1. Introduction
Cancer, characterized by the uncontrolled
proliferation and spread of abnormal cells, remains one of the
world's most pervasive and deadly diseases. While advancements in
therapy have improved outcomes for certain patients, several
patients exhibit recurrent or refractory disease, primarily due to
drug resistance. This phenomenon, where cancer cells evolve to
evade the effects of chemotherapy, targeted therapy or
immunotherapy, remains a major barrier to long-term treatment
success.
Among the several types of cancer, brain tumors are
particularly lethal. Annually, over 300,000 new cases of brain
cancer are diagnosed globally, leading to >250,000 deaths. In
the United States alone, an estimated 25,400 new brain tumor cases
and ~19,000 deaths were predicted in 2024(1). Brain tumors are also the leading
cause of cancer-related deaths in children and adolescents under
the age of 19(2). The 5-year
relative survival rate for brain and other nervous system cancers
in the U.S. stands at ~34%, highlighting the urgent need for
improved detection and more effective therapies (1).
Despite the ongoing development of therapeutics,
brain cancer remains among the most difficult malignancies to
treat. This is largely due to the aggressive biology of the disease
and the unique challenges posed by the brain's protective
environment. The blood-brain barrier (BBB), while essential for
maintaining neural homeostasis, limits the penetration of most
chemotherapeutic and biologic agents, posing a major hurdle for
effective drug delivery.
Treatment strategies for brain tumors include
chemotherapy, targeted therapy and immunotherapy. Temozolomide
(TMZ), an oral alkylating agent, remains the cornerstone of
chemotherapeutic regimens, supplemented by agents such as
carmustine, lomustine, vincristine, procarbazine and etoposide.
Targeted agents like vorasidenib [isocitrate dehydrogenase (IDH)1/2
inhibitor] and bevacizumab [BEV, a vascular endothelial growth
factor (VEGF) inhibitor] have shown utility in specific subtypes.
Immunotherapeutics, including anti-PD-1 antibodies pembrolizumab
and nivolumab, are under active investigation in clinical trials,
particularly for GBM.
GBM, the most common and aggressive malignant brain
tumor, accounts for ~50% of all primary malignant brain tumors. It
exhibits a particularly poor prognosis due to its rapid progression
and formidable resistance to existing therapies. The 5-year
survival rate for GBM remains at a dismal 7% (3,4),
reflecting the urgent need for novel therapeutic strategies.
The aim of the present review is to provide a
comprehensive and integrative analysis of the molecular and
cellular mechanisms driving therapeutic resistance in GBM, with a
focus on emerging and underappreciated pathways such as exosomal
non-coding RNAs (ncRNAs), metabolic reprogramming and dysregulated
signaling networks [such as the Wnt/β-catenin, Hippo, PI3K/Akt and
MAPK]. Importantly, beyond being merely a descriptive overview, the
therapeutic implications of these mechanisms, including how they
might be exploited for combination regimens, drug delivery
innovations and precision medicine approaches, are also discussed.
By bridging mechanistic insights with actionable strategies, the
present review seeks to support the development of more effective,
resistance-overcoming treatments for GBM, ultimately improving
clinical outcomes in this intractable disease.
2. BBB
BBB is a dynamic interface that regulates molecular
transport between systemic circulation and brain parenchyma. While
essential for neuroprotection, its structural and functional
complexity poses significant challenges for treating brain tumors.
The structural organization of the BBB plays a crucial role in its
function as a selective and protective interface between the
systemic circulation and the brain. Its key structural components
contribute to its function through the following ways:
Endothelial cells and tight junctions (TJs).
Brain microvascular endothelial cells form the primary barrier with
TJs composed of claudins, occludins and zonula occludens proteins.
These TJs create a high trans-endothelial electrical resistance
(>1,800 Ω·cm²), restricting paracellular passage of polar
molecules and toxins (5).
Bicellular (claudin-5) and tricellular (angulin-1) junctions
further regulate permeability.
Supporting cellular components. Pericytes
stabilize endothelial junctions via platelet-derived growth factor
receptor (PDGFR)-β signaling, and their loss leads to barrier
leakage, especially in gliomas (6). Astrocytes cover >99% of the
endothelial surface, and along with pericytes, maintain BBB
integrity. Microglia modulate the BBB but can also disrupt it by
secreting inflammatory cytokines.
Basement membrane. The 50-100 nm
extracellular matrix, rich in collagen IV and laminins, provides
structural support and regulates leukocyte trafficking, ensuring
vascular stability and selective permeability. These structural
elements collectively restrict solute exchange, regulate selective
transport and maintain homeostasis, which are essential for
neuroprotection. However, these same features significantly
contribute to drug resistance in GBM through several mechanisms
(7).
Heterogeneous BBB disruption. In brain
tumors, the BBB has leaky regions but retains an intact BBB in
non-enhancing tumor areas, creating sanctuaries for cancer cells
(8). In high-grade glioma (HGG)
tumors, surgical and imaging data show that tumor cells extend
beyond contrast-enhancing MRI regions, with PET tracers confirming
metabolic activity in these areas, indicating BBB integrity. Drug
penetration studies reveal poor delivery of several therapies to
non-enhancing tumor regions due to active efflux transporters.
Additionally, imaging-pathology mismatches highlight that
abnormalities on T2-weighted and fluid-attenuated inversion
recovery (FLAIR) MRI sequences, which are sensitive to edema and
infiltrative tumor regions, represent infiltrative tumors without
sufficient BBB disruption for effective drug delivery. Clinically,
most GBM recurrences originate near contrast-enhancing regions, but
infiltrative cells in non-enhancing areas contribute to
progression. Therefore, there is a need for therapies that can
effectively target tumor cells beyond contrast-enhancing regions,
addressing both enhancing and non-enhancing compartments for
improved treatment outcomes.
Efflux transporters. Overexpression of
ATP-dependent efflux transporters, such as P-glycoprotein (P-gp)
and breast-cancer-resistance protein (BCRP), in tumor endothelial
cells reduces intracellular drug concentrations. For example,
HER2+ breast cancer brain metastases exhibit upregulated
expression of BCRP, limiting trastuzumab efficacy (9).
Signaling pathways. The Wnt and sonic
hedgehog (Shh) pathways contribute to BBB integrity and, when
dysregulated in GBM, also affect drug penetration (10). Wnt signaling promotes BBB integrity
by maintaining the expression of key vascular stability proteins
and suppressing factors that increase permeability. β-catenin
activation, a key component of Wnt signaling, helps regulate
claudin-5 and Glut1, which are essential for BBB function.
Experimental knockout of β-catenin, as well as LRP5 and LRP6, leads
to increased expression of PLVAP, a protein associated with
vascular fenestrations, resulting in abnormal BBB leakiness.
Furthermore, paracrine Wnt inhibitors such as WIF1 and DKK1,
secreted in Wnt medulloblastomas, disrupt BBB integrity, but their
experimental inhibition restores normal vascular function. These
findings highlight that active Wnt signaling is crucial for
maintaining a tightly regulated and selective BBB by preventing the
formation of fenestrations and excessive permeability.
The Shh pathway plays a critical role in maintaining
BBB integrity through both structural and immunological mechanisms
(11). Astrocytes secrete Shh,
which binds to Patched-1 and Smoothened receptors on endothelial
cells. This binding enhances the expression of tight and adherens
junction proteins, strengthening the BBB and reducing permeability.
Additionally, Shh suppresses proinflammatory responses by
inhibiting chemokine secretion, preventing leukocyte adhesion and
modulating T cell activity. Disruption of Shh signaling can lead to
BBB breakdown and heightened inflammation. Therefore, Shh is
essential for both the development and therapeutic maintenance of
BBB function.
Several strategies to enhance drug delivery by
overcoming the BBB have been proposed and some have been evaluated
in clinical trials with various degrees of success (12). Direct drug administration via
intra-tumoral, intranasal or intrathecal routes allows localized
delivery (such as carmustine and trastuzumab) but faces challenges,
including neurotoxicity and inconsistent efficacy (13,14).
Chemical modifications, such as increasing lipophilicity or
encapsulating drugs in nanoparticles (for example, liposomal
doxorubicin), improve BBB penetration and targeted delivery
(15). Inhibition of P-gp and BCRP
efflux transporters enhanced brain penetration of doxorubicin and
vemurafenib in vitro and in vivo (16,17).
Physical disruption techniques, including focused
ultrasound, laser-induced thermal therapy and osmotic disruption
(mannitol), temporarily open the BBB to facilitate drug entry
(18). Ongoing clinical trials are
evaluating ultrasound-mediated BBB disruption systems, such as
Exablate and SonoCloud-9, which use focused or implantable
ultrasound with microbubbles to transiently and noninvasively open
the BBB, enhancing the delivery of carboplatin to GBM tumor sites
(NCT04417088; NCT03744026).
Tumor-tropic neural stem cells engineered to deliver
anticancer agents show promise in clinical trials (19). Ongoing studies are evaluating
BBB-penetrating analogs of approved drugs, such as Berubicin (a
doxorubicin analog) (NCT04762069) and Buparlisib (a PI3K inhibitor)
(NCT01349660), for their potential to improve outcomes in recurrent
or refractory GBM.
While these strategies demonstrate potential in
preclinical and early clinical studies, challenges like side
effects, invasiveness and variable efficacy hinder widespread
adoption, with future research focusing on refining combination
therapies and BBB modulation techniques.
3. Resistance to chemotherapy
Since 2005, the Stupp protocol has been the standard
of treatment for GBM. It consists of radiation combined with TMZ
following surgery (20). While
this protocol has improved 2-year survival rates from 10.4 to 26.5%
compared with radiation alone, >90% of patients with GBM still
develop resistance and experience relapse (21). Uncovering the molecular drivers of
chemotherapy resistance in GBM is therefore critical for advancing
more effective therapeutic strategies. The following sections
outline key resistance mechanisms and their clinical
implications.
Upregulation of
O6-methylguanine (O6MeG)-DNA
methyltransferase. TMZ functions by inducing O6MeG
adducts that cause DNA breaks, cell cycle arrest and apoptosis,
leading to cell death (22,23).
TMZ resistance in treatment-naïve patients with GBM is mediated
predominantly by the DNA repair enzyme
O6-methylguanine-DNA methyltransferase (MGMT), which
removes the methyl adducts formed by TMZ, thereby preventing DNA
damage and rescuing cancer cells from death (24). In GBM, the MGMT promoter is often
methylated, silencing its expression and rendering tumors more
sensitive to TMZ. However, TMZ treatment can apply selective
pressure, potentially leading to demethylation of the MGMT
promoter, increased MGMT expression and subsequent repair of
TMZ-induced lesions, resulting in acquired resistance (25). MGMT promoter methylation status
remains one of the most robust and clinically relevant predictive
biomarkers of the response to TMZ. Several clinical trials have
explored the prognostic and therapeutic relevance of MGMT
methylation status in both newly diagnosed and recurrent GBM. The
CENTRIC (NCT00689221) and Alliance A071102 (NCT02152982) trials
investigated the addition of cilengitide and veliparib,
respectively, to standard chemoradiotherapy in MGMT-methylated GBM.
Both trials failed to demonstrate survival benefits, highlighting
the challenges of improving outcomes beyond TMZ in this subgroup
(26,27).
Conversely, trials targeting MGMT-unmethylated GBM
have focused on TMZ-sparing strategies. A Phase II trial of VAL-083
with radiotherapy showed early promise by bypassing MGMT-mediated
resistance (NCT03050736). Similarly, a study comparing etoposide
and cisplatin to TMZ (NCT05694416) aims to identify more effective
chemotherapeutic options. Immunotherapy combinations, such as
nivolumab and ipilimumab with radiation (NCT04396860), are also
under evaluation in this difficult-to-treat population.
The GENOM 009 study (NCT01102595) reaffirmed the
prognostic value of tissue-based MGMT methylation but found limited
utility in serum-based assays (28). These trials collectively underscore
the critical role of MGMT status in GBM therapy and the ongoing
pursuit of personalized treatment approaches, particularly for
patients with unmethylated tumors.
Defective DNA mismatch repair (MMR). TMZ
causes O6MeG mismatches with thymine, which are normally
recognized by the MMR system. Functional MMR triggers apoptosis in
response to such damage. Deficiencies in the MMR system, including
mutations in genes such as MSH6, MLH1 and PMS2, impair DNA repair,
contributing to resistance (29-31).
MMR-deficient GBMs may appear responsive initially but often recur
with hypermutation phenotypes and TMZ resistance (32). Hypermutation phenotype
MMR-deficient GBMs with a high tumor mutational burden (TMB) are
excellent candidates for immunotherapy (33). Immunotherapy has shown remarkable
success in MMR-deficient tumors across various cancer types,
including colorectal cancer, leading to FDA approval of the PD-1
checkpoint inhibitor pembrolizumab for any microsatellite
instability-high/MMR-deficient solid tumors, a landmark
tumor-agnostic approval. However, early trials, including
KEYNOTE-028 (NCT02054806) and a Phase II window-of-opportunity
study (NCT02337686), found pembrolizumab to be safe but with
limited clinical benefit in recurrent GBM, showing modest 6-month
progression-free survival (PFS) rates (~40-44%) and a median OS of
14-20 months. Additionally, pembrolizumab was studied in a
multi-cancer, Phase II trial (NCT02886585) focused on patients with
brain metastases, including those with GBM. In this setting, the
intracranial benefit rate was 42.1%, and certain patients with GBM
exhibit survival exceeding two years (34). These findings suggest that a subset
of patients with GBM may derive durable benefits from PD-1
blockade, although predictive biomarkers remain undefined, and
support further studies to identify biomarkers and mechanisms of
resistance.
Glioma stem-like cells (GSCs). GSCs are a
quiescent subpopulation within the tumor that exhibits stem-like
properties, including self-renewal and the ability to repopulate
the tumor after treatment. Nestin, a marker of neural stem cells,
is also expressed in GSCs and can serve as a biomarker to identify
this resistant population. In preclinical models,
Nestin+ GSCs were shown to persist after TMZ therapy and
regenerate proliferative tumor cells (35). Remarkably, selective ablation of
these Nestin+ cells using genetic approaches
significantly impaired tumor growth. These findings underscore the
therapeutic potential of targeting GSCs to improve GBM treatment
outcomes.
Survivin is emerging as a functionally relevant and
potential marker of GSCs, particularly in the context of
therapeutic targeting. Survivin (BIRC5) is highly expressed
in GSCs compared with non-stem GBM cells. Its inhibition
selectively reduces the viability and self-renewal capacity of GSCs
(36). Furthermore, downregulation
of Survivin sensitizes these cells to chemotherapeutic agents,
highlighting its critical role in GSC maintenance and therapy
resistance.
Building on these preclinical findings, clinical
strategies targeting Survivin have begun to show promise. The
SurVaxM vaccine trial, a Phase II clinical study, evaluated a
vaccine for the Survivin protein in combination with TMZ and
granulocyte-macrophage colony-stimulating factor in patients with
newly diagnosed glioblastoma. Initial results have been
encouraging, with 96% of participants progression-free at 6 months
and 93% alive at 12 months, demonstrating substantial improvement
over historical benchmarks (37).
Efflux transporters and TMZ resistance in
GBM. Efflux transporters play a pivotal role in mediating
resistance to TMZ in GBM by actively reducing intracellular drug
concentrations, thereby diminishing its therapeutic efficacy. Munoz
et al (38) demonstrated
that TMZ is a substrate for P-gp. In GBM cells with upregulated
MDR1 mRNA levels (which encodes P-gp), there is increased efflux of
TMZ, lowering its intracellular concentration and reducing its
cytotoxicity. P-gp inhibitors restored TMZ sensitivity, confirming
its role in resistance. Another study found that hypomethylation of
the ABCB1 and ABCG2 promoters led to overexpression of these
transporters, facilitating drug efflux and decreasing TMZ efficacy
in GBM cells. Epigenetic regulation was a key driver of this
phenotype (39). Elevated levels
of ABCA1, a protein traditionally associated with lipid transport,
are linked to poor clinical outcomes. Silencing ABCA1 expression
increases the sensitivity of glioma cells to TMZ, indicating its
involvement in drug efflux and resistance mechanisms (40). Mechanistically, ABCA1 contributes
to TMZ resistance by altering the TME, specifically by promoting M2
macrophage infiltration, a phenotype associated with immune
suppression and tumor progression. The p53/E2F7 axis was shown to
transcriptionally upregulate ABCA8 and ABCB4, both contributing to
reduced intracellular accumulation of TMZ and enhanced resistance
in GBM (41). Silencing E2F7
resensitized cells to TMZ.
Additional support for TMZ being a substrate of
efflux transporters such as P-gp and BCRP comes from studies
showing that genetic deletion or pharmacological inhibition of
these proteins, using agents like elacridar (GF120918),
significantly enhanced brain penetration by 1.5-fold, and antitumor
efficacy of TMZ in mice with orthotopic or intracranial GBM tumors.
Similarly, Reversan, another efflux transporter inhibitor, also
markedly enhanced TMZ activity in patient-derived primary and
recurrent GBM models (42). These
observations suggest that inhibiting efflux transporters to enhance
intracellular TMZ levels may re-sensitize GBM cells to TMZ.
However, despite promising preclinical data, this approach has not
yet translated to clinical success, likely due to the complexity of
resistance mechanisms and safety concerns at effective doses.
Nonetheless, it remains an active area of research with potential
for identifying a clinically viable efflux transporter
inhibitor.
Hypoxia. Hypoxia, a defining feature of
aggressive gliomas such as GBM, arises when rapidly growing tumors
outpace their blood supply (43).
This oxygen-deficient microenvironment stabilizes hypoxia-inducible
transcription factors HIF-1α and HIF-2α, which orchestrate a broad
transcriptional program that enhances tumor cell survival,
therapeutic resistance and disease progression. Key downstream
targets of these factors include MGMT, which contributes to TMZ
resistance by reducing intracellular TMZ levels (44).
HIF-2α promotes the expression of GSC signature
genes, supporting the maintenance and expansion of this inherently
TMZ-resistant subpopulation (45).
Moreover, hypoxia induces SHOX2 expression, a gene associated with
TMZ resistance, particularly in MGMT-unmethylated gliomas and
TMZ-resistant cell lines (46).
Hypoxia also modulates the apoptotic machinery to
support resistance. For example, HIF-1α upregulates miR-26a under
hypoxic conditions, which in turn suppresses pro-apoptotic proteins
Bad and Bax, thereby inhibiting mitochondrial apoptosis and
contributing to TMZ resistance (47).
Collectively, these interconnected hypoxia-driven
mechanisms underscore the pivotal role of hypoxia in driving
therapeutic resistance in GBM. In response, several clinical trials
have been launched to explore therapeutic strategies that either
inhibit or leverage hypoxia in GBM. These approaches include
hypoxia-activated prodrugs, oxygen therapeutics and inhibitors
targeting HIF proteins, to enhance tumor sensitivity to
conventional treatments or disrupt hypoxia-mediated survival
pathways.
One notable trial, NCT01403610, evaluated
evofosfamide (TH-302), a hypoxia-activated prodrug, in combination
with BEV in patients with recurrent GBM who had failed prior BEV
treatment (48). PFS at 4 months
was 31%, which was a statistically significant improvement over the
historical rate of 3%, although the clinical significance of this
may be limited. Another trial, NCT03216499, investigated PT2385, a
selective HIF-2α inhibitor, in patients with first recurrent GBM
(49). While no radiographic
responses were observed, patients with higher systemic drug
exposure showed improved PFS, suggesting potential pharmacodynamic
relevance.
Addressing tumor oxygenation directly, NCT02189109
explored NVX-108, an oxygen therapeutic designed to improve tumor
oxygen levels and enhance the effects of radiotherapy and
chemotherapy in newly diagnosed GBM (50). Preliminary results demonstrated
effective tumor reoxygenation without affecting normal brain
tissue, with indications of improved survival outcomes. These
findings support further exploration of oxygenation strategies in
GBM therapy.
Several early-phase trials focused on inhibiting
HIF-1α, a central regulator of hypoxic adaptation. These
include EZN-2208 (NCT01251926), EZN-2698 (NCT01120288), PX-478
(NCT00522652), OKN-007 (NCT01672463) and Icaritin (NCT02496949).
These agents employ a range of mechanisms, from downregulating
HIF-1α mRNA to promoting its degradation, and were primarily
studied for safety and pharmacodynamic effects, with limited
efficacy data reported (51).
In conclusion, while targeting hypoxia in GBM
presents a compelling therapeutic avenue, clinical trials to date
have yielded mixed results, with most agents demonstrating safety
but limited efficacy in monotherapy settings. However, these
studies highlight the potential for hypoxia-directed therapies to
complement existing treatments. Ongoing research focusing on
patient stratification, drug combinations, and biomarkers of
hypoxic response will be key to unlocking the full therapeutic
potential of these strategies in GBM.
Dysregulated intracellular signaling
pathways. While DNA repair mechanisms, such as MGMT
upregulation and defective MMR, are well-established contributors
to TMZ resistance, dysregulated intracellular signaling pathways,
especially the PI3K/Akt and MAPK pathways, play an equally critical
role in mediating resistance in GBM.
PI3K/Akt/mTOR pathway. The PI3K/Akt/mTOR
signaling pathway plays a central role in GBM resistance to TMZ. It
is frequently dysregulated in GBM, in up to 88% of tumors,
primarily due to upstream receptor tyrosine kinase (RTK)
alterations such as epidermal growth factor receptor (EGFR)
amplification, activating mutations of PI3CA (p110) or PIK3R1
(P85), or loss of PTEN expression (52).
One mechanism through which PI3K/Akt/mTOR confers
TMZ resistance is metabolic reprogramming, known as the Warburg
effect. Akt upregulates PDK1, which inhibits pyruvate
dehydrogenase, suppressing oxidative phosphorylation and promoting
glycolysis (53). This metabolic
shift supports tumor cell survival under hypoxic conditions and
contributes to resistance. Dichloroacetate, a PDK1 inhibitor, can
reverse this effect, particularly in EGFRvIII-positive GBMs,
restoring TMZ sensitivity (54).
Another key resistance mechanism involves the
response to hypoxia and the promotion of angiogenesis. Akt
stabilizes HIF-1α, which enhances the transcription of VEGF and
other hypoxia-response genes, driving angiogenesis and increasing
tumor survival in hypoxic microenvironments (55). HIF-1 also induces the expression of
CXCL12 and its receptor CXCR4, promoting tumor cell proliferation
and invasiveness (56).
Additionally, NF-κB is activated via Akt-mediated
degradation of its inhibitor, IkB. This activation leads to
increased transcription of pro-survival genes and has been strongly
linked to both poor prognosis and TMZ resistance (57).
The PI3K/Akt pathway further promotes resistance by
inhibiting apoptosis. It modulates the upregulation of
anti-apoptotic proteins such as Bcl-2 and Mcl-1, which inhibit
apoptotic processes and contribute to chemoresistance. For example,
studies have shown that the inhibition of PI3K and Bcl-2 can
sensitize glioblastoma cells to apoptosis by downregulating Mcl-1
and phospho-BAD, highlighting the role of these proteins in therapy
resistance (58). Moreover, the
PI3K/Akt pathway influences the overexpression of other
anti-apoptotic factors such as MDM2, a negative regulator of the
tumor suppressor p53, thereby further inhibiting apoptotic pathways
(59,60). Akt also increases the expression of
survivin, an inhibitor of apoptosis, which blocks TMZ-induced cell
death. Inhibition of survivin has been shown to increase TMZ
sensitivity in GBM cells (61).
Furthermore, the activation of the PI3K/Akt/mTOR
pathway in general, and mTOR in particular, positively regulates
autophagy in response to TMZ-induced DNA damage, leading to tumor
cell survival under therapeutic stress (62). Moreover, the CaMKKβ/AMPKα/mTOR
signaling axis has been linked to the upregulation of TRPC5, a
protein that triggers autophagy and contributes to TMZ resistance.
Targeting mTOR-mediated TRPC5 expression is thus emerging as
another potential strategy to sensitize GBM cells to TMZ and
overcome therapeutic resistance (63).
In summary, the PI3K/Akt pathway orchestrates
multiple survival strategies in GBM, altering metabolism,
suppressing apoptosis, enhancing angiogenesis and modulating
transcription factors, all of which collectively foster resistance
to TMZ. In recent years, multiple clinical trials have evaluated
pharmacological inhibitors targeting this pathway as a strategy to
overcome TMZ resistance, with mixed results.
One of the more advanced clinical efforts involved
GDC-0084 (Paxalisib), a dual PI3K/mTOR inhibitor evaluated in a
Phase IIa trial (NCT03522298) in patients with newly diagnosed GBM
with unmethylated MGMT promoter status, an indicator of poor TMZ
response. Administered post-chemoradiation, GDC-0084 showed
manageable toxicity and modest clinical activity, though its
definitive efficacy remains to be confirmed in larger, controlled
studies (64).
Combination approaches have also been tested. A
Phase I study of temsirolimus (a mTOR inhibitor) with perifosine
(an AKT inhibitor) in recurrent malignant gliomas reported
acceptable safety but limited efficacy (NCT01051557), with disease
stabilization rather than regression being the most common outcome.
Similarly, the dual PI3K/mTOR inhibitor voxtalisib (XL765) was
combined with TMZ (± radiotherapy) in early-phase studies
(NCT00704080) (65). Although
biologically active, these agents were generally associated with
tolerable side effects but only modest improvements in
progression-free survival.
Other compounds, such as NVP-BEZ235, sapanisertib
(TAK-228) and CC-115 have shown encouraging preclinical efficacy,
particularly in inducing apoptosis and sensitizing GBM cells to TMZ
(66). However, their clinical
development in GBM has been limited by toxicity, lack of clear
efficacy signals, or logistical challenges in crossing the BBB
(NCT02133183).
In summary, while targeting the PI3K/AKT/mTOR
pathway remains a rational and biologically validated strategy to
circumvent TMZ resistance in GBM, clinical translation has thus far
yielded limited success. Future approaches may require improved
patient stratification, rational drug combinations and more
effective brain-penetrant inhibitors to fully exploit this pathway
therapeutically.
MAPK pathway. The MAPK signaling pathway
plays a central role in mediating resistance to TMZ in GBM,
operating through multiple mechanisms. Among these, the JNK isoform
MAPK8 is upregulated in TMZ-resistant GBM cells (67). This activation suppresses
apoptosis, thereby allowing tumor cells to evade drug-induced
death. Inhibition of MAPK8 restores TMZ sensitivity and increases
apoptotic activity, positioning it as a promising therapeutic
target to overcome resistance.
The ERK arm of the MAPK pathway is activated by
TMZ-induced reactive oxygen species (ROS). Upon activation, it
induces autophagy, which serves as a cytoprotective mechanism that
helps glioma cells survive the cytotoxic effects of TMZ (68). However, this pathway can be
disrupted by resveratrol, which inhibits ROS production and ERK
activation, leading to decreased autophagy and increased
apoptosis.
Another critical arm of the MAPK cascade is the p38
MAPK pathway, which enhances TMZ resistance through the
upregulation of Nrf2, a key regulator of antioxidant responses
(69). By promoting detoxification
and neutralization of ROS, Nrf2 enables glioma cells to survive the
oxidative stress induced by TMZ. Targeting the p38-Nrf2 axis offers
a potential approach to weaken this protective mechanism and
resensitize tumors to treatment.
Beyond its individual components, the MAPK pathway
functions as a convergent hub, integrating upstream signals from
RTKs [(such as EGFR and mesenchymal-epithelial transition (MET)] to
drive proliferation, suppress apoptosis and stabilize resistant
phenotypes. This convergence underscores the resilience of MAPK
signaling in maintaining cell survival under therapeutic pressure.
Furthermore, MAPK is tightly interconnected with other resistance
networks, including DNA repair pathways, autophagy and metabolic
reprogramming, and supports the persistence of GSCs, a
subpopulation that is crucially involved in failure of therapy and
in recurrence.
In addition to these intrinsic mechanisms, adaptive
resistance to TMZ also involves the MAPK pathway, particularly
through feedback activation of ERK and p38(70). p38 MAPK activation is associated
with a mesenchymal-like phenotypic shift in GBM cells,
characterized by enhanced migratory capacity, immune evasion and
resistance to apoptosis. This mesenchymal transition is a hallmark
of aggressive, therapy-resistant tumors. By promoting both stress
adaptation and phenotypic plasticity, the p38 MAPK pathway emerges
as a key enabler of TMZ resistance and tumor progression,
suggesting that targeting this pathway could help counteract
adaptive resistance mechanisms in GBM.
Taken together, these findings illustrate that the
MAPK pathway contributes at multiple levels, including apoptosis
evasion, redox balance, stem cell maintenance and adaptive
plasticity, making it a compelling target for combinatorial
strategies aimed at overcoming TMZ resistance in glioma and
GBM.
Several clinical trials have explored MAPK pathway
inhibitors as a strategy to overcome GBM resistance to TMZ, with
varying degrees of success. One of the early efforts was a Phase II
trial of TLN-4601 (NCT00730262), a Ras-MAPK pathway inhibitor,
tested as monotherapy in recurrent GBM. Unfortunately, the trial
reported no PFS at 6 months and a median overall survival (OS) of
just 130 days, indicating limited efficacy.
Sorafenib, a multi-kinase inhibitor targeting RAF,
was evaluated in multiple trials. In NCT00597493, sorafenib was
combined with TMZ in patients with recurrent GBM, yielding a median
PFS of 6.4 weeks and an OS of 41.5 weeks (71). Another trial, NCT00544817, tested
sorafenib with the standard Stupp protocol in an adjuvant setting,
showing a PFS of 6 months and an OS of 12 months (72). These studies suggested only modest
benefits.
More recent studies have focused on targeting
specific mutations in the MAPK pathway. The combination of
dabrafenib and trametinib, BRAF and MAPK inhibitors respectively,
is under investigation in NCT03919071, a Phase II study involving
patients with BRAF V600E-mutated HGGs, including GBM. Similarly,
NCT03973918 is assessing the efficacy of binimetinib and
encorafenib, another MEK and BRAF inhibitor pair, in BRAF
V600-mutated gliomas. Both trials are ongoing and aim to exploit
genetic vulnerabilities in the MAPK pathway.
LY2228820, a p38 MAPK inhibitor, was studied in
NCT02364206, a Phase II trial combined with the Stupp protocol. The
trial has been completed, but the results remain unpublished. This
agent represents an alternative mechanism of MAPK inhibition,
targeting stress-related kinases involved in TMZ resistance.
Finally, pazopanib, a broad-spectrum multikinase
inhibitor, is being tested in combination with TMZ in the ongoing
Pazoglio trial (NCT02331498). This Phase I/II study is currently
enrolling and aims to determine whether pazopanib can enhance the
effectiveness of standard TMZ therapy in patients with newly
diagnosed GBM.
Collectively, these studies highlight both the
challenges and evolving strategies in targeting the MAPK pathway to
combat TMZ resistance in GBM, with an increasing emphasis on
genetically defined patient subgroups.
Wnt/β-catenin signaling. Dysregulated Wnt
signaling, particularly through the canonical (β-catenin-dependent)
pathway, plays a central role in GBM resistance to TMZ and other
chemotherapies (73).
One of the key mechanisms involves the promotion of
GSC characteristics and maintenance of stemness (74). Wnt ligands such as Wnt3a are highly
expressed in GBM and drive this stem-like phenotype, which is
closely linked to therapeutic resistance. Activation of the
canonical pathway is marked by nuclear accumulation of β-catenin,
often facilitated by proteins like FoxO3a, and this nuclear
translocation is associated with increased resistance to TMZ
(75).
Wnt/β-catenin signaling also promotes
epithelial-mesenchymal transition (EMT), enhancing tumor cell
motility, invasiveness and plasticity, all of which contribute to
chemoresistance (76). Endothelial
cells within the TME can transdifferentiate into mesenchymal-like
cells via a c-Met-mediated axis that activates β-catenin, leading
to the expression of multidrug resistance-associated proteins and
further promoting TMZ resistance. This microenvironment-driven cell
plasticity adds another layer of complexity to therapy resistance
in GBM.
Another important mechanism is the positive
regulation of MGMT by Wnt signaling. (77). The pharmacological inhibition of
Wnt signaling using agents such as salinomycin, celecoxib and
Wnt-C59 has been shown to reduce MGMT expression and restore TMZ
sensitivity in resistant GBM cells. Furthermore, TMZ itself can
activate Wnt/β-catenin signaling through a
PI3K/Akt/GSK-3β-dependent cascade that operates independently of
the ATM/Chk2 DNA damage response pathway, suggesting that Wnt
pathway activation is a downstream effect of TMZ exposure,
potentially fueling resistance (78).
In addition, FERMT3 (also known as kindlin-3), an
integrin-activating adaptor protein, has been implicated in GBM
chemoresistance through its role in promoting integrin-mediated
activation of Wnt/β-catenin signaling (79). Knockdown of FERMT3 results in
decreased β1-integrin activity, reducing Wnt pathway activation and
sensitizing GBM cells to TMZ.
MicroRNAs (miRNAs or miRs) also play a critical role
in modulating Wnt/β-catenin signaling and influencing
chemoresistance in GBM. For example, downregulation of miR-126-3p
and miR-129-5p in TMZ-resistant GBM cells leads to constitutive Wnt
activity and enhanced tumor proliferation and resistance, while
their overexpression inhibits Wnt signaling and restores
chemosensitivity (80,81). By contrast, miR-21 is upregulated
in resistant GBM, promoting Wnt pathway activation and tumor
survival (82).
A regulatory circuit involving miR-125b/miR-20b
sustains Wnt activity in proneural GSCs by suppressing its negative
regulators, further contributing to tumor growth (83). Additionally, IGF-1 has been shown
to upregulate miR-513a-5p via PI3K signaling, which suppresses
NEDD4L, an inhibitor of Wnt/β-catenin signaling, thereby enhancing
GBM progression and TMZ resistance (84). These findings underscore the
complex, multilayered regulation of Wnt signaling by ncRNAs and
highlight additional potential therapeutic targets for overcoming
chemoresistance in GBM.
Collectively, these findings highlight the
multifaceted role of aberrant Wnt/β-catenin signaling in sustaining
GSCs, promoting EMT and invasive behavior, modulating MGMT and
other drug metabolism genes, and enabling microenvironmental
plasticity, all of which converge to promote TMZ resistance in GBM.
Targeting this pathway, therefore, offers a promising strategy to
overcome therapeutic resistance.
Currently, there are no clinical trials specifically
investigating the inhibition of the Wnt/β-catenin pathway in
patients with GBM. However, several Wnt pathway inhibitors,
originally developed and tested for other types of cancer, have
shown promise in preclinical models of GBM. These include LGK974
(WNT974), which has been shown to significantly reduce GBM cell
proliferation and stemness markers in vitro (85); XAV939, a tankyrase inhibitor that
enhances GBM cell radiosensitivity by promoting β-catenin
degradation (86); SEN461, which reduces glioma cell
viability and tumor volume in xenografts (87) and E7386, which is in Phase Ib/II
trials for other solid tumors and appears to eliminate
drug-resistant cancer stem cells in preclinical GBM models
(88).
Despite these encouraging results, translating Wnt
pathway inhibition into effective GBM therapies poses significant
challenges. BBB limits the ability of several systemic drugs,
including Wnt inhibitors, to reach therapeutic concentrations in
the brain. Additionally, the toxicity of Wnt pathway inhibition,
due to its essential role in tissue homeostasis, can lead to
undesirable side effects such as bone fragility. Another important
consideration is the role of Wnt/β-catenin signaling in adult
neurogenesis, raising concerns that pathway inhibition might impact
cognitive function or neuronal repair.
In conclusion, while no clinical trials have yet
targeted the Wnt/β-catenin pathway specifically in GBM, the pathway
remains an attractive target based on compelling preclinical
evidence. Continued efforts to develop brain-penetrant inhibitors
with favorable safety profiles are essential. Addressing the
challenges of BBB permeability and limiting systemic toxicity is
critical for the eventual success of Wnt-targeted therapies in the
treatment of GBM.
Hippo pathway. Dysregulation of the Hippo
pathway leads to overexpression of its key effectors, YAP and TAZ.
These proteins activate downstream target genes that promote cell
survival, proliferation, glioma growth and malignancy. Elevated
levels of TAZ and YAP are associated with poor outcomes in gliomas
(89). TAZ (both protein and mRNA)
is highly expressed in GBM and correlates with reduced survival.
Similarly, high YAP expression is observed across glioma grades and
predicts shorter survival (90).
Importantly, TAZ knockdown impairs tumor formation in mouse models,
underscoring its functional role in glioma pathogenesis.
The Hippo pathway also contributes to multidrug
resistance in GBM through dysregulated YAP/TAZ activity.
Overexpression of TAZ reduces TMZ cytotoxicity by upregulating the
anti-apoptotic protein MCL-1, thereby making glioma cells resistant
to apoptosis (91). In addition,
the YAP-TAZ-TEAD complex promotes TMZ resistance by inducing Hippo
target genes such as CTGF and Cyr61 through TGF-β1-mediated
Smad/ERK signaling (92). CD109,
another regulatory protein, further contributes to chemoresistance
by activating IL-6/STAT3 signaling and enhancing GSC stemness.
CD109 also stimulates the Hippo pathway; its loss leads to reduced
nuclear YAP, decreased STAT3 activity, diminished GSC stemness and
impaired tumorigenicity, highlighting its role in both chemo- and
radio-resistance (93). Finally,
the Hippo pathway interacts with other signaling networks such as
mTOR, Wnt/β-catenin and Notch, further reinforcing therapy
resistance in GBM (92).
As of April 2025, there are no clinical trials
specifically investigating Hippo pathway inhibitors for overcoming
GBM resistance to TMZ. However, considering the role of the Hippo
pathway in glioma tumor progression, chemoresistance and
immunosuppression in GBM, the targeting of the YAP/TAZ
transcriptional axis could be a potential therapeutic strategy.
Previous research has led to the identification of
small-molecule inhibitors aimed at disrupting YAP/TAZ-TEAD
interactions. Verteporfin, initially used in photodynamic therapy,
has demonstrated the ability to inhibit YAP/TAZ-TEAD binding,
induce apoptosis and suppress oncogenic gene expression in
EGFR-amplified/mutant GBM models (94). Clinical data from a Phase 0 trial
showed effective intra-tumoral delivery of liposomal Verteporfin
and reduced YAP/TAZ nuclear localization (NCT04590664) (95).
Other inhibitors under development and primarily
tested in other cancer models include GNE-7883, an allosteric
pan-TEAD inhibitor and ETS-003, a potent compound that blocks
YAP/TAZ-TEAD binding and downstream gene expression (96,97).
IAG933, currently in Phase I clinical trials, shows broad
inhibition across TEAD paralogs and is being assessed for antitumor
activity in solid tumors, including GBM (NCT04857372) (98).
Collectively, these agents represent a new class of
potential therapies designed to overcome YAP/TAZ-mediated
resistance mechanisms and enhance the effectiveness of standard
treatments such as TMZ. Ongoing research and clinical validation
will determine their translational potential in glioma therapy.
Exosomal ncRNAs and TMZ resistance in
glioblastoma. Exosomal ncRNAs [ncRNAs, including miRNAs, long
non-coding RNAs (lncRNAs) and circular RNAs (circRNAs)], are
increasingly being recognized as central mediators of
chemoresistance in GBM (99).
Encapsulated within exosomes and secreted into the TME, these
ncRNAs enable intercellular communication and orchestrate a range
of molecular processes that influence tumor progression, therapy
resistance and recurrence.
miRNAs as regulators of resistance and
sensitivity. miRNAs are small regulatory RNAs that control gene
expression post-transcriptionally and play crucial roles in
apoptosis, drug efflux, DNA repair and oncogenic signaling
pathways. Dysregulation of miRNAs in GBM contributes substantially
to TMZ resistance.
miR-21, one of the most prominent oncomiRs in GBM,
is upregulated in response to TMZ. It promotes resistance by
targeting tumor suppressors such as PTEN, PDCD4 and
RECK, thereby enhancing cell survival and impairing
apoptosis (100).
miR-328 is frequently downregulated in TMZ-resistant
cells and it targets ABCG2, a drug efflux transporter. Its
suppression leads to increased efflux of TMZ, reducing
intracellular drug accumulation and therapeutic efficacy (99).
miR-29c modulates TMZ sensitivity by targeting DNA
methyltransferases DNMT3A and DNMT3B, indirectly downregulating
MGMT, a DNA repair enzyme that confers TMZ resistance. In
GBM, miR-29c is often downregulated, contributing to elevated MGMT
levels and reduced TMZ efficacy (101).
Interestingly, certain miRNAs enhance
chemosensitivity to agents beyond TMZ. Members of the
miR-302-3p/372-3p/373-3p/520-3p family increase susceptibility to
tyrosine kinase inhibitors (TKIs) such as sunitinib and axitinib by
downregulating the PI3K/AKT and MAPK signaling pathways (102).
lncRNAs as multifaceted mediators of
resistance. lncRNAs contribute to drug resistance through a
range of mechanisms, including transcriptional regulation,
chromatin remodeling, miRNA sponging and modulation of signaling
cascades. H19, SBF2-AS1, MALAT1 and ADAMTS9-AS2 are overexpressed
in GBM and associated with TMZ resistance. These lncRNAs promote
EMT, enhance DNA repair and inhibit apoptotic signaling (99).
Specifically, H19 activates Wnt/β-catenin
signaling to maintain stemness and promote EMT (103) and SBF2-AS1 sponges miR-151a-3p,
leading to upregulation of XRCC4, a DNA repair factor (104). MALAT1 promotes TMZ
resistance in glioma through multiple mechanisms. Li et al
(105) demonstrated that
transfection of MALAT1 upregulates EMT and multidrug resistance
genes, such as ZEB1 and MDR1, respectively, both in vitro
and in vivo, collectively reducing glioma cell sensitivity
to TMZ. In addition, other studies have shown that MALAT1 promotes
chemoresistance and cell proliferation by suppressing miR-203,
which targets thymidylate synthase, and by sponging miR-101,
thereby blocking its inhibition of autophagy in glioma cells
(106). ADAMTS9-AS2 promotes TMZ
resistance by upregulating the FUS/MDM2 ubiquitination axis,
specifically by enhancing FUS expression, which increases
MDM2-mediated ubiquitination processes and leads to decreased
apoptosis and increased survival of GBM cells under TMZ treatment
(107).
By contrast, tumor-suppressive lncRNAs, such as
CASC2, can augment the sensitivity of malignant cells to TMZ by
enhancing its ability to suppress cell proliferation through the
regulation of miR-181a, which, in turn, upregulates PTEN expression
and downregulates phosphorylated AKT levels (108).
circRNAs as stable sponges shaping
resistance. circRNAs are covalently closed RNA loops with high
stability that function predominantly by sponging miRNAs or
modulating protein interactions. They are emerging as key
regulators of drug resistance in GBM.
circNFIX is overexpressed in glioma and promotes
tumor growth and resistance to TMZ by sponging miR-34a-5p, which
normally suppresses NOTCH1, thereby activating the Notch signaling
pathway. Downregulation of circNFIX or overexpression of miR-34a-5p
reduced glioma cell proliferation, migration and survival both
in vitro and in vivo, highlighting the role of
circ-NFIX in glioma progression (109). Additionally, exosomal circ-NFIX
can transfer TMZ resistance from resistant glioma cells to
sensitive glioma cells by promoting migration and invasion while
inhibiting apoptosis, and suppression of circ-NFIX restores TMZ
sensitivity by upregulating miR-132(110).
circHIPK3 is upregulated in TMZ-resistant glioma and
promotes tumor progression, metastasis and drug resistance by
acting as a competing endogenous RNA (ceRNA) for miRNAs. For
example, circHIPK3 sponges miR-421 to upregulate the oncogene ZIC5,
whose expression drives glioma cell proliferation, migration and
drug resistance; circHIPK3 knockdown reduces ZIC5 levels, inhibits
tumor growth and enhances TMZ sensitivity (111). In a parallel pathway, circHIPK3
sponges miR-524-5p to increase KIF2A expression, thereby activating
the oncogenic PI3K/AKT pathway and contributing to proliferation,
metastasis and resistance to TMZ (112).
circ_0043949 is overexpressed in TMZ-resistant GBM
cells and exosomes, where it contributes to chemoresistance by
functioning as a ceRNA. It sponges multiple miRNAs, including
miR-876-3p, miR-7161-3p and miR-6783-3p, thereby upregulating
targets like integrin α1 (ITGA1) and suppressing miR-140-mediated
inhibition of EMT (113). These
actions collectively enhance the proliferation, invasion and
survival of GBM cells, and exosomal circ_0043949 can transfer
resistance traits to other cells, as shown in xenograft models.
Among the additional circRNAs upregulated in
recurrent GBM tissues and TMZ-resistant cell lines are circASAP1
and circ_0000936. circASAP1 confers resistance by sponging
miR-502-5p, which leads to NRAS upregulation and activation of the
NRAS/MEK1/ERK1/2 signaling pathway (114). Of note, its depletion restores
TMZ sensitivity in resistant xenograft models. Likewise,
circ_0000936 promotes resistance by suppressing miR-1294, and its
downregulation sensitizes glioma cells to TMZ (115). These findings underscore both
circRNAs as potential therapeutic targets for overcoming TMZ
resistance in GBM.
Exosomal ncRNAs play pivotal roles in mediating
chemoresistance in GBM by regulating apoptosis, drug efflux, DNA
repair and key signaling pathways. Their dual function as
biomarkers and active effectors makes them compelling targets for
therapeutic intervention. Strategies aimed at inhibiting oncogenic
ncRNAs or restoring tumor-suppressive ones, either directly or by
blocking their exosomal transfer, offer promising avenues to
overcome TMZ resistance and improve patient outcomes.
Currently, no clinical trials specifically target
exosomal ncRNAs to counteract TMZ resistance in GBM. However,
preclinical studies, including those aforementioned, have
identified several exosomal ncRNAs involved in resistance
mechanisms, highlighting potential targets for future therapies.
One notable study employed CRISPR-Cas9 screening to identify
resistance-associated genes in the mesenchymal subtype of GBM. It
then used an exosome-based system to co-deliver siRNAs targeting
genes such as RASGRP1 and VPS28, the small-molecule
inhibitor EPIC-0412 and TMZ (116). This combination significantly
reduced tumor burden in vivo, though further validation is
needed for clinical translation. Future clinical efforts should
focus on developing exosome-based delivery systems for targeted
therapy, using circulating exosomal ncRNAs as biomarkers to predict
TMZ resistance, and combining exosomal ncRNA-targeted strategies
with standard treatments to overcome resistance
3. Resistance to radiotherapy
Radiotherapy for GBM has seen significant
technological advancements, evolving from 2D whole-brain
radiotherapy to 3D conformal radiotherapy, intensity-modulated
radiation therapy and volumetric arc therapy (117,118). These modern techniques offer
improved targeting and reduced damage to healthy tissues.
Stereotactic and hypofractionated approaches have also been
introduced, offering more precise delivery and shorter treatment
durations, respectively (119).
Despite these improvements, clinical outcomes remain poor due to
intrinsic and acquired resistance to radiation in patients with
GBM, especially in recurrent cases. Overall, technological progress
in radiotherapy has not translated into substantial survival
benefits for patients with GBM.
The Stupp protocol, which consists of maximal safe
surgical resection followed by concurrent radiation therapy and TMZ
chemotherapy, and subsequently adjuvant TMZ, represents a
multimodal treatment approach. This complexity makes it challenging
to isolate the specific contribution of radiation resistance in
clinical settings. Nonetheless, radiotherapy resistance in GBM is
broadly attributed to three key factors: Hypoxic niches,
dysregulated DNA damage response (DDR) and GSCs, each of which has
been extensively discussed in earlier sections.
A recent study using patient-derived xenograft (PDX)
models of GBM have further substantiated these resistance
mechanisms. A notable finding is the existence of
radiation-tolerant persister (RTP) cells, a subpopulation that
survives radiotherapy through enhanced DNA repair capabilities and
sustained GSC-like properties (120). These RTP cells exhibit elevated
activity in both homologous recombination (HR) and non-homologous
end joining pathways, rendering them highly resilient to
radiation-induced DNA breaks. Mechanistically, constitutive
activation of NF-κB signaling in RTP cells leads to increased
expression of the transcription factor YY1, which suppresses
miR-103a. This suppression, in turn, upregulates FGF2 and XRCC3,
promoting DNA repair and maintenance of stemness. Of note,
restoring miR-103a levels using transferrin-functionalized
nanoparticles has been shown to significantly enhance
radiosensitivity in PDX models, offering a promising therapeutic
strategy to overcome radio-resistance and prevent recurrence.
Fang et al (121) have demonstrated that
DNA-dependent protein kinase (DNA-PK) plays a critical role in
maintaining GSC properties by stabilizing the transcription factor
SOX2, a key regulator of stemness and therapeutic resistance in
GBM. Specifically, DNA-PK phosphorylates SOX2 at serine 251,
preventing its ubiquitination and subsequent degradation, thereby
sustaining its function. Pharmacological inhibition of DNA-PK using
NU7441 destabilizes SOX2, promotes GSC differentiation, enhances
radiosensitivity and leads to tumor regression and improved
survival in glioblastoma-bearing mice. These findings position
DNA-PK as a compelling therapeutic target for overcoming
radio-resistance in GBM.
In parallel, other studies have shown that repeated
radiation exposure induces metabolic and transcriptional
adaptations, including mitochondrial biogenesis and enhanced
oxidative stress tolerance. Quiescent CD133+ GSCs may be
reactivated following radiation, upregulating key self-renewal
genes such as BMI1 and SOX2, which contribute to tumor repopulation
(122). Additionally, radiation
promotes IGF1 secretion, which induces N-cadherin-mediated
cell-cell adhesion, enhancing GSC survival by suppressing
differentiation and protecting against apoptosis via Clusterin
secretion. A previous study showed that this IGF1-N-cadherin axis
drives the development of a radioresistant GSC phenotype marked by
reduced proliferation, increased stemness and membrane-localized
β-catenin accumulation, which suppresses Wnt signaling and
differentiation (123).
Crucially, this adaptive resistance can be reversed by
CRISPR-mediated knockout of N-cadherin or pharmacologic IGF1R
inhibition with picropodophyllin, underscoring its therapeutic
relevance.
A particularly comprehensive investigation created
radiation-selected (RTS) PDX models by serially irradiating
treatment-naïve GBMs (124).
These RTS models closely mimic clinical recurrence and revealed
significant alterations in gene expression and kinase activity.
Interestingly, specific lncRNAs were identified as regulators of
DNA repair and other resistance-associated pathways. Kinomic
profiling of RTS tumors uncovered elevated activity of several
kinases, including JAK, fibroblast growth factor receptor and
Ephrin kinases. Fortunately, small-molecule inhibitors targeting
these kinases were able to re-sensitize RTS tumors to radiation in
preclinical settings (124).
Collectively, these findings illustrate that GBM
resistance to radiation is a multifactorial and adaptive process,
involving intricate crosstalk between DNA repair, stemness,
metabolism, signaling and tumor microenvironmental cues. Targeting
these mechanisms, especially those unique to resistant
subpopulations, may offer novel therapeutic opportunities to
enhance the efficacy of radiation therapy in GBM.
In this context, radiosensitizers have emerged as a
promising strategy to improve the effectiveness of radiotherapy
without increasing radiation doses, thus reducing collateral damage
to healthy tissues. Radiosensitizers function by interfering with
cellular mechanisms that normally protect tumor cells from
radiation-induced damage. These mechanisms include inhibition of
DNA repair enzymes [such as poly ADP-ribose polymerase (PARP), ATM
and ATR] (125), disruption of
redox homeostasis via thiol depletion or pro-oxidant activity,
suppression of stemness pathways and mimicking the electrophilic
activity of oxygen to overcome hypoxia-associated resistance.
Several chemotherapeutic agents and targeted inhibitors have
demonstrated radio-sensitizing effects in preclinical GBM models
and early clinical studies including: i) Gemcitabine, which
disrupts DNA replication by incorporating into the DNA strand
(126); Talazoparib and veliparib
(PARP inhibitors) that impair single-strand break repair (127); iii) Gefitinib (an EGFR inhibitor)
that enhances radiation-induced cytotoxicity (128); iv) histone deacetylase inhibitors
(quisinostat, valproate and vorinostat) to affect chromatin
remodeling and DNA repair (129,130); v) Chloroquine (an autophagy
inhibitor), to enhance radiation-induced apoptosis (131); vi) Adavosertib (a WEE1 inhibitor)
that disrupts cell cycle checkpoints (125); and vii) Papaverine (a
mitochondrial complex I inhibitor) to improve tumor oxygenation and
radiosensitivity in hypoxic tumors (132).
Despite promising preclinical data, translation to
clinical success has been limited. Several radiosensitizers have
progressed to Phase I/II clinical trials, including ascorbate,
sulfasalazine, trans-sodium crocetinate, NVX-108 and others;
however, most have failed to show significant improvements in
progression-free or OS in patients with GBM (133).
Among radiosensitizer classes, oxygen mimetics and
metabolic modulators hold special promise for alleviating tumor
hypoxia, a known driver of radio-resistance. Compounds containing
nitro groups, hydrogen peroxide and mitochondrial inhibitors, such
as papaverine have shown potential in preclinical models but face
challenges in effective tumor delivery due to poor vascularization
(132).
Thus, while radiosensitizers remain a viable
therapeutic adjunct, there is an urgent need to identify biomarkers
predicting radiosensitizer response, improve drug delivery to
hypoxic tumor regions, develop combinatorial strategies targeting
both DNA repair and tumor metabolism, and validate novel agents in
robust GBM models, including PDX and radiation-selected
systems.
By addressing these challenges, radiosensitizers may
fulfill their therapeutic potential as key enhancers of
radiotherapy outcomes in glioblastoma.
4. Resistance to targeted therapy
Glioblastomas are classified based on genetic
features rather than just histology. GBMs are categorized into
three subgroups based on differential gene expression: Proneural,
classical and mesenchymal, each of which is driven by distinct
molecular alterations (134).
There has been increasing interest in developing targeted drugs to
address genomic abnormalities specific to subtypes, such as EGFR
amplification in the classical subtype, NF-κB hyperactivation in
the mesenchymal subtype and PDGFR mutation in the proneural
subtype.
Glioma cell lines and primary GBM tissues exhibit
upregulated expression of PDGF and PDGFRs, particularly in the
proneural subtype, characterized by a high rate (35%) of focal
PDGFRA amplification. PDGFRβ expression is specific to GBM
endothelial cells, being absent in normal brain vessels,
highlighting its potential for GBM-specific targeted therapy
(135).
Several anti-PDGFR agents, including olaratumab (an
anti-PDGFRα antibody with significant tumor growth inhibition
activity in xenograft models, but minimal clinical efficacy),
CP-673,451 (a small molecule targeting PDGFRα and PDGFRβ to induce
terminal differentiation of GBM cells) (136), crenolanib (CP-868,596, exhibiting
brain penetration and in vivo PDGFR phosphorylation
inhibition) (137), and
avapritinib and ripretinib (targeting PDGFRα mutations, with
avapritinib showing improved CNS penetrance) (138), have demonstrated promise in
preclinical GBM models, but their clinical efficacy remains
uncertain, with varying degrees of success and limitations observed
in patient trials.
BEV, a monoclonal antibody targeting VEGF isoforms,
has been extensively studied in regard to GBM. After promising
results from Phase II trials, the FDA approved BEV in 2009 for
recurrent GBM. In the BRAIN trial, BEV achieved a 6-month PFS rate
of 42% as a single agent and 50% when combined with irinotecan,
though OS was similar between groups. Despite exceeding benchmarks,
trials for BEV in combination therapies for primary GBM have shown
disappointing results, partly due to pseudo-responses that mimic
tumor shrinkage on imaging without true clinical benefit (139,140).
EGFR amplification is prevalent in 57% of primary
GBM cases, predominantly in the classical subtype and may involve
extracellular mutations, constitutively active EGFRvIII or
extrachromosomal DNA, which exacerbates intra-tumoral heterogeneity
and complicates drug targeting (141,142).
Although anti-EGFR therapies (such as dacomitinib,
gefitinib and osimertinib) have been explored, their clinical
efficacy is limited by tumor heterogeneity, resistance mechanisms
and poor BBB penetrance (143).
Resistance arises from compensatory signaling activation (such as
PI3K and MET), and mutations like C797S and G724S that hinder drug
binding (144). Emerging
approaches, such as antibody-drug conjugates (for example,
ABT-414), show promise but face challenges, including loss of EGFR
amplification in resistant clones and inefficient BBB penetration
(145).
Combination therapies targeting both EGFR and
downstream effectors (for example, DDR1 and KRAS-driven MAPK
activation) have demonstrated synergistic effects in overcoming
resistance. However, improved specificity of EGFR inhibitors for
GBM-associated mutations and combinatorial strategies are crucial
for advancing GBM treatment.
Efforts to inhibit the PI3K/AKT/mTOR pathway using
drugs such as GDC-0068 and GDC-0084 have shown potential, but
resistance often arises (146-148).
Studies focusing on the BRAF V600E mutation in a small fraction of
gliomas have demonstrated that second- and third-generation BRAF
inhibitors, combined with MEK inhibitors, may provide some benefit.
However, the limited presence of BRAF mutations in gliomas reduces
the overall impact of these findings.
In addition to the aforementioned small kinase
inhibitors, other inhibitors targeting MET, FGFR, vascular
endothelial growth factor receptor, MEK and PKCβ have been
evaluated in clinical trials for GBM (21,149). However, these agents demonstrated
limited efficacy, likely due to the acquisition of resistance.
Resistance mechanisms to kinase inhibitors in GBM
can be broadly categorized into four groups: Mutations,
coactivation of multiple RTKs, adaptation and alternative routes
(bypass).
Mutations. Patients with GBM often possess
mutations in the extracellular domain of kinases, unlike other
types of cancer. EGFR mutations in GBM primarily occur in the
extracellular domain, and this difference in mutation location may
contribute to primary resistance (150).
Coactivation of multiple RTKs. GBMs often
have multiple activated RTKs within a tumor, such as EGFR, ERBB3,
PDGFR and MET (151), granting
them resistance to single-agent inhibition.
Adaptation. Tumors can adapt to treatments
over time. As discussed previously, GBM cells can suppress EGFRvIII
protein expression in response to erlotinib treatment (152).
Alternative routes (bypass). Tumors can
activate alternative signaling pathways to bypass the inhibited
kinase. Examples include NF-κB pathway activation in response to
combined EGFR and MET inhibition (153) and IGF-1R activation mediates
resistance to EGFR inhibitors through activation of PI3K and AKT
signaling (154).
IDH mutations are highly prevalent in >80% of
WHO grade II/III gliomas, frequently occur in secondary
glioblastomas (73% of cases), and are rare in primary glioblastomas
(3.7%) (155). IDH-mutant GBM
cells show synthetic lethality with PARP inhibitors due to impaired
HR repair pathways. Several PARP inhibitors are under clinical
investigation for GBM treatment.
Olaparib can cross the BBB but did not meet
response-based thresholds in patients with IDH-mutant glioma
(156). Niraparib was shown to
exhibit improved tumor exposure and sustainability than olaparib,
with ongoing studies evaluating its combination with radiotherapy.
By contrast, IDH1/2-wildtype gliomas show limited tumor-suppressing
effects with PARP inhibitors due to functional BRCA1/2 recruitment
to the HR repair pathway.
IDH inhibitors such as ivosidenib and vorasidenib
have been developed to block the production of the oncometabolite
D-2-hydroxyglutarate (D-2-HG). D-2-HG drives gliomagenesis via
epigenetic alterations, including hypermethylation that silences
tumor suppressor genes and maintains stemness. While these
IDH1/IDH2 inhibitors show promise in early clinical trials
(157), resistance mechanisms
remain an active area of investigation.
In conclusion, targeted therapies have shown
success in other types of cancer, but not in GBM. New studies and
clinical trials should address and solve this problem using
preclinical models, improving BBB penetration and assessing
combination therapies.
6. Resistance to immunotherapy
Immune checkpoint inhibitors have shown success in
other types of cancer but have failed to show efficacy in GBM.
Nivolumab (anti-PD-1 antibody) plus radiotherapy
failed to improve OS compared with the standard treatment of
radiotherapy plus TMZ (158).
Neoadjuvant PD-1 blockade with pembrolizumab, though it induced T
cell-cell and cDC1 activation, failed to counteract the
immunosuppressive tumor-associated macrophages (TAMs) in recurrent
GBM (159). Both intrinsic and
acquired resistance mechanisms contribute to the absence of
efficacy of immunotherapy in GBM, as described below.
Immunosuppressive TME. GBM creates an
environment that supports immune evasion through TAMs, GSCs and
myeloid-derived suppressor cells (160,161). These suppress cytotoxic T cells
and promote immune evasion by upregulating immunosuppressive
proteins like CHI3L1(162).
Furthermore, the loss of S1P1 receptor on T-cell surface promotes
T-cell dysfunction due to the sequestering of T-cells within the
bone marrow (163).
Tumor heterogeneity. Tumor heterogeneity,
discussed in the previous sections, contributes to immunotherapy
resistance in GBM at both the molecular and cellular levels,
allowing a range of subclonal populations with distinct genetic
mutations and developmental states to coexist and evolve within the
same tumor (43,164). This diversity enables
therapy-resistant clones to survive treatment, adapt and repopulate
the tumor, often with new genetic alterations, ultimately driving
tumor recurrence and rendering monotherapies and some combination
therapies ineffective.
To improve outcomes, immune-based therapies should
be combined with approaches targeting the immunosuppressive
mechanisms of glioblastoma. For example, co-targeting chemokine
receptors such as CXCR4, often overexpressed in GBMs, alongside
immune-checkpoint inhibitors has shown potential in preclinical
studies (165). Rational
combinations of therapies addressing tumor-immune compositional
changes and leveraging immune-stimulatory mechanisms may maximize
therapeutic success.
7. Conclusion and future directions
Despite decades of research, GBM remains among the
most refractory of cancers. The current treatment paradigm is
consistently undermined by a constellation of resistance
mechanisms, ranging from poor drug penetration across the BBB and
intra-tumoral heterogeneity to robust compensatory signaling and
immune evasion.
Key mechanisms of drug resistance in GBM include
limited drug penetration due to the BBB and its variable
permeability, as well as active drug efflux by ATP-binding cassette
transporters. The extensive heterogeneity of GBM, including
molecular, genetic, cellular and spatial heterogeneity, gives rise
to therapy-resistant subpopulations, notably GSCs. The TME,
characterized by hypoxic niches and immunosuppressive myeloid
cells, further facilitates resistance to both targeted and immune
therapies. In addition, resistance is driven by intrinsic factors
such as DNA repair activity (for example, MGMT expression or MMR
defects), mutations in critical signaling pathways (such as p53, Rb
or RTK/Ras/PI3K) and metabolic rewiring. GBM cells also exhibit
antioxidant upregulation and dysregulated alternative splicing, all
of which contribute to a formidable capacity to evade therapy
(Fig. 1).
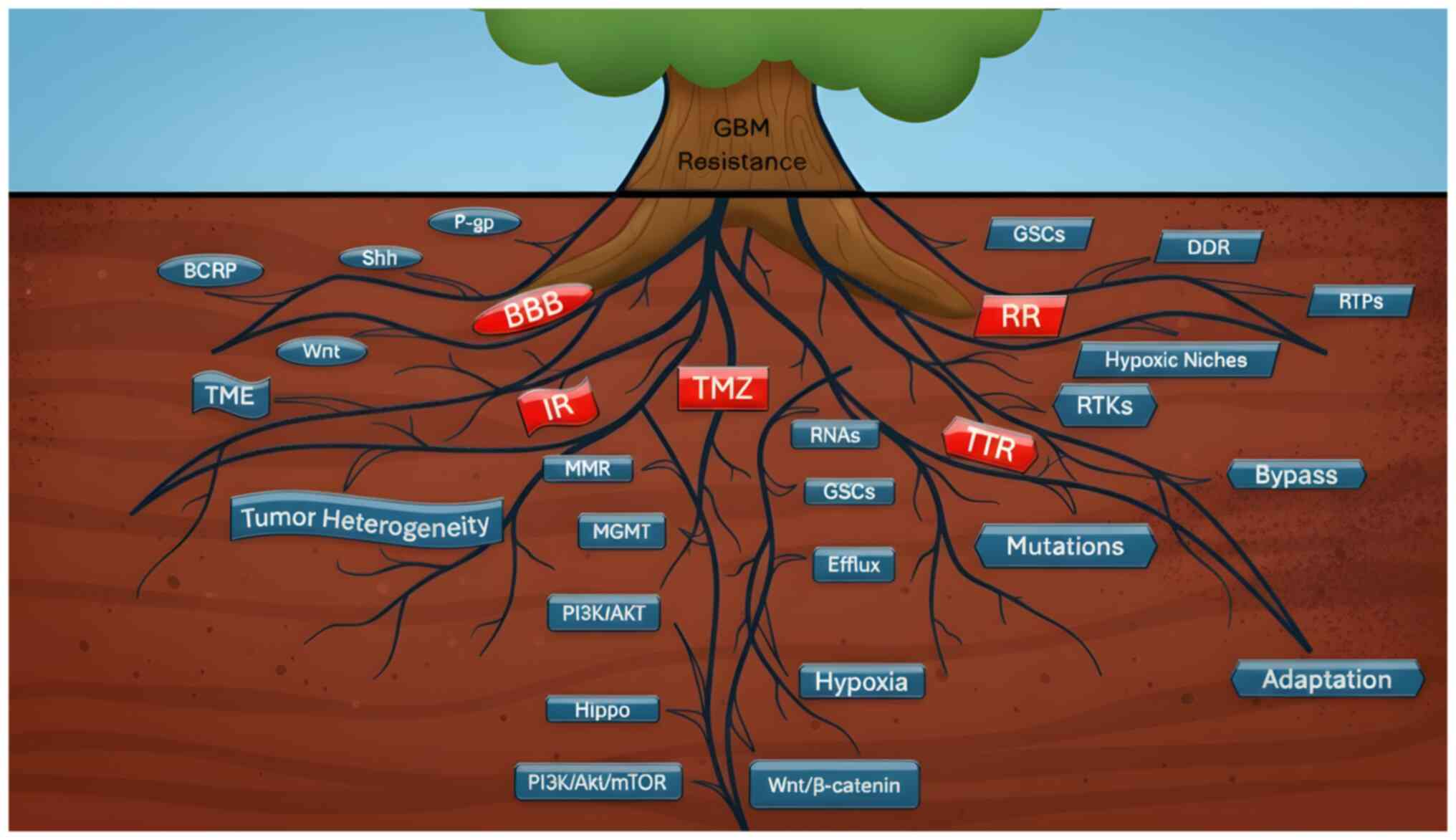 | Figure 1Root causes of therapeutic resistance
in GBM. Schematic representation of the interconnected mechanisms
that sustain GBM resistance, depicted as a tree where the visible
trunk represents clinical resistance and the hidden root system
illustrates its molecular and cellular drivers. Major resistance
categories are highlighted in red as BBB, IR, TMZ resistance, RR
and TTR. BBB serves as a physical barrier co-opted by the tumor to
reduce drug penetration. Overexpression of efflux transporters such
as P-gp and BCRP in tumor endothelial cells decreases intracellular
drug concentrations, while dysregulation of Wnt and Shh pathways
further limits permeability. IR is reinforced by the highly
immunosuppressive TME, which includes TAMs, GSCs and MDSCs that
suppress cytotoxic T-cell activity. Molecular heterogeneity further
sustains therapy-resistant clones. TMZ resistance is driven by MGMT
upregulation, DNA MMR defects and activation of PI3K/Akt, Hippo and
Wnt/β-catenin pathways. Additional contributors include efflux
transporters, GSCs, a hypoxic TME and exosomal RNAs. TTR reflects
the failure of precision approaches despite subtype-specific
alterations (EGFR in classical, PDGFR in proneural, NF-κB in
mesenchymal GBM). Resistance mechanisms include kinase domain
mutations, RTK coactivation (EGFR, ERBB3, PDGFR, MEK), adaptive
suppression of EGFRvIII and bypass signaling via NF-κB or
IGF1R-PI3K/AKT activation. RR arises from hypoxia, enhanced DDR and
GSC-mediated repair capacity. RTP cells survive through homologous
recombination and non-homologous end joining. Together, these
mechanisms form a deeply interconnected ‘root system’ that allows
GBM to withstand chemotherapy, radiotherapy, targeted therapy and
immunotherapy. This conceptual framework emphasizes that resistance
arises not from a single pathway but from a dynamic, adaptive
network, underscoring the need for rational combination regimens,
precision therapies, and innovative delivery strategies to overcome
therapeutic failure in GBM. GBM, glioblastoma multiforme; BBB,
blood-brain barrier; IR, immunotherapy resistance; TMZ,
temozolomide; RR, radiotherapy resistance; TTR, targeted therapy
resistance; TME, tumor microenvironment; TAM, tumor-associated
macrophage; GSC, glioma stem-like cell; MDSC, myeloid-derived
suppressor cell; MGMT, O6-methylguanine-DNA
methyltransferase; EGFR, epidermal growth factor; MMR, mismatch
repair; PDGFR, platelet-derived growth factor; RTL, receptor
tyrosine kinase; DDR, DNA damage response; RTP, radiation-tolerant
persister; P-gp, P-glycoprotein; BCRP, breast-cancer-resistance
protein; Shh, sonic hedgehog; MEK, mitogen-activated protein
kinase. |
The present review emphasizes that overcoming such
multifaceted resistance requires more than additive or sequential
strategies; it demands mechanistically informed, rational
combinations that account for the adaptive plasticity of GBM. For
example, combining PI3K/mTOR inhibitors with agents targeting
downstream anti-apoptotic proteins (such as Bcl-2 or Mcl-1) may
disrupt resistance circuits more effectively than monotherapy.
Similarly, dual inhibition of the MAPK and autophagy could thwart
adaptive survival mechanisms.
Rather than relying on generic combination
therapies, future strategies should emphasize biologically
justified, context-specific approaches tailored to the molecular
and cellular landscape of each patient's tumor. Progress will also
depend on bridging the translational gap between preclinical models
and clinical endpoints, particularly in evaluating therapies
targeting rare subpopulations or modulating the TME.
In summary, advancing GBM therapy demands a
paradigm shift, from reactive management of resistance to
proactive, mechanistically anchored intervention, with a relentless
focus on therapeutic precision and clinical feasibility.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
YKD drafted and wrote the manuscript. RP provided
critical review, edits, intellectual inputs, and overall guidance.
Both authors read and approved the final version of the manuscript.
Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
National Cancer Institute: Cancer Stat
Facts: Brain and Other Nervous System Cancer. https://seer.cancer.gov/statfacts/html/brain.html.
|
|
2
|
National Center for Health Statistics:
Declines in cancer death rates among youth: United States,
2001-2021. NCHS Data Brief No. 484, November 2023. https://www.cdc.gov/nchs/products/databriefs/db484.htm.
|
|
3
|
Schaff LR and Mellinghoff IK: Glioblastoma
and other primary brain malignancies in adults: A review. JAMA.
329:574–587. 2023.PubMed/NCBI View Article : Google Scholar
|
|
4
|
National Brain Tumor Society: Grade
4-Glioblastoma (GBM). https://braintumor.org/events/glioblastoma-awareness-day/about-glioblastoma/.
|
|
5
|
Butt AM, Jones HC and Abbott NJ:
Electrical resistance across the blood-brain barrier in
anaesthetized rats: A developmental study. J Physiol. 429:47–62.
1990.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Brown LS, Foster CG, Courtney JM, King NE,
Howells DW and Sutherland BA: Pericytes and neurovascular function
in the healthy and diseased brain. Front Cell Neurosci.
13(282)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Ou A, Yung WKA and Majd N: Molecular
mechanisms of treatment resistance in glioblastoma. Int J Mol Sci.
22(351)2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Sarkaria JN, Hu LS, Parney IF, Pafundi DH,
Brinkmann DH, Laack NN, Giannini C, Burns TC, Kizilbash SH, Laramy
JK, et al: Is the blood-brain barrier really disrupted in all
glioblastomas? A critical assessment of existing clinical data.
Neuro Oncol. 20:184–191. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Bendell JC, Domchek SM, Burstein HJ,
Harris L, Younger J, Kuter I, Bunnell C, Rue M, Gelman R and Winer
E: Central nervous system metastases in women who receive
trastuzumab-based therapy for metastatic breast carcinoma. Cancer.
97:2972–2977. 2003.PubMed/NCBI View Article : Google Scholar
|
|
10
|
McCord M, Mukouyama YS, Gilbert MR and
Jackson S: Targeting WNT signaling for multifaceted glioblastoma
therapy. Front Cell Neurosci. 11(318)2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Alvarez JI, Dodelet-Devillers A, Kebir H,
Ifergan I, Fabre PJ, Terouz S, Sabbagh M, Wosik K, Bourbonnière L,
Bernard M, et al: The Hedgehog pathway promotes blood-brain barrier
integrity and CNS immune quiescence. Science. 334:1727–1731.
2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Mo F, Pellerino A, Soffietti R and Rudà R:
Blood-brain barrier in brain tumors: Biology and clinical
relevance. Int J Mol Sci. 22(12654)2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ommaya AK: Subcutaneous reservoir and pump
for sterile access to ventricular cerebrospinal fluid. Lancet.
2:983–984. 1963.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Jabbour E, O'Brien S, Kantarjian H,
Garcia-Manero G, Ferrajoli A, Ravandi F, Cabanillas M and Thomas
DA: Neurologic complications associated with intrathecal liposomal
cytarabine given prophylactically in combination with high-dose
methotrexate and cytarabine to patients with acute lymphocytic
leukemia. Blood. 109:3214–3218. 2007.
|
|
15
|
Siegal T, Horowitz A and Gabizon A:
Doxorubicin encapsulated in sterically stabilized liposomes for the
treatment of a brain tumor model: Biodistribution and therapeutic
efficacy. J Neurosurg. 83:1029–1037. 1995.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Pinzón-Daza M, Garzón R, Couraud P, Romero
Ia, Weksler B, Ghigo D, Bosia A and Riganti C: The association of
statins plus LDL receptor-targeted Liposome-encapsulated
doxorubicin increases in vitro drug delivery across blood-brain
barrier cells. Br J Pharmacol. 167:1431–1447. 2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Durmus S, Sparidans RW, Wagenaar E,
Beijnen JH and Schinkel AH: Oral availability and brain penetration
of the B-RAFV600E inhibitor vemurafenib can be enhanced by the
P-GLYCOprotein (ABCB1) and breast cancer resistance protein (ABCG2)
inhibitor elacridar. Mol Pharm. 9:3236–3245. 2012.PubMed/NCBI View Article : Google Scholar
|
|
18
|
McDannold N, Zhang Y, Supko JG, Power C,
Sun T, Peng C, Vykhodtseva N, Golby AJ and Reardon DA: Acoustic
feedback enables safe and reliable carboplatin delivery across the
blood-brain barrier with a clinical focused ultrasound system and
improves survival in a rat glioma model. Theranostics. 9:6284–6299.
2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Portnow J, Synold TW, Badie B, Tirughana
R, Lacey SF, D'Apuzzo M, Metz MZ, Najbauer J, Bedell V, Vo T, et
al: Neural stem Cell-based anticancer gene therapy: A
First-in-Human study in recurrent High-grade glioma patients. Clin
Cancer Res. 23:2951–2960. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: Radiotherapy plus concomitant and adjuvant temozolomide
for glioblastoma. N Engl J Med. 352:987–996. 2005.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Shen Y, Thng DKH, Wong ALA and Toh TB:
Mechanistic insights and the clinical prospects of targeted
therapies for glioblastoma: A comprehensive review. Exp Hematol
Oncol. 13(40)2024.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Fu D, Calvo JA and Samson LD: Balancing
repair and tolerance of DNA damage caused by alkylating agents. Nat
Rev Cancer. 12:104–120. 2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Roos WP, Batista LF, Naumann SC, Wick W,
Weller M, Menck CF and Kaina B: Apoptosis in malignant glioma cells
triggered by the temozolomide-induced DNA lesion O6-methylguanine.
Oncogene. 26:186–197. 2007.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Kitange GJ, Carlson BL, Schroeder MA,
Grogan PT, Lamont JD, Decker PA, Wu W, James CD and Sarkaria JN:
Induction of MGMT expression is associated with temozolomide
resistance in glioblastoma xenografts. Neuro Oncol. 11:281–291.
2009.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Hegi ME, Diserens AC, Gorlia T, Hamou MF,
de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani
L, et al: MGMT gene silencing and benefit from temozolomide in
glioblastoma. N Engl J Med. 352:997–1003. 2005.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Sarkaria JN, Ballman KV, Kizilbash SH,
Sulman EP, Giannini C, Mashru SH, Piccioni DE, Friday BEB, Dixon
JG, Kabat B, et al: Randomized phase II/III trial of veliparib or
placebo in combination with adjuvant temozolomide in newly
diagnosed glioblastoma (GBM) patients with MGMT promoter
hypermethylation (Alliance A071102). J Clin Oncol. 40 (Suppl
16)(S2001)2022.
|
|
27
|
Stupp R, Hegi ME, Gorlia T, Erridge SC,
Perry J, Hong YK, Aldape KD, Lhermitte B, Pietsch T, Grujicic D, et
al: Cilengitide combined with standard treatment for patients with
newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC
EORTC 26071-22072 study): A multicentre, randomised, open-label,
phase 3 trial. Lancet Oncol. 15:1100–1108. 2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Balana C, De Las Penas R, Sepúlveda JM,
Gil-Gil MJ, Luque R, Gallego O, Carrato C, Sanz C, Reynes G,
Herrero A, et al: Bevacizumab and temozolomide versus temozolomide
alone as neoadjuvant treatment in unresected glioblastoma: The
GENOM 009 randomized phase II trial. J Neurooncol. 127:569–579.
2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Shinsato Y, Furukawa T, Yunoue S, Yonezawa
H, Minami K, Nishizawa Y, Ikeda R, Kawahara K, Yamamoto M, Hirano
H, et al: Reduction of MLH1 and PMS2 confers temozolomide
resistance and is associated with recurrence of glioblastoma.
Oncotarget. 4:2261–2270. 2013.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Cahill DP, Levine KK, Betensky RA, Codd
PJ, Romany CA, Reavie LB, Batchelor TT, Futreal PA, Stratton MR,
Curry WT, et al: Loss of the mismatch repair protein MSH6 in human
glioblastomas is associated with tumor progression during
temozolomide treatment. Clin Cancer Res. 13:2038–2045.
2007.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Stark AM, Doukas A, Hugo HH, Hedderich J,
Hattermann K, Maximilian Mehdorn H and Held-Feindt J: Expression of
DNA mismatch repair proteins MLH1, MSH2, and MSH6 in recurrent
glioblastoma. Neurol Res. 37:95–105. 2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Hunter C, Smith R, Cahill DP, Stephens P,
Stevens C, Teague J, Greenman C, Edkins S, Bignell G, Davies H, et
al: A hypermutation phenotype and somatic MSH6 mutations in
recurrent human malignant gliomas after alkylator chemotherapy.
Cancer Res. 66:3987–3991. 2006.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Leelatian N, Hong CS and Bindra RS: The
role of mismatch repair in glioblastoma multiforme treatment
response and resistance. Neurosurg Clin N Am. 32:171–180.
2021.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Brastianos PK, Kim AE, Giobbie-Hurder A,
Lee EQ, Lin NU, Overmoyer B, Wen PY, Nayak L, Cohen JV, Dietrich J,
et al: Pembrolizumab in brain metastases of diverse histologies:
Phase 2 trial results. Nat Med. 29:1728–1737. 2023.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Chen J, Li Y, Yu TS, McKay RM, Burns DK,
Kernie SG and Parada LF: A restricted cell population propagates
glioblastoma growth after chemotherapy. Nature. 488:522–526.
2012.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Warrier NM, Krishnan RK, Prabhu V,
Hariharapura RC, Agarwal P and Kumar P: Survivin inhibition by
piperine sensitizes glioblastoma cancer stem cells and leads to
better drug response. Int J Mol Sci. 23(7604)2022.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Ahluwalia MS, Reardon DA, Abad AP, Curry
WT, Wong ET, Figel SA, Mechtler LL, Peereboom DM, Hutson AD,
Withers HG, et al: Phase IIa study of SurVaxM plus adjuvant
temozolomide for newly diagnosed glioblastoma. J Clin Oncol.
41:1453–1465. 2023.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Munoz JL, Walker ND, Scotto KW and
Rameshwar P: Temozolomide competes for P-glycoprotein and
contributes to chemoresistance in glioblastoma cells. Cancer Lett.
367:69–75. 2015.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Oberstadt MC, Bien-Möller S, Weitmann K,
Herzog S, Hentschel K, Rimmbach C, Vogelgesang S, Balz E, Fink M,
Michael H, et al: Epigenetic modulation of the drug resistance
genes MGMT, ABCB1 and ABCG2 in glioblastoma multiforme. BMC Cancer.
13(617)2013.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Yan Y, Liu Y, Liang Q and Xu Z: Drug
metabolism-related gene ABCA1 augments temozolomide chemoresistance
and immune infiltration abundance of M2 macrophages in glioma. Eur
J Med Res. 28(373)2023.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Meng J, Qian W, Yang Z, Gong L, Xu D,
Huang H, Jiang X, Pu Z, Yin Y and Zou J: p53/E2F7 axis promotes
temozolomide chemoresistance in glioblastoma multiforme. BMC
Cancer. 24(317)2024.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Karve AS, Desai JM, Gadgil SN, Dave N,
Wise-Draper TM, Gudelsky GA, Phoenix TN, DasGupta B, Yogendran L,
Sengupta S, et al: A Review of approaches to potentiate the
activity of temozolomide against glioblastoma to overcome
resistance. Int J Mol Sci. 25(3217)2024.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Goenka A, Tiek D, Song X, Huang T, Hu B
and Cheng SY: The many facets of therapy resistance and tumor
recurrence in glioblastoma. Cells. 10(484)2021.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Persano L, Pistollato F, Rampazzo E, Della
Puppa A, Abbadi S, Frasson C, Volpin F, Indraccolo S, Scienza R and
Basso G: BMP2 sensitizes glioblastoma stem-like cells to
Temozolomide by affecting HIF-1α stability and MGMT expression.
Cell Death Dis. 3(e412)2012.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Seidel S, Garvalov BK, Wirta V, von
Stechow L, Schänzer A, Meletis K, Wolter M, Sommerlad D, Henze AT,
Nistér M, et al: A hypoxic niche regulates glioblastoma stem cells
through hypoxia inducible factor 2 alpha. Brain. 133:983–995.
2010.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Ren P, Wang JY, Zeng ZR, Li NX, Chen HL,
Peng XG, Bhawal UK and Guo WZ: A novel hypoxia-driven gene
signature that can predict the prognosis and drug resistance of
gliomas. Front Genet. 13(976356)2022.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Ge X, Pan MH, Wang L, Li W, Jiang C, He J,
Abouzid K, Liu LZ, Shi Z and Jiang BH: Hypoxia-mediated
mitochondria apoptosis inhibition induces temozolomide treatment
resistance through miR-26a/Bad/Bax axis. Cell Death Dis.
9(1128)2018.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Brenner AJ, Sun JD, Floyd J, Hart CP, Eng
C, Kroll S, Fichtel L, Gruslova A, Lodi A and Tiziani S: Phase 1/2
study of investigational hypoxia-targeted drug, TH-302, and
bevacizumab (bev) in recurrent glioblastoma (GBM) following bev
failure. J Clin Oncol. 32 (15_Suppl)(S2029)2014.
|
|
49
|
Strowd R, Ellingson B, Raymond C, Yao J,
Wen PY, Ahluwalia M, Piotrowski A, Desai A, Clarke JL, Lieberman
FS, et al: Activity of a first-in-class oral HIF2-alpha inhibitor,
PT2385, in patients with first recurrence of glioblastoma. J
Neurooncol. 165:101–112. 2023.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Lickliter JD, Ruben J, Kichenadasse G,
Jennens R, Gzell C, Mason RP, Zhou H, Becker J, Unger E and Stea B:
Dodecafluoropentane emulsion as a radiosensitizer in glioblastoma
multiforme. Cancer Res Commun. 3:1607–1614. 2023.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Jeong W, Park SR, Rapisarda A, Fer N,
Kinders RJ, Chen A, Melillo G, Turkbey B, Steinberg SM, Choyke P,
et al: Weekly EZN-2208 (PEGylated SN-38) in combination with
bevacizumab in patients with refractory solid tumors. Invest New
Drugs. 32:340–346. 2014.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Network TC: Corrigendum: Comprehensive
genomic characterization defines human glioblastoma genes and core
pathways. Nature. 494(506)2013.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Velpula KK and Tsung AJ: PDK1: A new
therapeutic target for glioblastoma? CNS Oncol. 3:177–179.
2014.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Velpula KK, Guda MR, Sahu K, Tuszynski J,
Asuthkar S, Bach SE, Lathia JD and Tsung AJ: Metabolic targeting of
EGFRvIII/PDK1 axis in temozolomide resistant glioblastoma.
Oncotarget. 8:35639–35655. 2017.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Kaur B, Khwaja FW, Severson EA, Matheny
SL, Brat DJ and Van Meir EG: Hypoxia and the
hypoxia-inducible-factor pathway in glioma growth and angiogenesis.
Neuro Oncol. 7:134–153. 2005.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Würth R, Bajetto A, Harrison JK, Barbieri
F and Florio T: CXCL12 modulation of CXCR4 and CXCR7 activity in
human glioblastoma stem-like cells and regulation of the tumor
microenvironment. Front Cell Neurosci. 8(144)2014.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Nogueira L, Ruiz-Ontañon P,
Vazquez-Barquero A, Moris F and Fernandez-Luna JL: The NFκB
pathway: A therapeutic target in glioblastoma. Oncotarget.
2:646–653. 2011.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Pareja F, Macleod D, Shu C, Crary JF,
Canoll PD, Ross AH and Siegelin MD: PI3K and Bcl-2 inhibition
primes glioblastoma cells to apoptosis through downregulation of
Mcl-1 and Phospho-BAD. Mol Cancer Res. 12:987–1001. 2014.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Valdés-Rives SA, Casique-Aguirre D,
Germán-Castelán L, Velasco-Velázquez MA and González-Arenas A:
Apoptotic signaling pathways in glioblastoma and therapeutic
implications. BioMed Res Int. 2017(7403747)2017.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Daniele S, Costa B, Zappelli E, Da Pozzo
E, Sestito S, Nesi G, Campiglia P, Marinelli L, Novellino E,
Rapposelli S and Martini C: Combined inhibition of AKT/mTOR and
MDM2 enhances Glioblastoma Multiforme cell apoptosis and
differentiation of cancer stem cells. Sci Rep.
5(9956)2015.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Song Z, Pan Y, Ling G, Wang S, Huang M,
Jiang X and Ke Y: Escape of U251 glioma cells from
temozolomide-induced senescence was modulated by CDK1/survivin
signaling. Am J Transl Res. 9:2163–2180. 2017.PubMed/NCBI
|
|
62
|
Josset E, Burckel H, Noël G and Bischoff
P: The mTOR inhibitor RAD001 potentiates autophagic cell death
induced by temozolomide in a glioblastoma cell line. Anticancer
Res. 33:1845–1851. 2013.PubMed/NCBI
|
|
63
|
Zou Y, Chen M, Zhang S, Miao Z, Wang J, Lu
X and Zhao X: TRPC5-induced autophagy promotes the TMZ-resistance
of glioma cells via the CAMMKβ/AMPKα/mTOR pathway. Oncol Rep.
41:3413–3423. 2019.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Wen PY, de Groot JF, Battiste J, Goldlust
SA, Garner JS, Friend J, Simpson JA, Damek D, Olivero A and
Cloughesy TF: Paxalisib in patients with newly diagnosed
glioblastoma with unmethylated MGMT promoter status: Final phase 2
study results. J Clin Oncol. 40 (16_suppl)(2047)2022.
|
|
65
|
Wen PY, Omuro A, Ahluwalia MS,
Fathallah-Shaykh HM, Mohile N, Lager JJ, Laird AD, Tang J, Jiang J,
Egile C and Cloughesy TF: Phase I dose-escalation study of the
PI3K/mTOR inhibitor voxtalisib (SAR245409, XL765) plus temozolomide
with or without radiotherapy in patients with high-grade glioma.
Neuro Oncol. 17:1275–1283. 2015.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Yu Z, Xie G, Zhou G, Cheng Y, Zhang G, Yao
G, Chen Y, Li Y and Zhao G: NVP-BEZ235, a novel dual PI3K-mTOR
inhibitor displays anti-glioma activity and reduces chemoresistance
to temozolomide in human glioma cells. Cancer Lett. 367:58–68.
2015.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Xu P, Zhang G, Hou S and Sha LG: MAPK8
mediates resistance to temozolomide and apoptosis of glioblastoma
cells through MAPK signaling pathway. Biomed Pharmacother.
106:1419–1427. 2018.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Lin CJ, Lee CC, Shih YL, Lin TY, Wang SH,
Lin YF and Shih CM: Resveratrol enhances the therapeutic effect of
temozolomide against malignant glioma in vitro and in vivo by
inhibiting autophagy. Free Radic Biol Med. 52:377–391.
2012.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Ma L, Liu J, Zhang X, Qi J, Yu W and Gu Y:
p38 MAPK-dependent Nrf2 induction enhances the resistance of glioma
cells against TMZ. Med Oncol. 32(69)2015.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Cheng HS, Chong YK, Lim EKY, Lee XY, Pang
QY, Novera W, Marvalim C, Lee JXT, Ang BT, Tang C and Tan NS: Dual
p38MAPK and MEK inhibition disrupts adaptive chemoresistance in
mesenchymal glioblastoma to temozolomide. Neuro Oncol.
26:1247–1261. 2024.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Reardon DA, Vredenburgh JJ, Desjardins A,
Peters K, Gururangan S, Sampson JH, Marcello J, Herndon JE II,
McLendon RE, Janney D, et al: Effect of CYP3A-inducing
anti-epileptics on sorafenib exposure: Results of a phase II study
of sorafenib plus daily temozolomide in adults with recurrent
glioblastoma. J Neurooncol. 101:57–66. 2011.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Hainsworth JD, Ervin T, Friedman E, Priego
V, Murphy PB, Clark BL and Lamar RE: Concurrent radiotherapy and
temozolomide followed by temozolomide and sorafenib in the
first-line treatment of patients with glioblastoma multiforme.
Cancer. 116:3663–3669. 2010.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Singh N, Miner A, Hennis L and Mittal S:
Mechanisms of temozolomide resistance in glioblastoma-a
comprehensive review. Cancer Drug Resist. 4:17–43. 2021.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Riganti C, Salaroglio IC, Caldera V,
Campia I, Kopecka J, Mellai M, Annovazzi L, Bosia A, Ghigo D and
Schiffer D: Temozolomide downregulates P-glycoprotein expression in
glioblastoma stem cells by interfering with the Wnt3a/glycogen
synthase-3 kinase/β-catenin pathway. Neuro Oncol. 15:1502–1517.
2013.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Xu K, Zhang Z, Pei H, Wang H, Li L and Xia
Q: FoxO3a induces temozolomide resistance in glioblastoma cells via
the regulation of β-catenin nuclear accumulation. Oncol Rep.
37:2391–2397. 2017.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Huang M, Zhang D, Wu JY, Xing K, Yeo E, Li
C, Zhang L, Holland E, Yao L, Qin L, et al: Wnt-mediated
endothelial transformation into mesenchymal stem cell-like cells
induces chemoresistance in glioblastoma. Sci Transl Med.
12(eaay7522)2020.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Wickström M, Dyberg C, Milosevic J, Einvik
C, Calero R, Sveinbjörnsson B, Sandén E, Darabi A, Siesjö P, Kool
M, et al: Wnt/β-catenin pathway regulates MGMT gene expression in
cancer and inhibition of Wnt signalling prevents chemoresistance.
Nat Commun. 6(8904)2015.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Tomar VS, Patil V and Somasundaram K:
Temozolomide induces activation of Wnt/β-catenin signaling in
glioma cells via PI3K/Akt pathway: Implications in glioma therapy.
Cell Biol Toxicol. 36:273–278. 2020.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Lu C, Cui C, Liu B, Zou S, Song H, Tian H,
Zhao J and Li Y: FERMT3 contributes to glioblastoma cell
proliferation and chemoresistance to temozolomide through integrin
mediated Wnt signaling. Neurosci Lett. 657:77–83. 2017.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Luo W, Yan D, Song Z, Zhu X, Liu X, Li X
and Zhao S: miR-126-3p sensitizes glioblastoma cells to
temozolomide by inactivating Wnt/β-catenin signaling via targeting
SOX2. Life Sci. 226:98–106. 2019.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Zeng A, Yin J, Li Y, Li R, Wang Z, Zhou X,
Jin X, Shen F, Yan W and You Y: miR-129-5p targets Wnt5a to block
PKC/ERK/NF-κB and JNK pathways in glioblastoma. Cell Death Dis.
9(394)2018.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Zhang KL, Han L, Chen LY, Shi ZD, Yang M,
Ren Y, Chen LC, Zhang JX, Pu PY and Kang CS: Blockage of a
miR-21/EGFR regulatory feedback loop augments anti-EGFR therapy in
glioblastomas. Cancer Lett. 342:139–149. 2014.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Huang T, Alvarez AA, Pangeni RP, Horbinski
CM, Lu S, Kim SH, James CD, J Raizer J, A Kessler J, Brenann CW, et
al: A regulatory circuit of miR-125b/miR-20b and Wnt signalling
controls glioblastoma phenotypes through FZD6-modulated pathways.
Nat Commun. 7(12885)2016.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Chen KC, Chen PH, Ho KH, Shih CM, Chou CM,
Cheng CH and Lee CC: IGF-1-enhanced miR-513a-5p signaling
desensitizes glioma cells to temozolomide by targeting the
NEDD4L-inhibited Wnt/β-catenin pathway. PLoS One.
14(e0225913)2019.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Kahlert UD, Suwala AK, Koch K, Natsumeda
M, Orr BA, Hayashi M, Maciaczyk J and Eberhart CG: Pharmacologic
wnt inhibition reduces proliferation, survival, and clonogenicity
of glioblastoma cells. J Neuropathol Exp Neurol. 74:889–900.
2015.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Kim Y, Kim KH, Lee J, Lee Y, Kim M, Lee
SJ, Park K, Yang H, Jin J, Joo KM, et al: Wnt activation is
implicated in glioblastoma radioresistance. Lab Invest. 92:466–473.
2012.PubMed/NCBI View Article : Google Scholar
|
|
87
|
De Robertis A, Valensin S, Rossi M, Tunici
P, Verani M, De Rosa A, Giordano C, Varrone M, Nencini A, Pratelli
C, et al: Identification and characterization of a small-molecule
inhibitor of Wnt signaling in glioblastoma cells. Mol Cancer Ther.
12:1180–1189. 2013.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Yoshino T, Ikeda M, Finn RS, Jeffry Evans
TR, Weng L, Saito K, Mody K, Tamai T, Paoletti C and Iwasa S:
Abstract CT523: An open-label, multicenter, phase 1b/2 Study of
E7386 (Wnt/β-catenin pathway inhibitor) + pembrolizumab in patients
with pretreated advanced solid tumors. Cancer Res. 82
(Suppl)(CT523)2022.
|
|
89
|
Bhat KP, Salazar KL, Balasubramaniyan V,
Wani K, Heathcock L, Hollingsworth F, James JD, Gumin J, Diefes KL,
Kim SH, et al: The transcriptional coactivator TAZ regulates
mesenchymal differentiation in malignant glioma. Genes Dev.
25:2594–2609. 2011.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Orr BA, Bai H, Odia Y, Jain D, Anders RA
and Eberhart CG: Yes-associated protein 1 is widely expressed in
human brain tumors and promotes glioblastoma growth. J Neuropathol
Exp Neurol. 70:568–577. 2011.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Tian T, Li A, Lu H, Luo R, Zhang M and Li
Z: TAZ promotes temozolomide resistance by upregulating MCL-1 in
human glioma cells. Biochem Biophys Res Commun. 463:638–643.
2015.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Casati G, Giunti L, Iorio AL, Marturano A,
Galli L and Sardi I: Hippo pathway in regulating drug resistance of
glioblastoma. Int J Mol Sci. 22(13431)2021.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Minata M, Audia A, Shi J, Lu S, Bernstock
J, Pavlyukov MS, Das A, Kim SH, Shin YJ, Lee Y, et al: Phenotypic
plasticity of invasive edge glioma stem-like cells in response to
ionizing radiation. Cell Rep. 26:1893–1905.e7. 2019.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Vigneswaran K, Boyd NH, Oh SY, Lallani S,
Boucher A, Neill SG, Olson JJ and Read RD: YAP/TAZ transcriptional
coactivators create therapeutic vulnerability to verteporfin in
EGFR-mutant glioblastoma. Clin Cancer Res. 27:1553–1569.
2021.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Read RD: Repurposing the drug verteporfin
as anti-neoplastic therapy for glioblastoma. Neuro Oncol.
24:708–710. 2022.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Hagenbeek TJ, Zbieg JR, Hafner M, Mroue R,
Lacap JA, Sodir NM, Noland CL, Afghani S, Kishore A, Bhat KP, et
al: An allosteric pan-TEAD inhibitor blocks oncogenic YAP/TAZ
signaling and overcomes KRAS G12C inhibitor resistance. Nat Cancer.
4:812–828. 2023.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Lu J, Gao M, He H, Feng L, Gao Z, Cui W,
Yang K, Lu J, Zheng Q, Zhu J, et al: Abstract 1671: Discovery of
ETS-003, a potent and selective YAP/TAZ-TEAD PPI inhibitor with
broad anti-tumor activity in Hippo-YAP aberrant cancers. Cancer
Res. 83 (Suppl)(S1671)2023.
|
|
98
|
Chapeau EA, Sansregret L, Galli GG, Chène
P, Wartmann M, Mourikis TP, Jaaks P, Baltschukat S, Barbosa IAM,
Bauer D, et al: Direct and selective pharmacological disruption of
the YAP-TEAD interface by IAG933 inhibits Hippo-dependent and
RAS-MAPK-altered cancers. Nat Cancer. 5:1102–1120. 2024.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Movahedpour A, Khatami SH, Khorsand M,
Salehi M, Savardashtaki A, Mirmajidi SH, Negahdari B, Khanjani N,
Naeli P, Vakili O, et al: Exosomal noncoding RNAs: Key players in
glioblastoma drug resistance. Mol Cell Biochem. 476:4081–4092.
2021.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Shea A, Harish V, Afzal Z, Chijioke J,
Kedir H, Dusmatova S, Roy A, Ramalinga M, Harris B, Blancato J, et
al: MicroRNAs in glioblastoma multiforme pathogenesis and
therapeutics. Cancer Med. 5:1917–1946. 2016.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Xiao S, Yang Z, Qiu X, Lv R, Liu J, Wu M,
Liao Y and Liu Q: miR-29c contribute to glioma cells temozolomide
sensitivity by targeting O6-methylguanine-DNA methyltransferases
indirectely. Oncotarget. 7:50229–50238. 2016.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Cunha PP, Costa PM, Morais CM, Lopes IR,
Cardoso AM, Cardoso AL, Mano M, Jurado AS and Pedroso de Lima MC:
High-throughput screening uncovers miRNAs enhancing glioblastoma
cell susceptibility to tyrosine kinase inhibitors. Hum Mol Genet.
26:4375–4387. 2017.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Xia Y, Pei T, Zhao J, Wang Z, Shen Y, Yang
Y and Liang J: Long noncoding RNA H19: Functions and mechanisms in
regulating programmed cell death in cancer. Cell Death Discov.
10(76)2024.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Zhang Z, Yin J, Lu C, Wei Y, Zeng A and
You Y: Exosomal transfer of long Non-coding RNA SBF2-AS1 enhances
chemoresistance to temozolomide in glioblastoma. J Exp Clin Cancer
Res. 38(166)2019.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Li H, Yuan X, Yan D, Li D, Guan F, Dong Y,
Wang H, Liu X and Yang B: Long Non-Coding RNA MALAT1 decreases the
sensitivity of resistant glioblastoma cell lines to temozolomide.
Cell Physiol Biochem. 42:1192–1201. 2017.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Amodio N, Raimondi L, Juli G, Stamato MA,
Caracciolo D, Tagliaferri P and Tassone P: MALAT1: A druggable long
non-coding RNA for targeted anti-cancer approaches. J Hematol
Oncol. 11(63)2018.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Yan Y, Xu Z, Chen X, Wang X, Zeng S, Zhao
Z, Qian L, Li Z, Wei J, Huo L, et al: Novel function of lncRNA
ADAMTS9-AS2 in promoting temozolomide resistance in glioblastoma
via upregulating the FUS/MDM2 ubiquitination axis. Front Cell Dev
Biol. 7(217)2019.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Liao Y, Shen L, Zhao H, Liu Q, Fu J, Guo
Y, Peng R and Cheng L: LncRNA CASC2 Interacts With miR-181a to
modulate glioma growth and resistance to TMZ through PTEN pathway.
J Cell Biochem. 118:1889–1899. 2017.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Ahmed SP, Castresana JS and Shahi MH: Role
of circular RNA in brain tumor development. Cells.
11(2130)2022.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Ding C, Yi X, Wu X, Bu X, Wang D, Wu Z,
Zhang G, Gu J and Kang D: Exosome-mediated transfer of circRNA
CircNFIX enhances temozolomide resistance in glioma. Cancer Lett.
479:1–12. 2020.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Han C, Wang S, Wang H and Zhang J:
Exosomal circ-HIPK3 facilitates tumor progression and temozolomide
resistance by regulating miR-421/ZIC5 axis in glioma. Cancer
Biother Radiopharm. 36:537–548. 2021.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Campelo MM, Reis-das-Mercês L, Vidal AF,
da Silva FRP, de Oliveira ACA, Monteiro JRS, Cabral CG, Noronha RCR
and Pereira AL: The dual role of circHIPK3 in cancer and its
implications for multiple drugs resistance: A systematic review and
computational approach. Front Oncol. 15(1547889)2025.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Li X, Wang N, Leng H, Yuan H and Xu L:
Hsa_circ_0043949 reinforces temozolomide resistance via
upregulating oncogene ITGA1 axis in glioblastoma. Metab Brain Dis.
37:2979–2993. 2022.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Wei Y, Lu C, Zhou P, Zhao L, Lyu X, Yin J,
Shi Z and You Y: EIF4A3-induced circular RNA ASAP1 promotes
tumorigenesis and temozolomide resistance of glioblastoma via
NRAS/MEK1/ERK1-2 signaling. Neuro Oncol. 23:611–624.
2021.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Hua L, Huang L, Zhang X and Feng H:
Downregulation of hsa_circ_0000936 sensitizes resistant glioma
cells to temozolomide by sponging miR-1294. J Biosci.
45(101)2020.PubMed/NCBI
|
|
116
|
Zhao J, Cui X, Zhan Q, Zhang K, Su D, Yang
S, Hong B, Wang Q, Ju J, Cheng C, et al: CRISPR-Cas9 library
screening combined with an exosome-targeted delivery system
addresses tumorigenesis/TMZ resistance in the mesenchymal subtype
of glioblastoma. Theranostics. 14:2835–2855. 2024.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Halasz LM, Soltys SG, Breneman JC, Chan
MD, Laack NN, Minniti G and Kirkpatrick JP: Treatment of gliomas: A
changing landscape. Int J Radiat Oncol Biol Phys. 98:255–258.
2017.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Shaffer R, Nichol AM, Vollans E, Fong M,
Nakano S, Moiseenko V, Schmuland M, Ma R, McKenzie M and Otto K: A
comparison of volumetric modulated arc therapy and conventional
intensity-modulated radiotherapy for frontal and temporal
High-grade gliomas. Int J Radiat Oncol Biol Phys. 76:1177–1184.
2010.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Wang TJC, Wu CC, Jani A, Estrada J, Ung T,
Chow DS, Soun JE, Saad S, Qureshi YH, Gartrell R, et al:
Hypofractionated radiation therapy versus standard fractionated
radiation therapy with concurrent temozolomide in elderly patients
with newly diagnosed glioblastoma. Pract Radiat Oncol. 6:306–314.
2016.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Gu J, Mu N, Jia B, Guo Q, Pan L, Zhu M,
Zhang W, Zhang K, Li W, Li M, et al: Targeting radiation-tolerant
persister cells as a strategy for inhibiting radioresistance and
recurrence in glioblastoma. Neuro Oncol. 24:1056–1070.
2022.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Fang X, Huang Z, Zhai K, Huang Q, Tao W,
Kim L, Wu Q, Almasan A, Yu JS, Li X, et al: Inhibiting DNA-PK
induces glioma stem cell differentiation and sensitizes
glioblastoma to radiation in mice. Sci Transl Med.
13(eabc7275)2021.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Huang ZL, Liu ZG, Lin Q, Tao YL, Li X,
Baxter P, Su JM, Adesina AM, Man C, Chintagumpala M, et al:
Fractionated radiation therapy alters energy metabolism and induces
cellular quiescence exit in patient-derived orthotopic xenograft
models of high-grade glioma. Transl Oncol.
45(101988)2024.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Osuka S, Zhu D, Zhang Z, Li C, Stackhouse
CT, Sampetrean O, Olson JJ, Gillespie GY, Saya H, Willey CD, et al:
N-cadherin upregulation mediates adaptive radioresistance in
glioblastoma. J Clin Invest. 131(e136098)2021.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Stackhouse CT, Anderson JC, Yue Z, Nguyen
T, Eustace NJ, Langford CP, Wang J, Rowland JR IV, Xing C, Mikhail
FM, et al: An in vivo model of glioblastoma radiation resistance
identifies long noncoding RNAs and targetable kinases. JCI Insight.
7(e148717)2022.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Huang RX and Zhou PK: DNA damage response
signaling pathways and targets for radiotherapy sensitization in
cancer. Signal Transduct Target Ther. 5(60)2020.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Bastiancich C, Bastiat G and Lagarce F:
Gemcitabine and glioblastoma: Challenges and current perspectives.
Drug Discov Today. 23:416–423. 2018.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Lesueur P, Chevalier F, El-Habr EA, Junier
MP, Chneiweiss H, Castera L, Müller E, Stefan D and Saintigny Y:
Radiosensitization effect of talazoparib, a parp inhibitor, on
glioblastoma stem cells exposed to low and high linear energy
transfer radiation. Sci Rep. 8(3664)2018.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Stea B, Falsey R, Kislin K, Patel J,
Glanzberg H, Carey S, Ambrad AA, Meuillet EJ and Martinez JD: Time
and dose-dependent radiosensitization of the glioblastoma
multiforme U251 cells by the EGF receptor tyrosine kinase inhibitor
ZD1839 (‘Iressa’). Cancer Lett. 202:43–51. 2003.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Lo Cascio C, Margaryan T, Luna-Melendez E,
McNamara JB, White CI, Knight W, Ganta S, Opachich Z, Cantoni C,
Yoo W, et al: Quisinostat is a brain-penetrant radiosensitizer in
glioblastoma. JCI Insight. 8(e167081)2023.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Galanis E, Anderson SK, Miller CR,
Sarkaria JN, Jaeckle K, Buckner JC, Ligon KL, Ballman KV, Moore DF
Jr, Nebozhyn M, et al: Phase I/II trial of vorinostat combined with
temozolomide and radiation therapy for newly diagnosed
glioblastoma: Results of Alliance N0874/ABTC 02. Neuro Oncol.
20:546–556. 2018.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Compter I, Eekers DBP, Hoeben A, Rouschop
KMA, Reymen B, Ackermans L, Beckervordersantforth J, Bauer NJC,
Anten MM, Wesseling P, et al: Chloroquine combined with concurrent
radiotherapy and temozolomide for newly diagnosed glioblastoma: A
phase IB trial. Autophagy. 17:2604–2612. 2021.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Benej M, Hong X, Vibhute S, Scott S, Wu J,
Graves E, Le QT, Koong AC, Giaccia AJ, Yu B, et al: Papaverine and
its derivatives radiosensitize solid tumors by inhibiting
mitochondrial metabolism. Proc Natl Acad Sci USA. 115:10756–10761.
2018.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Ali MY, Oliva CR, Noman ASM, Allen BG,
Goswami PC, Zakharia Y, Monga V, Spitz DR, Buatti JM and Griguer
CE: Radioresistance in glioblastoma and the development of
radiosensitizers. Cancers (Basel). 12(2511)2020.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Verhaak RG, Hoadley KA, Purdom E, Wang V,
Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al:
Integrated genomic analysis identifies clinically relevant subtypes
of glioblastoma characterized by abnormalities in PDGFRA, IDH1,
EGFR, and NF1. Cancer Cell. 17:98–110. 2010.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Szerlip NJ, Pedraza A, Chakravarty D, Azim
M, McGuire J, Fang Y, Ozawa T, Holland EC, Huse JT, Jhanwar S, et
al: Intratumoral heterogeneity of receptor tyrosine kinases EGFR
and PDGFRA amplification in glioblastoma defines subpopulations
with distinct growth factor response. Proc Natl Acad Sci USA.
109:3041–3046. 2012.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Lane R, Cilibrasi C, Chen J, Shah K,
Messuti E, Mazarakis NK, Stebbing J, Critchley G, Song E, Simon T,
et al: PDGF-R inhibition induces glioblastoma cell differentiation
via DUSP1/p38MAPK signalling. Oncogene. 41:2749–2763.
2022.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Cenciarelli C, Marei HE, Felsani A,
Casalbore P, Sica G, Puglisi MA, Cameron AJ, Olivi A and Mangiola
A: PDGFRα depletion attenuates glioblastoma stem cells features by
modulation of STAT3, RB1 and multiple oncogenic signals.
Oncotarget. 7:53047–53063. 2016.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Mayr L, Neyazi S, Schwark K, Trissal M,
Beck A, Labelle J, Eder SK, Weiler-Wichtl L, Marques JG, de
Biagi-Junior CAO, et al: Effective targeting of PDGFRA-altered
high-grade glioma with avapritinib. Cancer Cell. 43:740–756.e8.
2025.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Gilbert MR, Dignam JJ, Armstrong TS, Wefel
JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S,
Won M, et al: A randomized trial of bevacizumab for newly diagnosed
glioblastoma. N Engl J Med. 370:699–708. 2014.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Reardon DA, Brandes AA, Omuro A,
Mulholland P, Lim M, Wick A, Baehring J, Ahluwalia MS, Roth P, Bähr
O, et al: Effect of nivolumab vs bevacizumab in patients with
recurrent glioblastoma: The CheckMate 143 Phase 3 randomized
clinical trial. JAMA Oncol. 6:1003–1010. 2020.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Brennan CW, Verhaak RG, McKenna A, Campos
B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ,
Berman SH, et al: The somatic genomic landscape of glioblastoma.
Cell. 155:462–477. 2013.PubMed/NCBI View Article : Google Scholar
|
|
142
|
An Z, Aksoy O, Zheng T, Fan QW and Weiss
WA: Epidermal growth factor receptor and EGFRvIII in glioblastoma:
Signaling pathways and targeted therapies. Oncogene. 37:1561–1575.
2018.PubMed/NCBI View Article : Google Scholar
|
|
143
|
Ezzati S, Salib S, Balasubramaniam M and
Aboud O: Epidermal growth factor receptor inhibitors in
glioblastoma: Current status and future possibilities. Int J Mol
Sci. 25(2316)2024.PubMed/NCBI View Article : Google Scholar
|
|
144
|
Guo G, Gong K, Ali S, Ali N, Shallwani S,
Hatanpaa KJ, Pan E, Mickey B, Burma S, Wang DH, et al: Primary
resistance to EGFR inhibition in glioblastoma is mediated by a
TNF-JNK-Axl-ERK signaling axis. Nat Neurosci. 20:1074–1084.
2017.PubMed/NCBI View Article : Google Scholar
|
|
145
|
Marin BM, Porath KA, Jain S, Kim M,
Conage-Pough JE, Oh JH, Miller CL, Talele S, Kitange GJ, Tian S, et
al: Heterogeneous delivery across the blood-brain barrier limits
the efficacy of an EGFR-targeting antibody drug conjugate in
glioblastoma. Neuro Oncol. 23:2042–2053. 2021.PubMed/NCBI View Article : Google Scholar
|
|
146
|
Hottinger AF, Ben Aissa A, Espeli V,
Squiban D, Dunkel N, Vargas MI, Hundsberger T, Mach N, Schaller K,
Weber DC, et al: Phase I study of sorafenib combined with radiation
therapy and temozolomide as first-line treatment of high-grade
glioma. Br J Cancer. 110:2655–2661. 2014.PubMed/NCBI View Article : Google Scholar
|
|
147
|
Kim JY, Jo Y, Oh HK and Kim EH: Sorafenib
increases tumor treating Fields-induced cell death in glioblastoma
by inhibiting STAT3. Am J Cancer Res. 10:3475–3486. 2020.PubMed/NCBI
|
|
148
|
Schreck KC, Grossman SA and Pratilas CA:
BRAF mutations and the utility of RAF and MEK inhibitors in primary
brain tumors. Cancers (Basel). 11(1262)2019.PubMed/NCBI View Article : Google Scholar
|
|
149
|
Fabro F, Lamfers MLM and Leenstra S:
Advancements, challenges, and future directions in tackling
glioblastoma resistance to small kinase inhibitors. Cancers
(Basel). 14(600)2022.PubMed/NCBI View Article : Google Scholar
|
|
150
|
Marie Y, Carpentier AF, Omuro AM, Sanson
M, Thillet J, Hoang-Xuan K and Delattre JY: EGFR tyrosine kinase
domain mutations in human gliomas. Neurology. 64:1444–1445.
2005.PubMed/NCBI View Article : Google Scholar
|
|
151
|
Stommel JM, Kimmelman AC, Ying H,
Nabioullin R, Ponugoti AH, Wiedemeyer R, Stegh AH, Bradner JE,
Ligon KL, Brennan C, et al: Coactivation of receptor tyrosine
kinases affects the response of tumor cells to targeted therapies.
Science. 318:287–290. 2007.PubMed/NCBI View Article : Google Scholar
|
|
152
|
Nathanson DA, Gini B, Mottahedeh J,
Visnyei K, Koga T, Gomez G, Eskin A, Hwang K, Wang J, Masui K, et
al: Targeted therapy resistance mediated by dynamic regulation of
extrachromosomal mutant EGFR DNA. Science. 343:72–76.
2014.PubMed/NCBI View Article : Google Scholar
|
|
153
|
Day EK, Sosale NG, Xiao A, Zhong Q, Purow
B and Lazzara MJ: Glioblastoma cell resistance to EGFR and MET
inhibition can be overcome via blockade of FGFR-SPRY2 bypass
signaling. Cell Rep. 30:3383–3396.e7. 2020.PubMed/NCBI View Article : Google Scholar
|
|
154
|
Chakravarti A, Loeffler JS and Dyson NJ:
Insulin-like growth factor receptor I mediates resistance to
Anti-epidermal growth factor receptor therapy in primary human
glioblastoma cells through continued activation of phosphoinositide
3-kinase signaling. Cancer Res. 62:200–207. 2002.PubMed/NCBI
|
|
155
|
Han S, Liu Y, Cai SJ, Qian M, Ding J,
Larion M, Gilbert MR and Yang C: IDH mutation in glioma: Molecular
mechanisms and potential therapeutic targets. Br J Cancer.
122:1580–1589. 2020.PubMed/NCBI View Article : Google Scholar
|
|
156
|
Fanucci K, Pilat MJ, Shyr D, Shyr Y,
Boerner S, Li J, Durecki D, Drappatz J, Puduvalli V, Lieberman FS,
et al: Multicenter Phase II trial of the PARP inhibitor olaparib in
recurrent IDH1- and IDH2-mutant glioma. Cancer Res Commun.
3:192–201. 2023.PubMed/NCBI View Article : Google Scholar
|
|
157
|
Lin MD, Tsai AC, Abdullah KG, McBrayer SK
and Shi DD: Treatment of IDH-mutant glioma in the INDIGO era. NPJ
Precis Oncol. 8(149)2024.PubMed/NCBI View Article : Google Scholar
|
|
158
|
Omuro A, Brandes AA, Carpentier AF, Idbaih
A, Reardon DA, Cloughesy T, Sumrall A, Baehring J, van den Bent M,
Bähr O, et al: Radiotherapy combined with nivolumab or temozolomide
for newly diagnosed glioblastoma with unmethylated MGMT promoter:
An international randomized phase III trial. Neuro Oncol.
25:123–134. 2023.PubMed/NCBI View Article : Google Scholar
|
|
159
|
Lee AH, Sun L, Mochizuki AY, Reynoso JG,
Orpilla J, Chow F, Kienzler JC, Everson RG, Nathanson DA, Bensinger
SJ, et al: Neoadjuvant PD-1 blockade induces T cell and cDC1
activation but fails to overcome the immunosuppressive tumor
associated macrophages in recurrent glioblastoma. Nat Commun.
12(6938)2021.PubMed/NCBI View Article : Google Scholar
|
|
160
|
Lin YJ, Wu CY, Wu JY and Lim M: The role
of myeloid cells in GBM immunosuppression. Front Immunol.
13(887781)2022.PubMed/NCBI View Article : Google Scholar
|
|
161
|
Sesé B, Íñiguez-Muñoz S, Ensenyat-Mendez
M, Llinàs-Arias P, Ramis G, Orozco JIJ, Fernández de Mattos S,
Villalonga P and Marzese DM: Glioblastoma Embryonic-like stem cells
exhibit immune-evasive phenotype. Cancers (Basel).
14(2070)2022.PubMed/NCBI View Article : Google Scholar
|
|
162
|
Chen A, Jiang Y, Li Z, Wu L, Santiago U,
Zou H, Cai C, Sharma V, Guan Y, McCarl LH, et al: Chitinase-3-like
1 protein complexes modulate macrophage-mediated immune suppression
in glioblastoma. J Clin Invest. 131(e147552)2021.PubMed/NCBI View Article : Google Scholar
|
|
163
|
Chongsathidkiet P, Jackson C, Koyama S,
Loebel F, Cui X, Farber SH, Woroniecka K, Elsamadicy AA, Dechant
CA, Kemeny HR, et al: Sequestration of T cells in bone marrow in
the setting of glioblastoma and other intracranial tumors. Nat Med.
24:1459–1468. 2018.PubMed/NCBI View Article : Google Scholar
|
|
164
|
Kang W, Mo Z, Li W, Ma H and Zhang Q:
Heterogeneity and individualized treatment of microenvironment in
glioblastoma (review). Oncol Rep. 50(217)2023.PubMed/NCBI View Article : Google Scholar
|
|
165
|
Wu A, Maxwell R, Xia Y, Cardarelli P,
Oyasu M, Belcaid Z, Kim E, Hung A, Luksik AS, Garzon-Muvdi T, et
al: Combination anti-CXCR4 and anti-PD-1 immunotherapy provides
survival benefit in glioblastoma through immune cell modulation of
tumor microenvironment. J Neurooncol. 143:241–249. 2019.PubMed/NCBI View Article : Google Scholar
|














