Introduction
Angiosarcomas demonstrate diverse clinical
presentations and can be frequently misinterpreted as benign
vascular tumors or other non-vascular malignancies (1). Splenic angiosarcomas are
exceptionally rare tumors with a poor prognosis (2). Anemia and bone marrow fibrosis are
typically associated with advanced stages of myeloproliferative
neoplasms and other secondary causes (3). Additionally, splenomegaly is a common
hallmark of hematological disorders. The present study describes
the case of a patient, in whom the initial symptoms of anemia and
bone marrow fibrosis, suggestive of primary myelofibrosis,
culminated in the ultimate diagnosis of splenic angiosarcoma
following a splenectomy.
Case report
Clinical presentation
In September 2021, a 35-year-old woman was admitted
to the First Affiliated Hospital of Guangxi Medical University with
progressively worsening fatigue and bone pain. A complete blood
count upon admission revealed anemia, with a white blood cell count
of 6.48x109/l, hemoglobin at 69 g/l and platelet count
of 156x109/l. Serum chemistry tests indicated elevated
erythropoietin levels [156 mIU/ml; normal range, 4.3-29 mIU/ml,
chemiluminescent immunoassay (Siemens Healthcare Diagnostics
Products Limited)], elevated lactate dehydrogenase [356 U/l; normal
range, 109-245 U/l, lactate substrate method (Zhongsheng Beikong
Biotechnology Co., Ltd.)] and increased ferritin levels [1,524.67
ng/ml; normal range, 4.06-204 ng/ml, chemiluminescent immunoassay
(Abbott Ireland Diagnostics Division)]. Total bilirubin levels were
increased at 32.6 µmol/l (normal range, 3.4-20.5 µmol/l), as were
direct bilirubin levels at 20.8 µmol/l (normal range, 0-6.8 µmol/l)
[vanadate oxidation method (Zhongsheng Beikong Biotechnology Co.,
Ltd.)], and albumin levels were reduced at 26.7 g/l [normal range,
40-55 g/l, bromocresol green method (Siemens Healthcare Diagnostics
Inc.)]. Coagulation profiles demonstrated normal prothrombin time
and activated partial thromboplastin time [coagulation method
(Instrumentation Laboratory Co.)], decreased fibrinogen [0.79 g/l;
normal range, 2-5 g/l, Clauss method (Instrumentation Laboratory
Co.)], and elevated levels of fibrinogen degradation products
[251.26 µg/ml; normal range, 0-5 µg/ml, immunoturbidimetry (Biokit
S.A.)] and D-Dimer [40,346 ng/ml; normal range, 0-450 ng/ml,
immunoturbidimetry (Instrumentation Laboratory Co.)]. The
peripheral blood and bone marrow smears were stained using the
Wright stain (Tianjin Guangfu Fine chemical Research Institute) for
20 min at room temperature, washed under running water, dried up at
room temperature, and observed under a microscope (Olympus BX43,
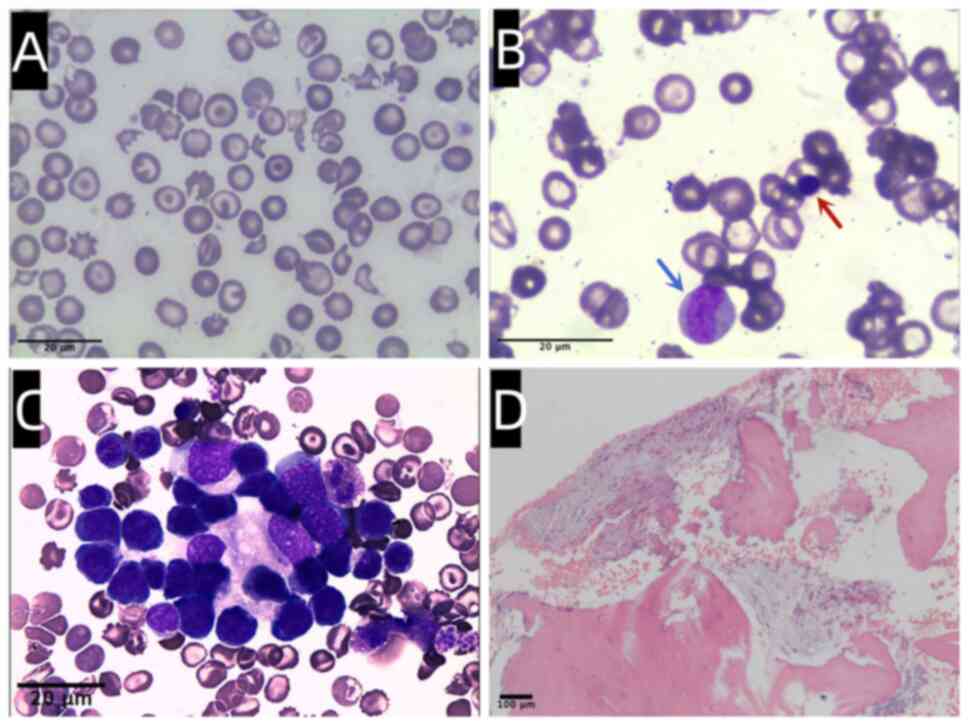
Olympus Corporation). Peripheral blood smear examination revealed a
variety of irregularly shaped erythrocytes, including
target-shaped, elliptic, teardrop-shaped, helmet-shaped, and
spherical red blood cells (Fig.
1A). Erythroblasts were frequently observed in peripheral
blood, with a nucleated red cell to white cell ratio of 66 to 100.
Additionally, immature granulocytes were presented in peripheral
blood (Fig. 1B). Bone marrow
smears exhibited 28% granulocytes and 56% erythrocytes, which were
mainly late juvenile erythrocytes. There was no obvious
proliferation or abnormal morphology of megakaryocytes. Bone marrow
biopsy demonstrated no blast cell proliferation and mild
myelofibrosis. Flow cytometry [flow cytometer DxFLEX (Beckman
Coulter, Inc.), data analysis by CytExpert, Gate setting: CD45-SSC
Gates] of the bone marrow revealed no abnormal immunophenotype
cells. Bone marrow aspiration was sent to a qualified third-party
company (Kindstar Globalgene Technology, Inc.) for related gene
testing and karyotype analysis. The BCR-ABL1 fusion gene,
JAK2 V617F and MPL W515L/K gene mutation were
detected using polymerase chain reaction. CALR exon 9 and
JAK2 exon12 were detected using sequencing. Fusion gene
analysis of BCR/ABL and mutational analysis of JAK2,
CALR and MPL genes yielded negative results. The
karyotype of bone marrow was normal (data not shown). Normal levels
of glucoencephalosidase, Lyso-GL-1 biomarkers [tandem mass
spectrometry (Suzhou PerkinElmer Medical Laboratory)] and GBA
genetic testing [LR-PCR and sequencing (Suzhou PerkinElmer Medical
Laboratory)] excluded Gaucher disease.
The positron emission tomography (PET)/computed
tomography (CT)(GE Medical Systems, LLC) scan revealed diffuse
fluorodeoxyglucose (FDG) accumulation in the enlarged liver and
spleen. The maximum cross-section of the spleen was 146x109 mm,
with an upper and lower diameter of 275 mm, and a maximum standard
unit value (SUVmax) of 4.7 (Fig.
2A). Mixed bone destruction and slightly active metabolism in
the vertebral bodies and sacroiliac bone were also observed.
Consequently, a fine needle puncture of the liver tissue guided by
B ultrasound (Mindray Resona7, Mindray Co., Ltd.) was performed.
However, the patient experienced massive abdominal bleeding after
the puncture, leading to an urgent laparotomy for hemostasis. The
histopathological analysis of the liver (data not shown) revealed
small patches of scattered small lymphocyte infiltration in the
portal area and hepatic sinusoids, mixed hyperplasia of T- and
B-lymphocytes, and edema and degeneration of liver cells. There was
no evidence of lymphoma, leukemia or extramedullary hematopoiesis.
The initial diagnosis was suspected to be primary myelofibrosis and
the patient was treated with prednisone (40 mg/qd, Shandong Xinhua
Pharmaceutical Co., Ltd.) and thalidomide (100 mg/qd, Changzhou
Pharmaceutical Co., Ltd.). However, 3 months later, fatigue and
bone pain worsened, the liver and spleen became enlarged,
hemoglobin levels decreased, and the need for red blood cell
transfusions continued. Due to the lack of response to the therapy,
a second bone marrow aspiration and biopsy were performed in
January, 2022. Erythropoiesis was active and hematopoiesis islands
were easily visible in the aspirate (Fig. 1C). Bone marrow biopsy was fixed
with 10% formalin at room temperature over 6 h and stained with
hematoxylin and eosin (H&E), with a protocol of 5 min of
hematoxylin (Beijing Solarbio Science & Technology Co., Ltd.)
and 1 min of pure eosin (Beijing Solarbio Science & Technology
Co., Ltd.), and subsequently observed under a microscope (Axio
Imager A2, Carl Zeiss AG). The biopsy specimen revealed aggravated
fibrotic changes, with extensive collagen fibrosis in the bone
marrow stroma (Fig. 1D),
highlighted by reticulin stains. Subsequently, ruxolitinib (5
mg/bid, Novartis Pharma Stein AG) was added to the treatment
regimen. A repeat of a PET/CT examination indicated multiple bone
lytic destruction with increased glucose metabolism and
intramedullary hemorrhage, hepatosplenomegaly and internal
hemorrhage. The maximum cross-section of the spleen was 164x112 mm,
with an upper and lower diameter of ~283 mm, and an SUVmax of 6.3
(Fig. 2B).
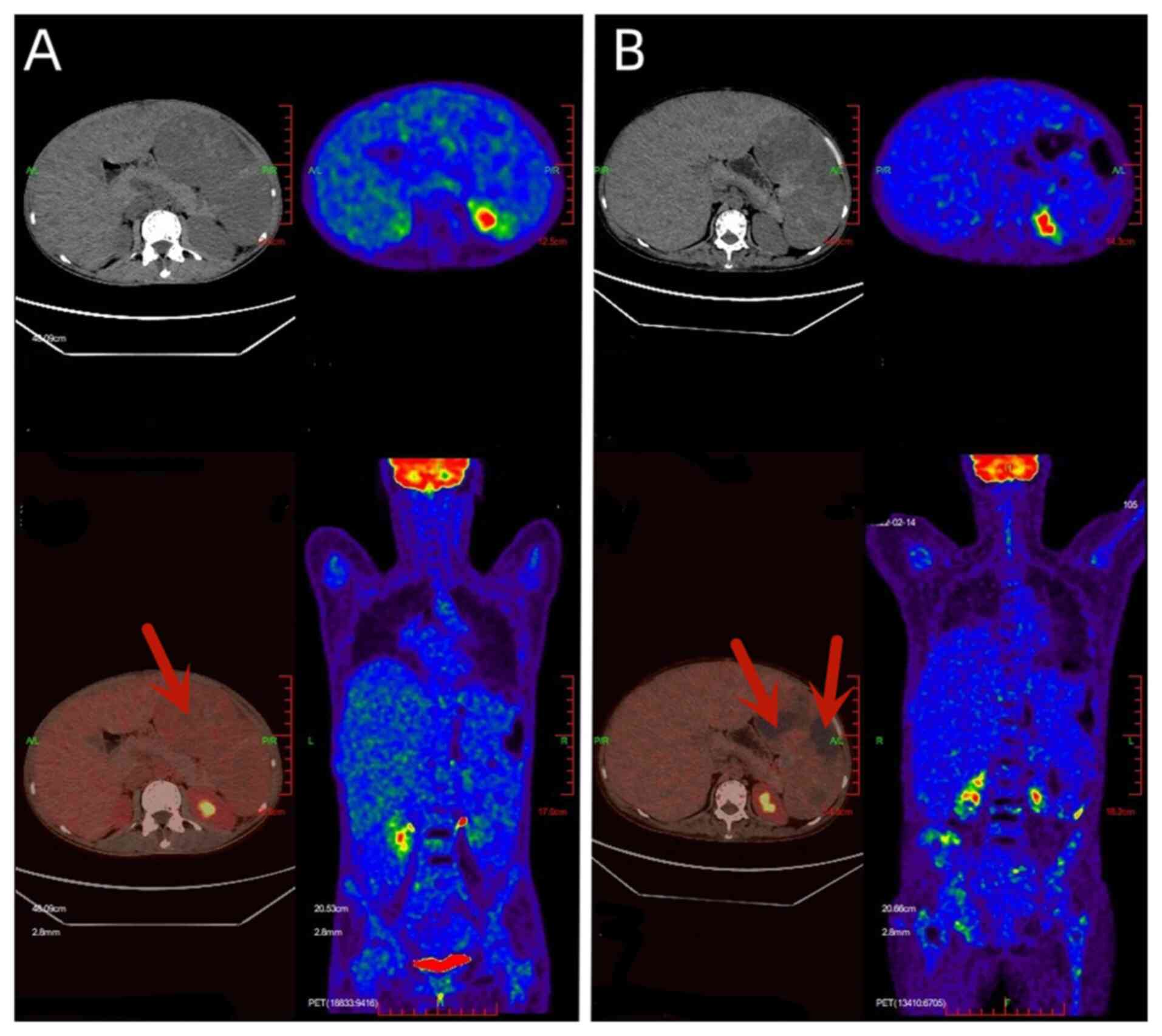 | Figure 2The images in the upper panels
represent CT scans, and the images in the lower panels exhibit the
fusion of PET and CT. The volume of the spleen increases and
multiple necrotic areas without glucose uptake are observed (red
arrows). (A) PET/CT performed on September, 2021 revealed maximum
cross-section of the spleen was 146x109 mm, with upper and lower
diameters of 275 mm, and an SUVmax of 4.7. (B) A PET/CT scan
performed on February, 2022 showed maximum cross-section of the
spleen was 164x112 mm, with an upper and lower diameter of ~283 mm,
and an SUVmax of 6.3. PET, positron emission tomography; CT,
computed tomography; SUVmax, maximum standard unit value. |
Operative and pathological
findings
Symptoms of spleen compression were deteriorating.
At 6 months after the onset, the patient underwent splenectomy,
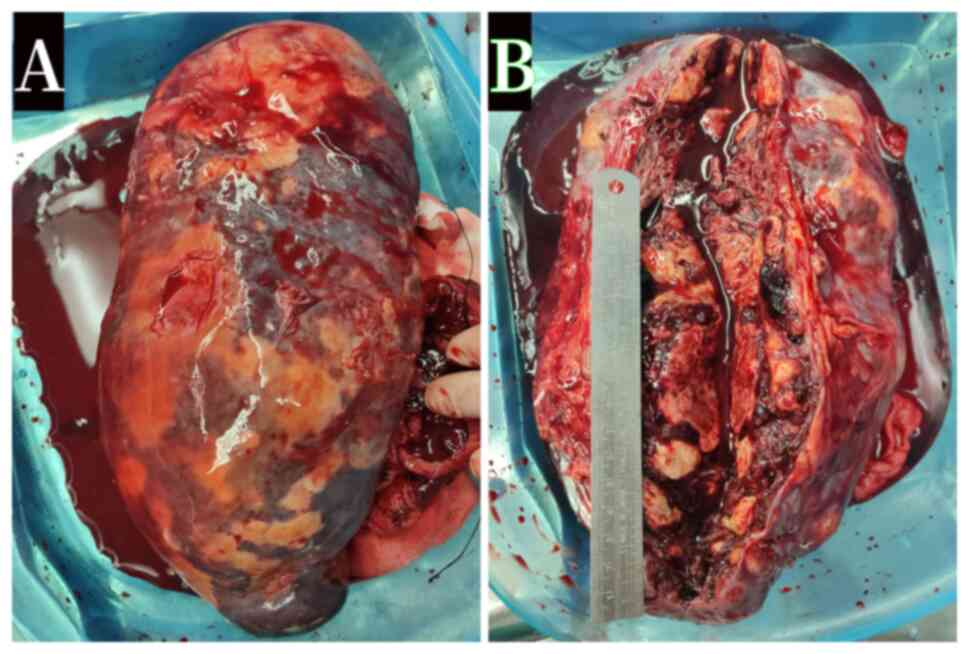
with the spleen weighing 1,200 g. Nodules and hemorrhage were
observed in the spleen specimens (Fig.
3A and B) and were also
visible in the liver during the surgery. The splenic parenchyma was
barely visible, almost completely replaced by hyperplastic and
intensified connective tissue. The splenic specimens were examined
using H&E staining 6 min at room temperature and
immunohistochemistry according to the protocol. The procedure of
immunohistochemical examination was performed. Firstly, a
paraffin-embedded section (4-µm-thick) was acquired after fixing a
tissue sample by 10% formalin at room temperature over 6 h. The
samples were then incubated with 3% H2O2 for
the inactivation of endogenous peroxidase at room temperature for
15 min. Subsequently, 50 µl primary antibody were added and
incubated in a wet box at 37˚C for 90 min. Secondary antibodies
were then added and incubated at room temperature for 25 min.
Freshly prepared DAB color developer (Beijing Solarbio Science
& Technology Co., Ltd.) was diluted x20 for color development.
The section samples were final observed under a microscope (Axio
Imager A2, Carl Zeiss AG). Various morphological patterns of
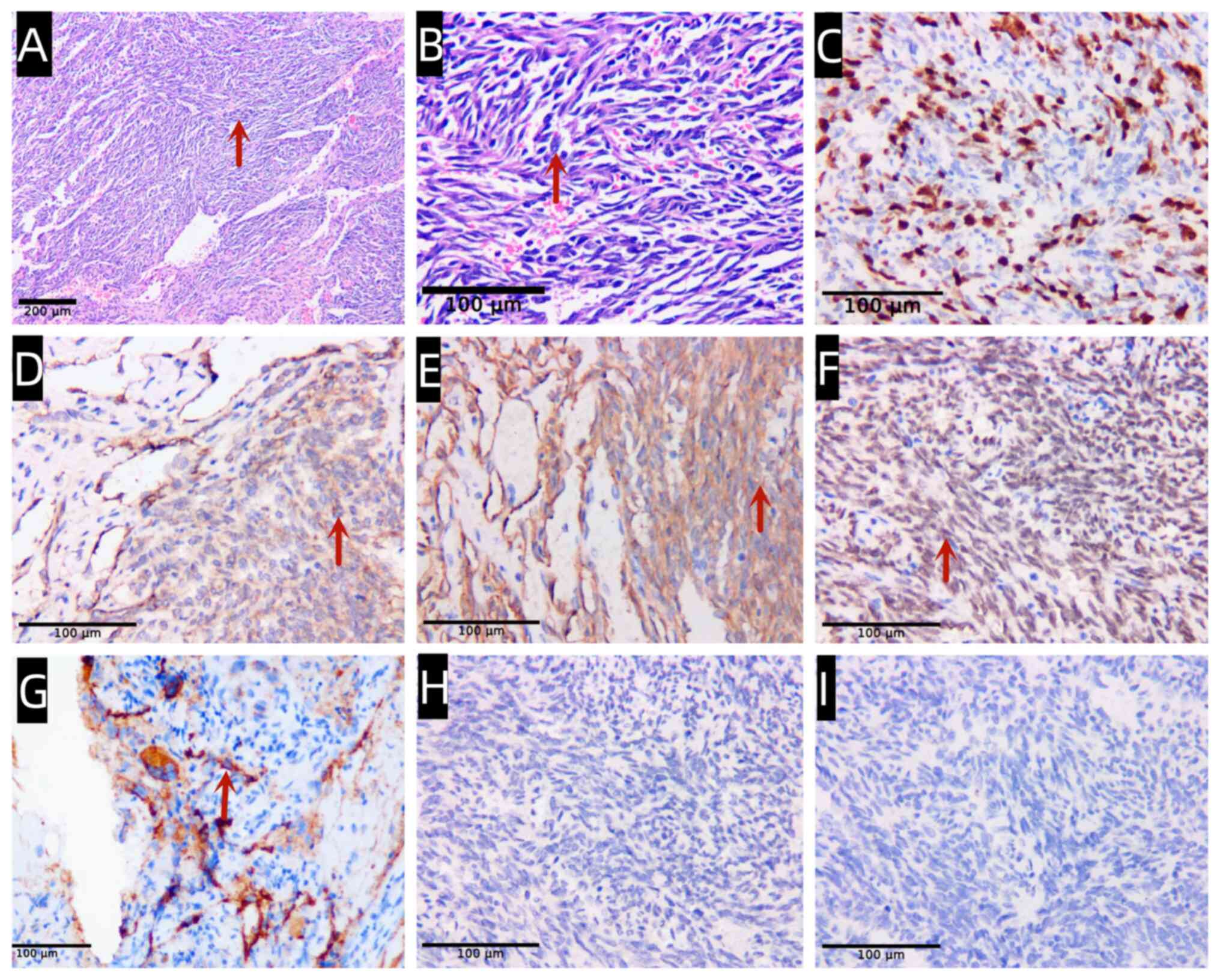
vascular neoplasms were presented in a background of hemorrhage,
along with solid flaky spindle cells (Fig. 4A and B). The Ki67 index was 60% (Fig. 4C). Immunohistochemical results
indicated that the neoplastic cells were positive for CD31, CD34,
ERG and Factor VIII (Fig. 4D-G),
and negative for human herpesvirus 8 (HHV-8) and CD8 (Fig. 4H and I), supporting an endothelial origin
hypothesis. The final diagnosis was splenic angiosarcoma. However,
the patient was not suitable for receiving chemotherapy due to her
poor medical condition after the surgery. Intermittent red blood
cell transfusions were required and the patient passed away 11
months after the surgery.
Discussion
Angiosarcomas, rare soft-tissue sarcomas originating
from endothelial cells, often exhibit a high metastatic rate and
present with a poor prognosis (1,4,5).
They can manifest in various locations throughout the body, with
cutaneous presentations being most commonly detected. Prior
information on these uncommon tumors has been primarily derived
from case series, suggesting that tumor behavior may be influenced
by the site of origin (6).
Notably, secondary breast angiosarcomas can result from therapeutic
radiation or chronic lymphedema (1). By contrast, splenic angiosarcoma is
exceedingly rare and highly aggressive, presenting in diverse ways
across cases (5,7). Common symptoms include abdominal
pain, distension and splenomegaly (8). Additionally, patients may experience
anemia, leucopenia, elevated lactate dehydrogenase levels and
thrombocytopenia; however, these symptoms are occasionally reported
(9). However, these vague
presentations can lead to a delayed diagnosis, and such cases may
initially resemble blood disorders, characterized by hematological
issues and splenomegaly (10). In
a subset of cases, splenic rupture occurs as an initial
presentation, usually signifying a poor prognosis (9,11-13).
Throughout the course of the disease, metastases are common, often
occurring early and extensively (14). The liver, lungs, lymph nodes, bone
marrow and bone are frequent sites of metastasis (4,10). A
previous study suggested that the median overall survival time of
patients with splenic angiosarcoma is ~8.1 months (15). Splenic rupture is an independent
indicator of adverse outcomes, while early diagnosis and surgical
resection prior to rupture are associated with an improved
prognosis (15).
In the present study, a case was reported which
initially presented with anemia and bone marrow fibrosis, mimicking
primary myelofibrosis, ultimately being diagnosed as splenic
angiosarcoma through splenectomy. The patient had no history of
prior disease, and no other risk factors were identified. The young
woman initially reported fatigue and bone pain. Following
admission, anemia, myelofibrosis and splenomegaly were confirmed.
The initial diagnosis was that of early myelofibrosis, and the
patient received prednisone and thalidomide treatment. However, the
symptoms of splenic compression did not alleviate following
treatment, and the progression of cachexia was evident. Therefore,
the purpose of splenectomy was to relieve the symptoms of splenic
compression and define the diagnosis of the primary disease.
Surgery was recommended after the multidisciplinary consultation,
and the final diagnosis was of splenic angiosarcoma following
splenectomy. A previous study reported varying FDG accumulation
levels in the primary angiosarcoma of the spleen, with different
distribution patterns, including diffuse, peripheral, or multiple
nodular type (16). In the case
described herein, PET-CT indicated hepatosplenomegaly and internal
hemorrhage, accompanied by multiple bone lytic destruction. In the
vast majority of previous splenic angiosarcoma case reports,
patients initially presented with gastrointestinal symptom or
spontaneous splenic rupture (5,13,17).
The initial manifestation of anemia and bone marrow fibrosis was a
noteworthy observation in the patient in the present case report,
since this specific case type may resemble blood diseases with
hematological disorders and splenomegaly.
Notably, persistently high ferritin levels were
observed in the present case. A previous retrospective study
suggested that patients with angiosarcoma with elevated ferritin
levels had a significantly poorer overall survival in comparison
with patients with normal ferritin levels (18). Additionally, the patient in the
present study exhibited a significant decrease in fibrinogen, which
aligns with reports in the literature indicating that patients with
angiosarcoma have a higher risk of bleeding, and low plasma
fibrinogen levels are predictive markers of a poor prognosis
(19,20). In the case reported in the present
study, it was hypothesized that excess splenic hemorrhage resulted
in substantial fibrinogen consumption, with a subsequent marked
reduction in fibrinogen levels. Furthermore, liver invasion may
also contribute to reduced fibrinogen synthesis. The present case
report highlights the importance of considering bleeding risks for
patients with splenic angiosarcoma complicated by
hypofibrinogenemia. Therefore, prior to performing a puncture or
surgery, it is essential to replenish exogenous fibrinogen to
normal levels to prevent hemorrhage.
The accurate diagnosis of splenic angiosarcoma
requires a triple assessment, combining clinical examination,
imaging findings and pathology (21). Nonetheless, the heterogeneity of
clinical presentations and non-specific imaging findings render the
definitive diagnosis of splenic angiosarcoma challenging, with
histopathological analysis remaining crucial. Splenic angiosarcoma
cells typically express multiple markers of vascular
differentiation (including CD31, CD34, factor VIII and vascular
endothelial growth factor 3) and at least one marker of histiocytic
differentiation (CD68 or lysozyme) (4). In the case in the present study,
neoplastic cells tested positive for CD31, CD34, ERG and Factor
VIII, and negative for HHV8 and CD8. Given the rarity of primary
angiosarcoma of the spleen, diagnosis often involves excluding
other malignant diseases (8). In
the patient described herein, an initial fine needle liver biopsy
did not yield a definitive diagnosis, prompting further assessment
of its origin. Additional tissue obtained during splenectomy
provided additional effective insight into the histological
features of a vascular neoplasm, facilitating subsequent
immunohistochemical analyses that confirmed the diagnosis of
splenic angiosarcoma. Ultimately, a histopathological examination
disclosed the diagnosis of primary angiosarcoma of the spleen, with
spreading to the liver and bone. The present case report
underscores the importance of using a panel of vascular
differentiation makers for the confirmation of the diagnosis when
encountering unexplained splenomegaly without hematological
malignancy.
Given the variability in symptomatology and the
potential for life-threatening complications, early diagnosis is
crucial. Biopsy acquisition prior to surgery for diagnostic
purposes is risky, due to the potential for bleeding and seeding.
As a result, histological diagnosis is generally only feasible
following splenectomy, which serves both diagnostic and therapeutic
purposes, as splenectomy is the treatment of choice for this
disease. A previous retrospective review of 145 patients with
angiosarcoma demonstrated that primary surgery resulted in an
improved overall and progression-free survival (22). Considering the aggressive nature
and high mortality rate of the disease, it is worth considering
that splenectomy without rupture may significantly extend patient
survival. Moreover, PET-CT is able to reveal characteristic changes
and detect any metastatic disease (16,23).
Metastasis at the time of diagnosis is a common finding and is
indicative of a poor prognosis. Therefore, splenectomy, when
performed after early diagnosis in the absence of metastatic
disease, is associated with a comparably improved prognosis. In the
present case, chemotherapy was not a suitable option due to the
poor condition of the patient at the time of diagnosis, ultimately
passing away 11 months after the surgery. Consequently, it is
suggested by the authors that splenectomy should be considered both
for diagnosis and treatment when appropriate in cases with a high
suspicion of primary splenic angiosarcoma.
The exact pathogenic mechanisms underlying
angiosarcoma remain incompletely understood. Yamamoto et al
(24) reported that tumor
cell-derived stem cell factor may potentially influence the
increased presence of mast cells, which in turn, could contribute
to the proliferation of tumor cells, ultimately driving the
progression of angiosarcoma. Primary myelofibrosis usually occurs
in elderly individuals, with a median age of onset of 60 years.
Additionally, mutations in JAK2, MPL and CALR
genes have been identified in the majority of primary
myelofibrosis, and patients negative for these three mutations are
relatively rare (25). In this
present study, the patient was 35 years of age without driver
mutations, thus being definitely diagnosed with splenic
angiosarcoma. Therefore, it was speculated that the myelofibrosis
in this patient was most likely secondary to angiosarcoma. It is
conceivable that the anemia may have resulted from hypersplenism or
extensive secondary myelofibrosis, triggered by angiosarcoma. In
addition, abnormally elevated peripheral blood nucleated red cells
were observed. The destruction of red blood cells was considered,
due to intrasplenic hemorrhage, compensation for erythroid
proliferation in the bone marrow, and extramedullary hematopoiesis
as possible causes of this type of abnormally increased numbers of
nucleated erythrocytes.
In conclusion, the present study describes the case
of a patient in whom the initial symptoms were suggestive of anemia
and bone marrow fibrosis, mimicking primary myelofibrosis. However,
this was eventually attributed to splenic angiosarcoma. The present
case underscores the importance of considering splenectomy for the
acquisition of histopathological evidence, further highlighting the
value of employing a panel of markers for vascular differentiation,
in order to aid in the diagnosis of angiosarcoma.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by The China
Postdoctoral Science Foundation (Grant no. 2020M673097).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
All authors (MW, ZL, LL, WZ and JL) contributed to
the conception and design of the study. Data collection was
performed by MW and ZL. Data analysis was performed by LL and WZ.
The first draft of the manuscript was written by MW and JL. ZL, LL
and WZ confirm the authenticity of all the raw data. All authors
have read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from the
patient described in the present case report.
Patient consent for publication
Written informed consent was obtained from the
patient for the publication of personal information-related data
and any related images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Young RJ, Brown NJ, Reed MW, Hughes D and
Woll PJ: Angiosarcoma. Lancet Oncol. 11:983–991. 2010.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Wheelwright M, Spartz EJ, Skubitz K,
Yousaf H, Murugan P and Harmon JV: Primary angiosarcoma of the
spleen, a rare indication for splenectomy: A case report. Int J
Surg Case Rep. 82(105929)2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Guglielmelli P, Rotunno G, Pacilli A, Rumi
E, Rosti V, Delaini F, Maffioli M, Fanelli T, Pancrazzi A, Pieri L,
et al: Prognostic impact of bone marrow fibrosis in primary
myelofibrosis. A study of the AGIMM group on 490 patients. Am J
Hematol. 91:918–922. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Neuhauser TS, Derringer GA, Thompson LD,
Fanburg-Smith JC, Miettinen M, Saaristo A and Abbondanzo SL:
Splenic angiosarcoma: A clinicopathologic and immunophenotypic
study of 28 cases. Mod Pathol. 13:978–987. 2000.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kania BE and Vasani S: A case report of
splenic rupture secondary to underlying angiosarcoma. Cureus.
12(e9439)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Fury MG, Antonescu CR, Van Zee KJ, Brennan
MF and Maki RG: A 14-year retrospective review of angiosarcoma:
Clinical characteristics, prognostic factors, and treatment
outcomes with surgery and chemotherapy. Cancer J. 11:241–247.
2005.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Gorzelak-Pabis P, Zuszek-Frynas A and
Broncel M: Primary splenic angiosarcoma: A very rare and aggressive
neoplasm with a poor prognosis. Pol Arch Intern Med. 130:142–144.
2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Damouny M, Mansour S and Khuri S: Primary
Angiosarcoma of the Spleen: An aggressive neoplasm. World J Oncol.
13:337–342. 2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Xu B, Xie X, Zhou X, Zhai M and Yang W:
Spontaneous rupture of primary splenic angiosarcoma: A case report.
Oncol Lett. 10:3271–3273. 2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Khorzhevskii VA, Ermachenko DA, Gappoev SV
and Levkovich LG: Metastatic lesion of the bone marrow caused by
primary angiosarcoma of the spleen. Arkh Patol. 84:52–55.
2022.PubMed/NCBI View Article : Google Scholar : (In Russian).
|
|
11
|
Badiani R, Schaller G, Jain K, Swamy R and
Gupta S: Angiosarcoma of the spleen presenting as spontaneous
splenic rupture: A rare case report and review of the literature.
Int J Surg Case Rep. 4:765–767. 2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Koutelidakis IM, Tsiaousis PZ, Papaziogas
BT, Patsas AG, Atmatzidis SK and Atmatzidis KS: Spleen rupture due
to primary angiosarcoma: A case report. J Gastrointest Cancer.
38:74–77. 2007.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ozcan B, Cevener M, Kargi AO, Dikici H,
Yıldız A, Özdoğan M and Gürkan A: Primary splenic angiosarcoma
diagnosed after splenectomy for spontaneous rupture. Turk J Surg.
34:68–70. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Levy ACJ, DeFilipp M, Blakely M, Asiry S,
Jormark S and Goodman A: Splenic angiosarcoma diagnosed on bone
marrow biopsy: Case report and literature review. Radiol Case Rep.
14:390–395. 2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Li R, Li M, Zhang LF, Liu XM, Hu TZ, Xia
XJ, Chi JS, Jiang XX and Xu CX: Clinical characteristics and
prognostic factors of primary splenic angiosarcoma: A retrospective
clinical analysis from China. Cell Physiol Biochem. 49:1959–1969.
2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Takahashi H, Hara T, Suzuki H, Hashimoto R
and Minami M: FDG-PET/CT demonstrates splenic angiosarcoma bone
marrow metastasis. Clin Nucl Med. 45:e20–e23. 2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Abdallah RA, Abdou AG, Asaad NY,
Al-Sharaky DR and Alhanafy AM: Primary epithelioid angiosarcoma of
spleen: A case report and review of literature. J Clin Diagn Res.
10:ED05–ED07. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chen D, Tang M, Lv S, Wang H, Du W, Zhao
X, Lin L, Zhu Y, Wang G, Zhu H and Zhao K: Prognostic usefulness of
clinical features and pretreatment (18)F-FDG PET/CT metabolic
parameters in patients with angiosarcoma. Quant Imaging Med Surg.
12:2792–2804. 2022.PubMed/NCBI View Article : Google Scholar
|
|
19
|
An R, Ma JY, Ni XH and Wang CL:
Angiosarcoma of the breast with hypofibrinogenemia: A rare case
report and review of the literature. Front Oncol.
12(1047935)2022.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Mori S, Taki T, Murakami Y, Urata T,
Okumura M, Akanabe H, Ebata A, Imai S, Yokota K and Akiyama M: Low
plasma fibrinogen levels are associated with poor prognosis in
cutaneous angiosarcoma of the head and neck. Cancer Sci.
112:3924–3927. 2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Juin Hsien BL and Shelat VG: Spleen
angiosarcoma: A world review. Expert Rev Gastroenterol Hepatol.
15:1115–1141. 2021.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Smrke A, Hamm J, Karvat A, Simmons C and
Srikanthan A: A retrospective review of 145 patients with
angiosarcoma: Radiation therapy, extent of resection and
chemotherapy are important predictors of survival. Mol Clin Oncol.
13:179–185. 2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zhao Q, Dong A, Wang Y, He T and Zuo C:
FDG PET/CT in primary splenic angiosarcoma with diffuse involvement
of the spleen. Clin Nucl Med. 42:815–817. 2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Yamamoto T, Umeda T and Nishioka K:
Immunohistological distribution of stem cell factor and kit
receptor in angiosarcoma. Acta Derm Venereol. 80:443–445.
2000.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Shantzer L, Berger K and Pu JJ: Primary
myelofibrosis and its targeted therapy. Ann Hematol. 96:531–535.
2017.PubMed/NCBI View Article : Google Scholar
|