1. Introduction
Tumorigenesis is a complex biological process
fundamentally driven by the accumulation of genetic lesions,
leading to dysregulated cellular growth and proliferation. This
process involves well-established mechanisms, including oncogene
activation, tumor suppressor gene inactivation, deficiencies in DNA
repair machinery, and dysregulation of apoptotic pathways.
Inactivation of tumor suppressor genes plays a pivotal regulatory
role in tumorigenesis, with key epigenetically silenced exemplars
including TP53, RB1, CDKN2A (encoding p16),
and members of the semaphorin (SEMA) gene family, notably
axon guidance regulators recurrently dysregulated in malignancies
(1). Recent research demonstrates
SEMA3B, a soluble ligand within the SEMA family members, induces
tumor cell apoptosis while also playing a pivotal role in
suppressing tumor angiogenesis and tumorigenesis (2). The present review focuses
specifically on SEMA3B due to its unique and frequent inactivation
across diverse cancers through mechanisms such as promoter
hypermethylation and loss of heterozygosity (LOH) at 3p21.3, a
genomic region notably enriched with tumor suppressor genes. While
other SEMA3 (including SEMA3A and SEMA3F) also exhibit
tumor-modulatory roles, SEMA3B stands out for its dual capacity to
directly induce apoptosis and competitively inhibit vascular
endothelial growth factor (VEGF)-mediated angiogenesis (3), positioning it as a multifaceted tumor
suppressor with broad therapeutic potential. To advance the
mechanistic understanding and therapeutic application of SEMA3B
across various malignancies, this review comprehensively explores
its structural features and receptor characteristics, functional
interactions with other genes and signaling pathways, inactivation
mechanisms, and recent research progress in human cancers.
Clinically, SEMA3B holds promise as a diagnostic and
prognostic biomarker, given its detectable serum levels and
correlation with tumor progression in cancers such as
hepatocellular carcinoma (HCC) and esophageal squamous cell
carcinoma (ESCC) (4). Although no
clinical trials targeting SEMA3B are currently underway, its role
in regulating key pathways such as phosphatidylinositol
3-kinase/protein kinase B (PI3K/Akt) and its interaction with
VEGF/neuropilin (NRP)1 highlight its potential as a target for
epigenetic therapy or in combination with anti-angiogenic agents.
Translational studies exploring SEMA3B restoration, via
demethylating agents or gene therapy, represent an emerging
frontier in oncology.
Originally described as axonal guidance and neural
development molecules that control migration during central nervous
system (CNS) development, SEMAs are a large family of transmembrane
and secreted proteins that play key antitumor and pro-tumor roles
in cancer initiation, progression and metastasis (5). This family contains 28 genes, among
which 21 are present in vertebrates. SEMA3 constitutes a subfamily
of seven vertebrate SEMAs, defined by their unique secreted status
among vertebrate SEMAs and distinguished by a signature basic
domain within their C-terminus (6). Previous research (7) established that SEMAs function as axon
guidance molecules, directing axon pathfinding (as well as the
motility of other cell types) by altering the cytoskeleton and
adhesion components essential for determining cellular morphology.
SEMA proteins are now recognized as key regulators of morphology
and motility across diverse cell types, spanning neural,
cardiovascular, immune, endocrine, hepatic, renal, reproductive,
respiratory, and musculoskeletal systems, as well as cancer cells.
Furthermore, they play crucial roles in multiple fundamental
biological processes, including organogenesis, tissue repair,
immune cell activation and proliferation, and tumorigenesis
(7,8). In recent years, a large amount of
research has been conducted on the regulatory role of SEMAs in the
tumor microenvironment (TME). Studies have shown that in addition
to the intrinsic genetic and epigenetic changes that drive the
behavior of cancer cells, the TME plays a crucial role in
supporting disease progression and treatment resistance. The
nervous system and its related mediators are now regarded as
important components of the TME. Due to their potential as novel
therapeutic targets, interest in them is increasing. As our
understanding of cancer biology further highlights the functional
role of SEMAs, growing evidence indicates that different SEMAs can
stimulate or limit tumor progression. However, how the aberrant
expression of SEMAs and their interaction with major receptors
affect the main components of the TME still requires further study
(9-11).
SEMAs are classified into eight subfamilies based on
origin, species distribution, and structural characteristics.
Subfamilies 1 and 2 are found exclusively in invertebrates, whereas
subfamilies 3 to 7 are primarily vertebrate-specific, with the
notable exception of SEMA-5C (subfamily 5), which also occurs in
invertebrates. Subfamily 5 is additionally present in certain
viruses. Structurally, subfamilies 1, 4, 5, and 6 are transmembrane
proteins; members of subfamilies 2, 3, and 5 are secreted; and
subfamily 7 members are membrane-anchored via
glycosylphosphatidylinositol. Furthermore, members of subfamilies
4, 5, and 7, and potentially others, undergo proteolytic cleavage
and are released extracellularly. Notably, SEMA3 constitutes a key
subfamily of secreted glycoproteins, encompassing multiple isoforms
including SEMA3A through SEMA3G (12). Mounting evidence demonstrates that
SEMA3 regulates tumor angiogenesis, cancer cell proliferation,
invasiveness, and metastatic dissemination, positioning this
subfamily as a promising therapeutic target in oncology research
(6,13).
2. Structure and receptors of SEMA3B
SEMA3B, a member of the SEMA3 subfamily, maps to
chromosomal locus 3p21.3 and serves as a candidate tumor suppressor
gene that undergoes frequent inactivation in multiple cancers
(13,14). SEMA3B is a protein of 749 amino
acids. Its gene contains 17 exons, covering an exonic region of 3.4
kb and a genomic length of 8-10 kb. The mRNA encodes an N-terminal
SEMA domain that binds to NRPs and plexins, forming the NRP pathway
involved in apoptotic regulation (15). SEMA3B contains distinctive
accessory sequences, including an immunoglobulin-like domain and a
signal peptide, which facilitate its secretion. SEMAs mediate
intercellular signaling through specific interactions with NRPs,
plexins, and five additional receptor classes. NRPs are 130-140 kDa
transmembrane glycoproteins that serve as receptors for both VEGF
and SEMA3 families. In humans, the NRP family comprises NRP1 and
NRP2, each containing cytoplasmic, transmembrane, and extracellular
regions. The extracellular domain features three structural
subdomains: a1/a2, b1/b2, and c. Crucially, the a1/a2 subdomains
enhance binding affinity between b1/b2 and VEGF165,
thereby promoting tumor angiogenesis and increasing microvessel
density (16). SEMA3B exhibits
broad expression and serves as an indispensable regulator in neural
development. Plexins, transmembrane receptors for SEMAs, feature
extracellular domains containing three plexin-semaphorin-integrin
motifs and three Ig-like, plexin, transcription factor domains.
Their intracellular regions harbour a conserved sema-plexin domain.
Previous studies reveal that SEMA3B activates plexin receptors upon
binding, inducing tyrosine phosphorylation within their cytoplasmic
domains. This triggers kinase pathway activation that modulates
cellular behaviours. Notably, the SEMA domain must first form
heterodimers with NRP receptors to achieve functional activation,
subsequently recruiting plexin co-receptors to execute downstream
signaling (9,17). In summary, SEMA3 family members
engage distinct NRPs, with NRPs providing the binding platform for
SEMA3B to form heterodimeric complexes. These complexes
subsequently associate with plexin co-receptors to assemble
signaling-competent SEMA3 receptor complexes, ultimately
suppressing tumour cell proliferation, metastasis, and angiogenesis
through tumour-suppressor mechanisms (6,8).
Notably, SEMA3 proteins exploit NRP and plexin receptors to engage
cadherins (18), integrins
(19), and VEGF receptors (VEGFRs)
(7) for signal transduction,
thereby demonstrating their capacity to activate and modulate
multiple divergent signaling pathways. These signaling complexes
critically regulate axon and dendrite growth, cell migration,
angiogenesis, proliferation, invasion, and epithelial-mesenchymal
transition processes within the CNS (18,20).
Within tumor biology, SEMA3 proteins exhibit complex mechanisms of
action. Their functional outcomes, dictated by the distinct
signaling states of their receptor complexes, drive tumor
progression towards either promotion or suppression, resulting in
dualistic pro-tumorigenic or anti-tumorigenic effects (21).
The tumor-suppressive function of SEMA3B is exerted
through the formation of complexes with NRP and plexin receptors,
and its structural features provide the basis for its functional
diversity. However, the composition of SEMA3B receptor complexes in
different tumor types and their dynamic regulatory mechanisms
remain unclear. Furthermore, whether SEMA3B exhibits receptor
preference in different cellular contexts, and how this preference
influences its function, represent important directions for future
research. A schematic diagram summarizing the SEMA3B receptor
complex, its key inhibited pathways (PI3K/Akt and VEGF), and its
major inactivation mechanisms is presented in Fig. 1.
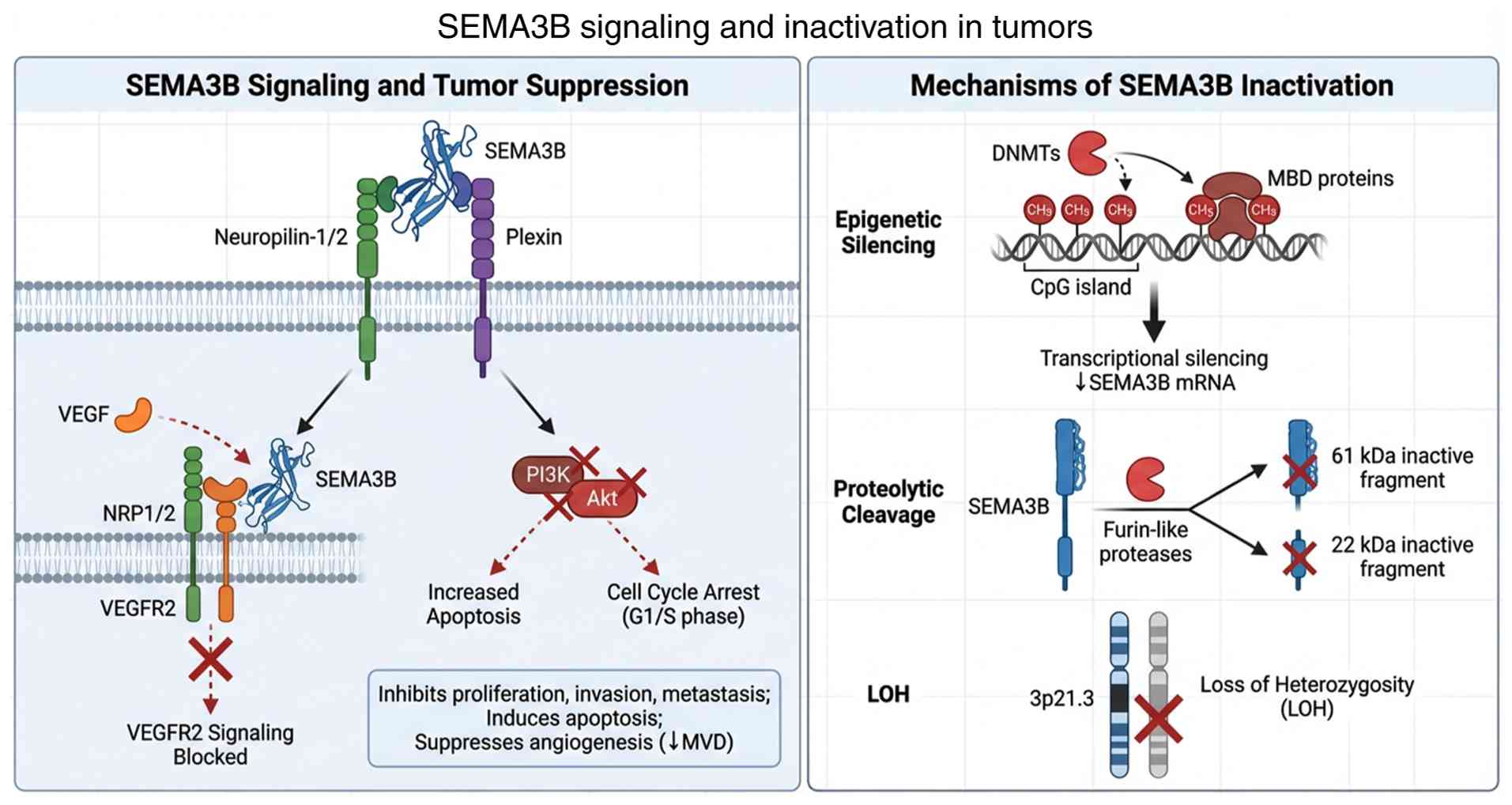 | Figure 1Schematic overview of SEMA3B
signaling and inactivation mechanisms in tumors. Left panel: SEMA3B
binds to NRP1/2 and plexin receptors, inhibiting PI3K/Akt signaling
and competing with VEGF for NRP1 binding, leading to suppression of
tumor progression. Right panel: SEMA3B is inactivated through
promoter hypermethylation, proteolytic cleavage by furin-like
proteases, and loss of heterozygosity at 3p21.3. SEMA3B, semaphorin
3B; NRP, neuropilin; VEGF, vascular endothelial growth factor;
VEGFR2, VEGF receptor 2; MVD, microvessel density; DNMTs, DNA
methyltransferases; MBD, methyl-CpG binding domain. |
3. Interactions of SEMA3B with other genes
and signaling pathways
Functional interplay between SEMA3B
and TP53
TP53 is a critical tumor suppressor gene that
encodes a transcription factor regulating cell-cycle initiation and
facilitating DNA repair to constrain neoplastic growth. Under
physiological conditions, TP53 maintains genomic stability by
orchestrating cell-cycle control, DNA damage repair, and apoptosis.
During tumorigenesis, cumulative genetic and epigenetic
alterations, including gene mutations and promoter methylation,
dysregulate growth, differentiation, and apoptotic pathways,
frequently resulting in constitutively elevated TP53 expression in
malignant tissues (22-24).
SEMA3B expression is transcriptionally regulated by TP53.
Extracellular stimuli such as UV irradiation induce TP53
expression, thereby upregulating SEMA3B to constrain mitotic entry.
This functional synergy suggests SEMA3B may partially share
cell-cycle regulatory mechanisms associated with TP53 in mediating
tumor-suppressive effects (25).
Both TP53 and SEMA3B function as critical tumor suppressor genes,
exhibiting complex regulatory crosstalk. TP53 exerts direct
transcriptional control over SEMA3B expression and further
modulates downstream signaling effectors in its pathway (26). Additional research demonstrates
that SEMA3B induces tumor cell cycle arrest at the G1/S phase by
upregulating expression of TP53 and CDKN1A (encoding p21), while
suppressing phosphorylation of Akt at Ser473(27). Elucidating the specific molecular
mechanisms underlying their reciprocal regulation will shed light
on tumorigenesis mechanisms, thereby providing a theoretical
foundation for early diagnosis, intervention, and clinical
applications in oncology.
Although numerous studies support that SEMA3B is
regulated by TP53 and plays a role in cell cycle regulation, the
feedback mechanisms between them and their synergistic effects in
tumorigenesis remain unclear. Particularly in TP53-mutant tumors,
it is still unknown whether SEMA3B retains its tumor-suppressive
function or if its role is compensated by other pathways,
warranting further investigation.
Interplay between SEMA3B and the
PI3K/Akt signaling pathway
Activation of the PI3K/Akt signaling pathway
represents a key mechanism in the pathogenesis and progression of
various tumors, promoting tumor invasion and metastasis.
Furthermore, this pathway suppresses apoptosis, adhesion, and
transformation processes in tumor cells, thereby modulating the
proliferative and invasive capabilities of cancer cells. Akt, also
known as PKB, serves as a pivotal regulatory molecule within the
PI3K pathway, orchestrating cell survival, cell cycle progression,
and cell growth. Phosphorylation mediated by Akt leads to the
inactivation of several pro-apoptotic factors, including the
apoptosis regulator Bad, procaspase-9, and specific Forkhead family
members that induce Fas expression (28). Furthermore, phosphorylated Akt
activates mouse double minute 2 homolog (MDM2), which targets P53
for degradation; Akt is activated downstream of PI3K upon receptor
stimulation (29). Insulin and
other growth and survival factors activate the Akt signaling
pathway. The Akt mutation abolishes its ability to suppress tumor
proliferation and induce apoptosis (30). Relevant studies reveal that in lung
and breast carcinomas, SEMA3B binding to NRP1 inhibits the
phosphorylation of key Akt pathway-associated proteins, including
p85, PDK1, PTEN, GSK-3β, FKHR, and MDM2. This blockade of
downstream signaling consequently suppresses tumor progression
(28,31). Rolny et al (32) demonstrated that SEMA3B induces IL-8
production in tumor cells by activating the p38 mitogen-activated
protein kinase (MAPK) pathway in an NRP1-dependent manner.
Silencing endogenous SEMA3B expression in tumor cells impaired IL-8
transcription. Conversely, the release of IL-8 induced the
recruitment of tumor-associated macrophages (TAMs) and facilitated
metastatic dissemination to the lungs; this effect was rescued by
blocking IL-8 with a neutralizing antibody (32). These findings indicate that SEMA3B
unexpectedly promotes a pro-metastatic environment by coupling
enhanced IL-8 secretion, which drives macrophage recruitment, with
tumor growth inhibition.
The inhibition of the Akt pathway by SEMA3B via NRP1
is a significant component of its tumor-suppressive mechanism.
However, the discovery that SEMA3B can promote tumor metastasis via
IL-8 under certain conditions highlights the complexity of its
functions. Whether this dual role depends on tumor type,
microenvironment, or expression levels currently lacks a consensus.
Future studies need to clarify the upstream and downstream
regulatory network of SEMA3B within the PI3K/Akt pathway and its
interactions with other inflammatory factors.
Interplay between SEMA3B and VEGF in
angiogenesis
VEGF is a highly specific mitogen for vascular
endothelial cells. It signals through specific receptors to
regulate angiogenesis. The VEGFA gene resides at chromosome 6p21 in
humans, encoding a ~45 kDa glycoprotein. This family comprises
several members, with VEGFA, the first identified and most
extensively studied, exhibiting at least six isoforms
(VEGF121, VEGF145, VEGF165,
VEGF183, VEGF189, VEGF206) defined
by amino acid sequence length. Among these, VEGF121,
VEGF145 and VEGF165 are implicated in
angiogenesis. VEGF165, the most abundant and pivotal
isoform, exhibits the highest biological activity and represents
the predominant form secreted by both benign and malignant cells
(33). VEGF exerts its biological
effects by binding receptors on endothelial cell membranes,
initiating signaling cascades that critically regulate
angiogenesis, vascular permeability, and cell survival and
migration. Three VEGF receptors are established: VEGFR1 (FLT1), the
first identified; VEGFR2 (KDR/FLK1), the principal signaling
receptor for VEGF; and VEGFR3 (FLT4), expressed predominantly in
lymphatic endothelial cells. These receptors are essential for the
development, maintenance, and function of blood and lymphatic
vessels. NRP1 and NRP2 function as VEGF co-receptors, and research
indicates that VEGF signaling can occur directly through NRPs,
independent of VEGFR binding. Notably, most VEGFA downstream
effects are mediated by VEGFR2(34). VEGF acts primarily on vascular
endothelial cells, exhibiting marked specificity for those within
tumors while exerting minimal effects on benign non-tumor cells. In
the molecular mechanisms of tumor angiogenesis, the binding of
VEGFA to VEGFR1 and VEGFR2 is a central event. This binding
activates multiple signaling pathways, including PI3K/AKT and MAPK,
promoting endothelial cell proliferation, survival, adhesion, and
the formation of new vessels from preexisting vasculature. The
VEGFA/VEGFR network is tightly regulated by diverse mechanisms,
including transcriptional and post-transcriptional control
(35,36). Apte et al (37) demonstrated that VEGF binding to
vascular endothelial cells elevates vascular permeability, thereby
inducing excessive leakage of intravascular components; this
process concurrently disrupts apoptotic machinery. Moreover, it has
been established that VEGF activates proteases, resulting in the
progressive dismantling of the extracellular matrix (ECM). This ECM
remodeling facilitates neovascularization and ultimately creates a
permissive microenvironment for tumor cell invasion and metastasis
(38,39). In the study by Wang et al
(40), SEMA3B and NRP1
co-localized on vascular endothelium within colorectal cancer
tissues. Both SEMA3B and VEGF bound NRP1, yet exhibited no direct
interaction with each other. Compared with controls, NRP1
demonstrated increased association with SEMA3B, but reduced binding
to VEGF in SEMA3B-AS1-overexpressing cells. Notably, elevating
exogenous VEGF concentrations induced a concomitant increase in
SEMA3B levels within conditioned media. This study established
functional competition between SEMA3B and VEGF for NRP1 binding,
thereby suppressing VEGF pathway activation and ultimately
inhibiting tumor neovascularization (40). This aligns with prior findings by
Castro-Rivera et al (41),
who demonstrated that VEGF165 markedly attenuates the
pro-apoptotic and anti-mitogenic activities of transfected or
secreted SEMA3B in lung and breast carcinoma cells. Specifically,
SEMA3B induces apoptosis in these prevalent human cancers, an
effect overridden by VEGF165.
The competitive mechanism between SEMA3B and VEGF
for binding to NRP1 provides strong evidence for its
anti-angiogenic effect. However, the phenomenon where the SEMA3B
function is ‘overridden’ in high VEGF environments suggests it
might be at a disadvantage in some tumors. Furthermore, whether the
competition efficiency between SEMA3B and different VEGF isoforms
(such as VEGF165) varies, and whether this competition
is regulated by other factors in the tumor microenvironment, remain
unresolved issues.
4. Mechanisms of SEMA3B inactivation
The inactivation mechanisms of tumor suppressor
genes primarily comprise DNA methylation, LOH, dominant-negative
effects, and haploinsufficiency. Research has established that
SEMA3B exhibits promoter methylation and 3p21.3 LOH across multiple
malignancies, including non-small cell lung cancer, gastric cancer,
hepatocellular carcinoma, oral squamous cell carcinoma,
cholangiocarcinoma, and breast cancer. Notably, 3p21.3 LOH occurs
in 60% of ovarian carcinomas and 90% of small-cell lung cancers.
SEMA3B inactivation likely occurs through a two-hit mechanism
involving epigenetic alterations and allelic loss, thereby ablating
its tumor-suppressive function (14,26,42,43).
Specifically, this epigenetic modification is catalyzed by DNA
methyltransferases, which add methyl groups to the cytosine bases
of CpG dinucleotides. The methylation of specific CpG sites can
directly prevent the recruitment of essential transcription factors
and RNA polymerase II. Moreover, methyl-CpG binding domain proteins
recognize methylated DNA and recruit additional inhibitory
complexes. The cooperation between DNA methylation and histone
modification leads to an inhibitory chromatin state, effectively
compressing the chromatin and preventing the SEMA3B promoter from
being accessed by the transcriptional machinery (44). The frequency of SEMA3B promoter
methylation and its association with gene silencing vary depending
on different tumor types, as summarized in Table I. Researchers have found that tumor
cells expressing endogenous or recombinant SEMA3B failed to
effectively repel endothelial cells. SEMA3B detected in the culture
medium was almost entirely cleaved by furin-like proprotein
convertases, generating inactive 61- and 22-kDa fragments. These
findings suggest that upregulation of furin-like proprotein
convertases in malignant cells may enable tumors to evade the
anti-angiogenic effects of SEMA3B. Consequently, proteolytic
cleavage by these convertases represents a potential mechanism for
SEMA3B inactivation (16).
 | Table IFrequency and significance of SEMA3B
promoter methylation in different tumors. |
Table I
Frequency and significance of SEMA3B
promoter methylation in different tumors.
| Tumor type | Promoter
methylation frequency | Biological or
clinical consequences | (Refs.) |
|---|
| Gastric
carcinoma | 88% | The high-frequency
methylation of the promoter region of the SEMA3B gene leads to its
expression inactivation, thereby promoting the proliferation of
tumor cells. | (26) |
| Lung cancers | 44% in
adenocarcinomas, 45% in squamous cell carcinomas | Correlates with
reduced SEMA3B mRNA; associated with advanced stage | (46) |
| Esophageal squamous
cancer | Significantly
higher than those in corresponding normal tissues | Associated with
progression and poor prognosis; correlates with TNM stage and lymph
node metastasis | (14) |
| Oral squamous cell
carcinoma | 77.8% | The methylation of
the SEMA3B promoter is significantly associated with the advanced
stage of the disease or lymph node metastasis. | (42) |
| Glioma | 48.8% | A general
increasing trend in the methylation of the SEMA3B gene was observed
with increasing pathological grade of the samples | (1) |
| Breast cancer | 46% | A significant
correlation between hyper-methylation and mRNA downregulation | (43) |
Promoter methylation and LOH are classical
mechanisms for SEMA3B inactivation, but our understanding of other
inactivation pathways remains limited. Proteolytic cleavage is an
important mechanism, but it remains unclear whether proteases
beyond furin are involved? Similarly, how the activity of these
cleavage enzymes is upregulated in tumors has yet to be fully
elucidated. Additionally, whether the two inactivation mechanisms,
epigenetic silencing (methylation) and proteolytic inactivation,
occur independently or synergistically during tumor progression
remains unresolved Exploring the crosstalk between these different
inactivation mechanisms and developing SEMA3B variants or analogs
resistant to proteolysis could represent novel therapeutic
directions.
5. Research progress on SEMA3B in malignant
tumors
In the study by Dong et al (14), immunohistochemistry (IHC) and
reverse transcription-quantitative PCR (RT-qPCR) revealed
significantly reduced SEMA3B expression in ESCC cells and tissues
compared with normal esophageal cells and adjacent non-tumorous
tissues. SEMA3B expression was significantly correlated with TNM
stage and lymph node metastasis. Analysis indicated that expression
within the CpG island of the SEMA3B promoter region may be
regulated by promoter methylation status. These findings
collectively suggest that SEMA3B functions as a tumor suppressor
and represents a potential therapeutic target. Guo et al
(44) employed RT-qPCR and IHC to
assess SEMA3B expression in gastric cancer tissues, associating it
with clinicopathological parameters. Using bisulfite genomic
sequencing and bisulfite-specific methylation PCR, methylation
status was determined and the biological effects of SEMA3B in
vitro were investigated. Their findings confirm SEMA3B acts as
a tumor suppressor in gastric carcinogenesis, with its expression
co-regulated by promoter hypermethylation and histone
modifications. Pang et al (1) assessed SEMA3B expression using
RT-qPCR, revealing significantly lower levels in glioma tissues vs.
normal brain tissues, with progressive reduction correlating with
advanced pathological grades. Methylation analysis detected SEMA3B
promoter methylation in gliomas but not in normal tissue. These
findings demonstrate a significant association between SEMA3B
downregulation and glioma progression, suggesting its potential as
a therapeutic target. Li et al (4) measured serum SEMA3B levels in
patients with HCC using ELISA, revealing inverse correlations with
tumor size, capsule status, and TNM stage. These findings indicate
that serum SEMA3B, readily detectable in peripheral blood, holds
potential value for diagnosing HCC and predicting patient
prognosis. Li et al (45)
evaluated the expression of SEMA3 via IHC in 198 prostate biopsies
from patients with low- and intermediate-risk localized prostate
cancer. Their results indicated that SEMA3A, SEMA3B, SEMA3C, and
SEMA3E expression levels represent potential indicators for
predicting the risk of biochemical recurrence after radical
prostatectomy in survival analyses. Furthermore, their
immunostaining may complement standard clinicopathological
parameters.
Numerous studies in this section consistently
demonstrate that SEMA3B expression is downregulated in various
cancers and associated with poor prognosis, strongly supporting its
role as a broad-spectrum tumor suppressor and potential biomarker.
However, current research is largely correlative, lacking direct
functional restoration experiments demonstrating that re-expressing
SEMA3B in vivo can reverse malignant phenotypes. Future
research priorities should shift towards: i) Utilizing gene editing
and overexpression techniques to validate the therapeutic potential
of SEMA3B in various animal models; and ii) integrating SEMA3B
expression with broader molecular subtypes (such as gene mutation
profiles, immune microenvironment characteristics) to identify
patient populations most likely to benefit from SEMA3B-related
therapies.
6. Conclusions and future directions
Studying tumor suppressor genes provides crucial
insights into the molecular etiology of cancer, with fundamental
implications for diagnosis, therapy, and prognosis assessment.
Although substantial advances have elucidated molecular
characteristics and mechanisms of SEMA3B, its pathophysiological
functions and roles in tumor biology remain to be fully elucidated.
Notably, the frequent epigenetic silencing of SEMA3B across diverse
malignancies, coupled with its roles in apoptosis induction,
angiogenesis suppression, and cell cycle arrest, underscores its
potential not only as a tumor suppressor but also as a promising
diagnostic and prognostic biomarker. Future research should
prioritize translational applications of SEMA3B. For example,
exploring whether SEMA3B methylation status or expression levels
can stratify patients for targeted therapy or predict outcomes in
combination with conventional treatments represents a compelling
direction. Moreover, given its role in modulating the TME,
particularly through IL-8-mediated macrophage recruitment, SEMA3B
may interface with immuno-oncology mechanisms. Investigating its
interplay with immune checkpoint molecules, T-cell infiltration, or
response to immunotherapy could unveil novel combination strategies
to enhance antitumor immunity. In summary, continued in-depth
investigation of SEMA3B promises to unravel its complex mechanisms,
facilitate the discovery of novel biomarkers, and inspire
innovative therapeutic approaches that integrate its
tumor-suppressive functions with emerging modalities in precision
oncology and immuno-oncology.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
XZ and XX conceived the study. XX, XZ and RF
performed the formal analysis and data interpretation, conducted
the literature analysis, wrote the original draft and supervised
the study. HZ, XX, XZ and JW reviewed and edited of the manuscript.
RF and JW were responsible for project administration. All authors
read and approved the final version of the manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pang CH, Du W, Long J and Song LJ:
Mechanism of SEMA3B gene silencing and clinical significance in
glioma. Genet Mol Res: 15, 2016 doi: 10.4238/gmr.15017664.
|
|
2
|
Qiuju W, Liya Z, Yan S and Yuanyuan W:
Effects of miR-335-5p targeting SEMA3B on proliferation and
apoptosis of endometrial cancer cells. Shaanxi Med J. 52:798–802.
2023.
|
|
3
|
Zhang X, Klamer B, Li J, Fernandez S and
Li L: A pan-cancer study of class-3 semaphorins as therapeutic
targets in cancer. BMC Med Genomics. 13(45)2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Li GZ, Shen D, Li GH, Wei M, Zheng LJ, Liu
ZL, Sun RQ, Zhou SJ, Zhang ZL and Gao YC: Decreased expression of
serum semaphorin 3B is associated with poor prognosis of patients
with hepatocellular carcinoma. Exp Ther Med. 21(236)2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Mastrantonio R, You H and Tamagnone L:
Semaphorins as emerging clinical biomarkers and therapeutic targets
in cancer. Theranostics. 11:3262–3277. 2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Toledano S, Nir-Zvi I, Engelman R, Kessler
O and Neufeld G: Class-3 semaphorins and their receptors: Potent
multifunctional modulators of tumor progression. Int J Mol Sci.
20(556)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Alto LT and Terman JR: Semaphorins and
their signaling mechanisms. Methods Mol Biol. 1493:1–25.
2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Valiulyte I, Steponaitis G, Kardonaite D,
Tamasauskas A and Kazlauskas A: A SEMA3 signaling pathway-based
multi-biomarker for prediction of glioma patient survival. Int J
Mol Sci. 21(7396)2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Fernández-Nogueira P, Linzoain-Agos P,
Cueto-Remacha M, De la Guia-Lopez I, Recalde-Percaz L, Parcerisas
A, Gascon P, Carbó N, Gutierrez-Uzquiza A, Fuster G and Bragado P:
Role of semaphorins, neuropilins and plexins in cancer progression.
Cancer Lett. 606(217308)2024.PubMed/NCBI View Article : Google Scholar
|
|
10
|
de Visser KE and Joyce JA: The evolving
tumor microenvironment: From cancer initiation to metastatic
outgrowth. Cancer Cell. 41:374–403. 2023.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Chen T, Li S and Wang L: Semaphorins in
tumor microenvironment: Biological mechanisms and therapeutic
progress. Int Immunopharmacol. 132(112035)2024.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wagner W, Ochman B and Wagner W:
Semaphorin 6 Family-an important yet overlooked group of signaling
proteins involved in cancerogenesis. Cancers (Basel).
15(5536)2023.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Meng Z, Li FL, Fang C, Yeoman B, Qiu Y,
Wang Y, Cai X, Lin KC, Yang D, Luo M, et al: The Hippo pathway
mediates Semaphorin signaling. Sci Adv. 8(eabl9806)2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Dong Z, Liang X, Wu X, Kang X, Guo Y, Shen
S, Liang J and Guo W: Promoter hypermethylation-mediated
downregulation of tumor suppressor gene SEMA3B and lncRNA
SEMA3B-AS1 correlates with progression and prognosis of esophageal
squamous cell carcinoma. Clin Exp Metastasis. 36:225–241.
2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Gaur P, Bielenberg DR, Samuel S, Bose D,
Zhou Y, Gray MJ, Dallas NA, Fan F, Xia L, Lu J and Ellis LM: Role
of class 3 semaphorins and their receptors in tumor growth and
angiogenesis. Clin Cancer Res. 15:6763–6770. 2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Varshavsky A, Kessler O, Abramovitch S,
Kigel B, Zaffryar S, Akiri G and Neufeld G: Semaphorin-3B is an
angiogenesis inhibitor that is inactivated by furin-like
pro-protein convertases. Cancer Res. 68:6922–6931. 2008.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Jialing Z, Qun H, Lifei B and Xiulan S:
Progress in the study of Semaphorin3 antitumor mechanism. J Modern
Oncol. 16:312–314. 2008.
|
|
18
|
Zhou Y, Gunput RA and Pasterkamp RJ:
Semaphorin signaling: Progress made and promises ahead. Trends
Biochem Sci. 33:161–170. 2008.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Schwarz Q, Waimey KE, Golding M, Takamatsu
H, Kumanogoh A, Fujisawa H, Cheng HJ and Ruhrberg C: Plexin A3 and
plexin A4 convey semaphorin signals during facial nerve
development. Dev Biol. 324:1–9. 2008.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Tam KJ, Hui DHF, Lee WW, Dong M, Tombe T,
Jiao IZF, Khosravi S, Takeuchi A, Peacock JW, Ivanova L, et al:
Semaphorin 3 C drives epithelial-to-mesenchymal transition,
invasiveness, and stem-like characteristics in prostate cells. Sci
Rep. 7(11501)2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Bica C, Tirpe A, Nutu A, Ciocan C, Chira
S, Gurzau ES, Braicu C and Berindan-Neagoe I: Emerging roles and
mechanisms of semaphorins activity in cancer. Life Sci.
318(121499)2023.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Tingzhe S: Synergistic effect of
hyperthermia combined with radiotherapy in tumor therapy based on
mathematical model of p53 signal transduction network. J China
Pharmaceutical Univ. 52:361–370. 2021.
|
|
23
|
Rui S, Peixin X and Lin D: Establishment
of a human lung adenocarcinoma A549 cell model knockdown by tumor
suppressor gene p53 and its functional study. Chin J Diagnost
Pathol. 29:323–325. 2022.
|
|
24
|
Jing Y: Expression of p16, p53, Ki-67
protein in cervical carcinoma and its correlation with
clinicopathological features. Clin Med. 44:42–44. 2024.
|
|
25
|
Ochi K, Mori T, Toyama Y, Nakamura Y and
Arakawa H: Identification of semaphorin3B as a direct target of
p53. Neoplasia. 4:82–87. 2002.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Chen R, Zhuge X, Huang Z, Lu D, Ye X, Chen
C, Yu J and Lu G: Analysis of SEMA3B methylation and expression
patterns in gastric cancer tissue and cell lines. Oncol Rep.
31:1211–1218. 2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Tang H, Wu Y, Liu M, Qin Y, Wang H, Wang
L, Li S, Zhu H, He Z, Luo J, et al: SEMA3B improves the survival of
patients with esophageal squamous cell carcinoma by upregulating
p53 and p21. Oncol Rep. 36:900–908. 2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Castro-Rivera E, Ran S, Brekken RA and
Minna JD: Semaphorin 3B inhibits the phosphatidylinositol
3-kinase/Akt pathway through neuropilin-1 in lung and breast cancer
cells. Cancer Res. 68:8295–8303. 2008.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Fumarola C, Bonelli MA, Petronini PG and
Alfieri RR: Targeting PI3K/AKT/mTOR pathway in non small cell lung
cancer. Biochem Pharmacol. 90:197–207. 2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Huang GS, Brouwer-Visser J, Ramirez MJ,
Kim CH, Hebert TM, Lin J, Arias-Pulido H, Qualls CR, Prossnitz ER,
Goldberg GL, et al: Insulin-like growth factor 2 expression
modulates Taxol resistance and is a candidate biomarker for reduced
disease-free survival in ovarian cancer. Clin Cancer Res.
16:2999–3010. 2010.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Sherr CJ and Weber JD: The ARF/p53
pathway. Curr Opin Genet Dev. 10:94–99. 2000.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Rolny C, Capparuccia L, Casazza A, Mazzone
M, Vallario A, Cignetti A, Medico E, Carmeliet P, Comoglio PM and
Tamagnone L: The tumor suppressor semaphorin 3B triggers a
prometastatic program mediated by interleukin 8 and the tumor
microenvironment. J Exp Med. 205:1155–1171. 2008.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Yunyan G and Xilong O: Progress in the
study of the relationship between VEGF165 and VEGF 165b and Tumor.
Modern Med J. 34:447–450. 2006.
|
|
34
|
de Castro Junior G, Puglisi F, de Azambuja
E, El Saghir NS and Awada A: Angiogenesis and cancer: A cross-talk
between basic science and clinical trials (the ‘do ut des’
paradigm). Crit Rev Oncol Hematol. 59:40–50. 2006.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Abou Faycal C, Gazzeri S and Eymin B: A
VEGF-A/SOX2/SRSF2 network controls VEGFR1 pre-mRNA alternative
splicing in lung carcinoma cells. Sci Rep. 9(336)2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Wiszniak S and Schwarz Q: Exploring the
intracrine functions of VEGF-A. Biomolecules. 11(128)2021.DOI:
10.3390/biom11010128.
|
|
37
|
Apte RS, Chen DS and Ferrara N: VEGF in
signaling and disease: Beyond discovery and development. Cell.
176:1248–1264. 2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Chen Y, Mathy NW and Lu H: The role of
VEGF in the diagnosis and treatment of malignant pleural effusion
in patients with non-small cell lung cancer (Review). Mol Med Rep.
17:8019–8030. 2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Min Z, Liqiang X, Huizhi Y, Nan Y and Guo
W: Expression of VEGF in non-small cell lung cancer and its
correlation with pathological characteristics. J Modern Oncol.
24:3386–3389. 2016.
|
|
40
|
Wang YQ, Chen H, Xu S, Liao CR, Xu A, Han
Y, Yang MH, Zhao L, Hu SS, Wang L, et al: SEMA3B-AS1 suppresses
colorectal carcinoma progression by inhibiting Semaphorin
3B-dependent VEGF signaling pathway activation. MedComm (2020).
4(e365)2023.PubMed/NCBI View
Article : Google Scholar
|
|
41
|
Castro-Rivera E, Ran S, Thorpe P and Minna
JD: Semaphorin 3B (SEMA3B) induces apoptosis in lung and breast
cancer, whereas VEGF165 antagonizes this effect. Proc Natl Acad Sci
USA. 101:11432–11437. 2004.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Wang K, Ling T, Wu H and Zhang J:
Screening of candidate tumor-suppressor genes in 3p21.3 and
investigation of the methylation of gene promoters in oral squamous
cell carcinoma. Oncol Rep. 29:1175–1182. 2013.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Pronina IV, Loginov VI, Burdennyy AM,
Fridman MV, Kazubskaya TP, Dmitriev AA and Braga EA: Expression and
DNA methylation alterations of seven cancer-associated 3p genes and
their predicted regulator miRNAs (miR-129-2, miR-9-1) in breast and
ovarian cancers. Gene. 576:483–491. 2016.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Guo W, Liang X, Liu L, Guo Y, Shen S,
Liang J and Dong Z: MiR-6872 host gene SEMA3B and its antisense
lncRNA SEMA3B-AS1 function synergistically to suppress gastric
cardia adenocarcinoma progression. Gastric Cancer. 22:705–722.
2019.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Li K, Chen MK, Li LY, Lu MH, Shao ChK, Su
ZL, He D, Pang J and Gao X: The predictive value of semaphorins 3
expression in biopsies for biochemical recurrence of patients with
low- and intermediate-risk prostate cancer. Neoplasma. 60:683–689.
2013.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Loginov VI, Dmitriev AA, Senchenko VN,
Pronina IV, Khodyrev DS, Kudryavtseva AV, Krasnov GS, Gerashchenko
GV, Chashchina LI, Kazubskaya TP, et al: Tumor suppressor function
of the SEMA3B gene in human lung and renal cancers. PLoS One.
10(e0123369)2015.PubMed/NCBI View Article : Google Scholar
|














