and RB1 mutations for
unilateral retinoblastoma: The importance of genetic testing.
Ophthalmic Genet. 39:407–409. 2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Brody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
den Dunnen JT, Dalgleish R, Maglott DR,
Hart RK, Greenblatt MS, McGowan-Jordan J, Roux AF, Smith T,
Antonarakis SE and Taschner PE: HGVS recommendations for the
description of sequence variants: 2016 update. Hum Mutat.
37:564–569. 2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Landrum MJ, Lee JM, Riley GR, Jang W,
Rubinstein WS, Church DM and Maglott DR: ClinVar: Public archive of
relationships among sequence variation and human phenotype. Nucleic
Acids Res. 42:D980–D985. 2014.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Béroud C, Collod-Béroud G, Boileau C,
Soussi T and Junien C: UMD (Universal mutation database): A generic
software to build and analyze locus-specific databases. Hum Mutat.
15:86–94. 2000.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Fokkema IF, Taschner PE, Schaafsma GC,
Celli J, Laros JF and den Dunnen JT: LOVD v.2.0: The next
generation in gene variant databases. Hum Mutat. 32:557–563.
2011.PubMed/NCBI View Article : Google Scholar
|
|
40
|
ARUP Scientific Resource for Research and
Education. Available from: https://arup.utah.edu/.
|
|
41
|
Vaser R, Adusumalli S, Leng SN, Sikic M
and Ng PC: SIFT missense predictions for genomes. Nat Protoc.
11:1–9. 2016.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Kopanos C, Tsiolkas V, Kouris A, Chapple
CE, Aguilera MA, Meyer R and Massouras A: VarSome: The human
genomic variant search engine. Bioinformatics. 35:1978–1980.
2019.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Li Q and Wang K: InterVar: Clinical
interpretation of genetic variants by the 2015 ACMG-AMP guidelines.
Am J Hum Genet. 100:267–280. 2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
McLaren W, Gil L, Hunt SE, Riat HS,
Ritchie GS, Thormann A, Flicek P and Cunningham F: The ensembl
variant effect predictor. Genome Biol. 17(122)2016.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Le VS, Tran KT, Bui HTP, Le HTT, Nguyen
CD, Do DH, Ly HTT, Pham LTD, Dao LTM and Nguyen LT: A Vietnamese
human genetic variation database. Hum Mutat. 40:1664–1675.
2019.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Rentzsch P, Witten D, Cooper GM, Shendure
J and Kircher M: CADD: Predicting the deleteriousness of variants
throughout the human genome. Nucleic Acids Res. 47:D886–D894.
2019.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Social Science Statistics. Available from:
https://www.socscistatistics.com/.
|
|
50
|
Schoonjans F, Zalata A, Depuydt CE and
Comhaire FH: MedCalc: A new computer program for medical
statistics. Comput Methods Programs Biomed. 48:257–262.
1995.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Li WL, Buckley J, Sanchez-Lara PA,
Maglinte DT, Viduetsky L, Tatarinova TV, Aparicio JG, Kim JW, Au M,
Ostrow D, et al: A rapid and sensitive next-generation sequencing
method to detect RB1 mutations improves care for retinoblastoma
patients and their families. J Mol Diagn. 18:480–493.
2016.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Singh J, Mishra A, Pandian AJ, Mallipatna
AC, Khetan V, Sripriya S, Kapoor S, Agarwal S, Sankaran S,
Katragadda S, et al: Next-generation sequencing-based method shows
increased mutation detection sensitivity in an Indian
retinoblastoma cohort. Mol Vis. 22:1036–1047. 2016.PubMed/NCBI
|
|
53
|
Grotta S, D'Elia G, Scavelli R, Genovese
S, Surace C, Sirleto P, Cozza R, Romanzo A, De lois MA, Valente P,
et al: Advantages of a next generation sequencing targeted approach
for the molecular diagnosis of retinoblastoma. BMC Cancer.
15(841)2015.PubMed/NCBI View Article : Google Scholar
|
|
54
|
He MY, An Y, Gao YJ, Qian XW, Li G and
Qian J: Screening of RB1 gene mutations in Chinese patients with
retinoblastoma and preliminary exploration of genotype-phenotype
correlations. Mol Vis. 20:545–552. 2014.PubMed/NCBI
|
|
55
|
Rojanaporn D, Boontawon T,
Chareonsirisuthigul T, Thanapanpanich O, Attaseth T, Saengwimol D,
Anurathapan U, Sujirakul T, Kaewkhaw R and Hongeng S: Spectrum of
germline RB1 mutations and clinical manifestations in
retinoblastoma patients from Thailand. Mol Vis. 24:778–788.
2018.PubMed/NCBI
|
|
56
|
Mohd Khalid MK, Yakob Y, Md Yasin R, Teik
K, Siew CG, Rahmat J, Ramasamy S and Alagaratnam J: Spectrum of
germ-line RB1 gene mutations in Malaysian patients with
retinoblastoma. Mol Vis. 21:1185–1190. 2015.PubMed/NCBI
|
|
57
|
Kircher M, Witten DM, Jain P, O'Roak BJ,
Cooper GM and Shendure J: A general framework for estimating the
relative pathogenicity of human genetic variants. Nat Genet.
46:310–315. 2014.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Devarajan B, Prakash L, Kannan TR, Abraham
AA, Kim U, Muthukkaruppan V and Vanniarajan A: Targeted next
generation sequencing of RB1 gene for the molecular diagnosis of
Retinoblastoma. BMC Cancer. 15(320)2015.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Chen Z, Moran K, Richards-Yutz J, Toorens
E, Gerhart D, Ganguly T, Shields CL and Ganguly A: Enhanced
sensitivity for detection of low-level germline mosaic RB1
mutations in sporadic retinoblastoma cases using deep semiconductor
sequencing. Hum Mutat. 35:384–391. 2014.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Rodríguez-Martín C, Robledo C,
Gómez-Mariano G, Monzón S, Sastre A, Abelairas J, Sábado C,
Martín-Begué N, Ferreres JC, Fernández-Teijeiro A, et al: Frequency
of low-level and high-level mosaicism in sporadic retinoblastoma:
Genotype-phenotype relationships. J Hum Genet. 65:165–174.
2020.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Parsam VL, Kannabiran C, Honavar S,
Vemuganti GK and Ali MJ: A comprehensive, sensitive and economical
approach for the detection of mutations in the RB1 gene in
retinoblastoma. J Genet. 88:517–527. 2009.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Parma D, Ferrer M, Luce L, Giliberto F and
Szijan I: RB1 gene mutations in Argentine retinoblastoma patients.
Implications for genetic counseling. PLoS One.
12(e0189736)2017.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Lan X, Xu W, Tang X, Ye H, Song X, Lin L,
Ren X, Yu G, Zhang H and Wu S: Spectrum of RB1 germline mutations
and clinical features in unrelated Chinese patients with
retinoblastoma. Front Genet. 11(142)2020.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Berge EO, Knappskog S, Lillehaug JR and
Lønning PE: Alterations of the retinoblastoma gene in metastatic
breast cancer. Clin Exp Metastasis. 28:319–326. 2011.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Kato MV, Ishizaki K, Toguchida J, Kaneko
A, Takayama J, Tanooka H, Kato T, Shimuzu T and Sasaki MS:
Mutations in the retinoblastoma gene and their expression in
somatic and tumor cells of patients with hereditary retinoblastoma.
Hum Mutat. 3:44–51. 1994.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Fabian ID, Reddy A and Sagoo MS:
Classification and staging of retinoblastoma. Community Eye Health.
31:11–13. 2018.PubMed/NCBI
|