Introduction
Normally, proliferation ceases with a G0/G1 phase
arrest of cell cycle, when cells physically contact neighboring
cells and upon reaching confluence. This fundamental regulatory
process is termed contact inhibition of proliferation (CIP), or
simply, contact inhibition (1).
This intrinsic control mechanism ensures that cells within a tissue
proliferate in a coordinated manner and maintain appropriate
spatial organization, prevent uncontrolled cell division and
excessive accumulation, which could lead to the formation of
disorganized masses or tumors. Cell losses of CIP are more
susceptible to malignant transformation and hyperplasia, and such
phenomenon has widely been observed in cancer cells (1).
E-Cad and its downstream signaling play a crucial
role in regulating CIP (2). The
failure to respond appropriately of E-Cad in cancer cells
contributes to dysregulation of CIP, while it could be reversed by
restoring E-Cad expression (3).
Meanwhile, disruption of E-Cad binding between cells using
antibodies also reversed CIP (4).
Studies have revealed not only E-Cad/catenin adhesion complex, but
also E-Cad homophilic ligation are sufficient to regulate CIP
(5,6). Despite incomplete understanding of
how E-Cad ligation and cytoskeletal tension collaborate to achieve
CIP, Hippo pathway, the intracellular signal cascade of E-Cad, has
been thoroughly studied (7). Upon
activation by E-Cad, YAP undergoes phosphorylation, which retains
it in the cytoplasm and thereby suppresses cell proliferation.
Inactive Hippo signaling allows YAP to migrate into the nucleus,
interact with TEADs, and stimulate target gene transcription and
cell proliferation (2).
Numerous oncogenes have been documented to enhance
cell proliferation, including notably, CD276. CD276, also known as
B7-H3, is a member of the B7 immune checkpoint family of membrane
protein and was initially characterized as a costimulatory molecule
on T-cells activation (8).
However, CD276 promotes tumor progression either through inducing
immunosuppression or non-immunological mechanisms.
Non-immunologically, it promotes cancer cell proliferation,
invasion and metastasis through activating PI3K/AKT, Jak2/Stat3 and
MEK pathway and induces epithelial-to-mesenchymal transition (EMT)
via Jak2/Stat3/Slug (9-15).
Additionally, it facilitates tumor progression by enhancing tumor
resistance to radiotherapy and chemotherapy, augmenting
angiogenesis, enhancing aerobic glycolysis, and acting as an active
component of tumor-derived exosomes (16,17).
With high prevalence of CD276 on malignant cells, inhibitors
targeting CD276 are becoming a new class of antineoplastic agents,
which show early promising results in solid tumor malignancies
(18,19).
SIRT1, an NAD-dependent histone deacetylase, has
been implicated in various stages of cancer, particularly playing a
pivotal role in regulating cellular proliferation (20). A signal axis of SIRT1-p27KIP was
reported to control CIP (21).
Recently, Liao et al (22)
demonstrated that SIRT1 acts as a downstream effector molecule in
CD276-induced epithelial-mesenchymal transition process.
Considering CD276's established role in facilitating cancer cell
proliferation, the present study aimed to elucidate its potential
function in enabling cancer cells to overcome CIP, and
specifically, whether this mechanism is orchestrated via the
activation of SIRT1.
Consequently, it was found that CD276 inversely
correlates with E-Cad and positively correlates with overcoming
CIP. Increased CD276 reduces G0/G1 arrest under high density by
modulating SIRT1 via PI3K signaling. This defined a
CD276/PI3K/SIRT1/E-Cad pathway critical for hepatocellular
carcinoma cells (HCC) to evade CIP.
Materials and methods
Cell culture
Immortalized human hepatocyte cell line (HepLi5) was
kindly provided by Prof. Li (23).
Human HCC cell lines (SNU-449, HuH-7, Hep 3B2.1-7 and PLC/PRF/5)
were obtained from Cell Bank of the Chinese Academy of Sciences
(Shanghai, China). The cells were maintained in high-glucose DMEM
(cat. no. L100-500; BDBIO; https://www.biocode.cn/product-L100-500) supplemented
with 10% fetal calf serum (cat. no. F101-01; Vazyme Biotech Co.,
Ltd.; https://bio.vazyme.com/product/732.html) and incubated
at 37˚C in a humidified chamber containing 5% CO2.
Cell seeded for spares or confluent
culture
The number of cells seeded was calculated based on
the total cell count when the monolayer reached 100% confluence for
each cell line. For sparse cultures, cells were seeded at 30% of
this density, while for confluent cultures, the seeding density was
set at 80%. Then, the cells were cultured for ~36 h following
seeding and subsequently harvested for processing or analysis.
Cell cycle analysis
Adherent cells were detached using Trypsin-EDTA
solution (cat. no. A300-100; BDBIO; https://www.biocode.cn/product-A300-100), rinsed once
with phosphate-buffered saline (PBS), and pelleted at 300 x g for 5
min. After supernatant removal, cells were resuspended in 1 ml of
PBS. Thereafter, 3 ml of pre-chilled ethanol was added stepwise,
and the suspension was stored at -20˚C overnight. The next day,
cells underwent centrifugation at 600 x g for 10 min, followed by
supernatant disposal. This washing procedure was repeated by adding
1 ml of PBS, vortex, centrifuging, and discarding the supernatant.
Afterward, cells were stained with Cell Cycle Staining Buffer (cat.
no. CCS012; Multi Sciences Biotech) in darkness for 30 min.
Following that, cells were rinsed with PBS, and data acquisition
was carried out using a flow cytometer (Canto II; BD Biosciences).
Collected data was analyzed using ModFit LT software (version 5.0,
Verity Software House, Inc.) for further analysis.
CD276 overexpression
The open reading frame for overexpressing CD276 was
generated based on the sequence NM_001024736.2. The coding sequence
was cloned into the pcDNA3.1-3xFlag-C vector to generate the
overexpression construct. The empty pcDNA3.1-3xFlag-C vector was
utilized as a negative control. These molecular cloning procedures
were conducted by Guangzhou RiboBio Co., Ltd. On Day 0, a total of
2x106 cells were seeded into a 35 mm dish with 2 ml of
serum-supplemented DMEM. Proceeding to Day 1, once cell confluence
reached ~50%, 2 µg of plasmid DNA were diluted in 200 µl of
jetPRIME buffer (cat. no. 101000046, POLYPLUS-TRANSFECTION S.A;
https://shop.sartorius.com.cn/cn/p/jetprime-dnasirna-co-transfection/jetPRIME_DNA_siRNA_co_transfection),
followed by vortex and brief centrifugation. Next, 4 µl of jetPRIME
reagent were added, vortexed again and centrifuged at 4˚C and 200 g
for 30 sec before allowing a 10 min incubation at room temperature
to stabilize the mixture. This transfection mixture was
subsequently introduced to the cells in serum-containing DMEM and
left to incubate for 48 h at 37˚C in a humidified chamber
containing 5% CO2. On Day 3, the cells were harvested
for sparse and confluent cultures.
CD276 knockdown
Small interfering RNA (siRNA) sequences were as
follows: CD276 siRNA forward, 5'-GUGUGCUGGAGAAAGAUCAdTdT-3' and
reverse, 5'-UGAUCUUUCUCCAGCACACdTdT-3'; and negative control
forward, 5'-UUCUCCGAACGUGUCACGUdTdT-3' and reverse,
5'-ACGUGACACGUUCGGAGAAdTdT-3'. siRNAs were synthesized by Guangzhou
RiboBio Co., Ltd. On Day 0, a total of 1x106 cells were
seeded into a 35 mm dish with 2 ml of serum-supplemented DMEM.
Proceeding to Day 1, once cell confluence reached ~50%, 22 pmol of
siRNA were diluted in 200 µl of jetPRIME buffer, followed by vortex
and centrifuged at 4˚C and 200 g for 30 sec. Next, 4 µl of jetPRIME
reagent were added, vortexed again, and centrifuged at 4˚C and 200
g for 30 sec before allowing a 10 min incubation at room
temperature to stabilize the mixture. This transfection mixture was
subsequently introduced to the cells in serum-containing DMEM and
left to incubate for 48 h at 37˚C in a humidified chamber
containing 5% CO2. On Day 3, the cells were harvested
for sparse and confluent cultures.
Protein extraction, immunoblotting
assay and quantification of protein expression levels
Cell cultures were washed with pre-chilled PBS, then
lysed using RIPA buffer (cat. no. P0013B; Beyotime Institute of
Biotechnology). The lysate was incubated on ice for 10 min prior to
centrifugation at 14,000 x g at 4˚C for 15 min. Then the
supernatant was collected, and protein concentrations were
quantified utilizing a BCA assay kit (cat. no. 23225; Pierce;
Thermo Fisher Scientific, Inc.). Equivalent amounts of protein (20
µg per lane) were subjected to 4-12% SurePAGE (cat. no. M00656;
GenScript Biotech Corporation) and subsequently transferred to PVDF
membranes (cat. no. ISEQ00010; MilliporeSigma) utilizing the eBlot
L1 Fast Wet Transfer System (GenScript Biotech Corporation).
Membranes were then blocked with 5% non-fat milk for 1 h at room
temperature and incubated overnight at 4˚C with primary antibodies:
CD276 (1:1,000; cat. no. A17216), E-Cad (1:2,000; cat. no. A24874),
p27 (1:1,000; cat. no. A0290), cyclin D1 (1:1,000; cat. no.
A11310), GAPDH (1:50,000; cat. no. A19056) and SIRT1 (1:2,000; cat.
no. A0230; all from ABclonal Biotech Co., Ltd.). After the primary
antibody incubation, membranes were incubated with HRP-conjugated
secondary antibody (1:10,000; cat. no. RS0002; ImmunoWay
Biotechnology Company) at room temperature for 1 h. Finally, ECL
enhanced kit (cat. no. RM00021; ABclonal Biotech Co., Ltd.) was
used for detecting protein bounds, and signals were captured by
images capturer (ChemiScope, Clinx).
To ensure consistency in protein expression trends,
an additional independent biological replicate or cell transfection
was performed in duplicate. The resultant data were subsequently
employed to validate the previously documented expression patterns
(Fig. S1). Protein expression
levels were quantified using AlphaView software (ProteinSimple).
The loading control proteins were first normalized, followed by
calculation of fold change for the target proteins.
Small-molecule inhibitor
The PI3K inhibitor LY294002 (cat. no. HY-10108; MCE;
https://www.medchemexpress.cn/LY294002.html) was
prepared as a 20 mmol stock solution in dimethyl sulfoxide (DMSO).
For further studies, cells were treated with 20 µmol LY294002 for
24 h, while control cells received an equivalent volume of
DMSO.
Statistical analysis
Quantitative results are expressed as the mean,
accompanied by the standard deviation, calculated from three
independent experiments. Statistical analyses were carried out
using GraphPad Prism (Version 9.5.0; GraphPad Software, Inc.;
Dotmatics). Comparisons between two groups were made using an
unpaired t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
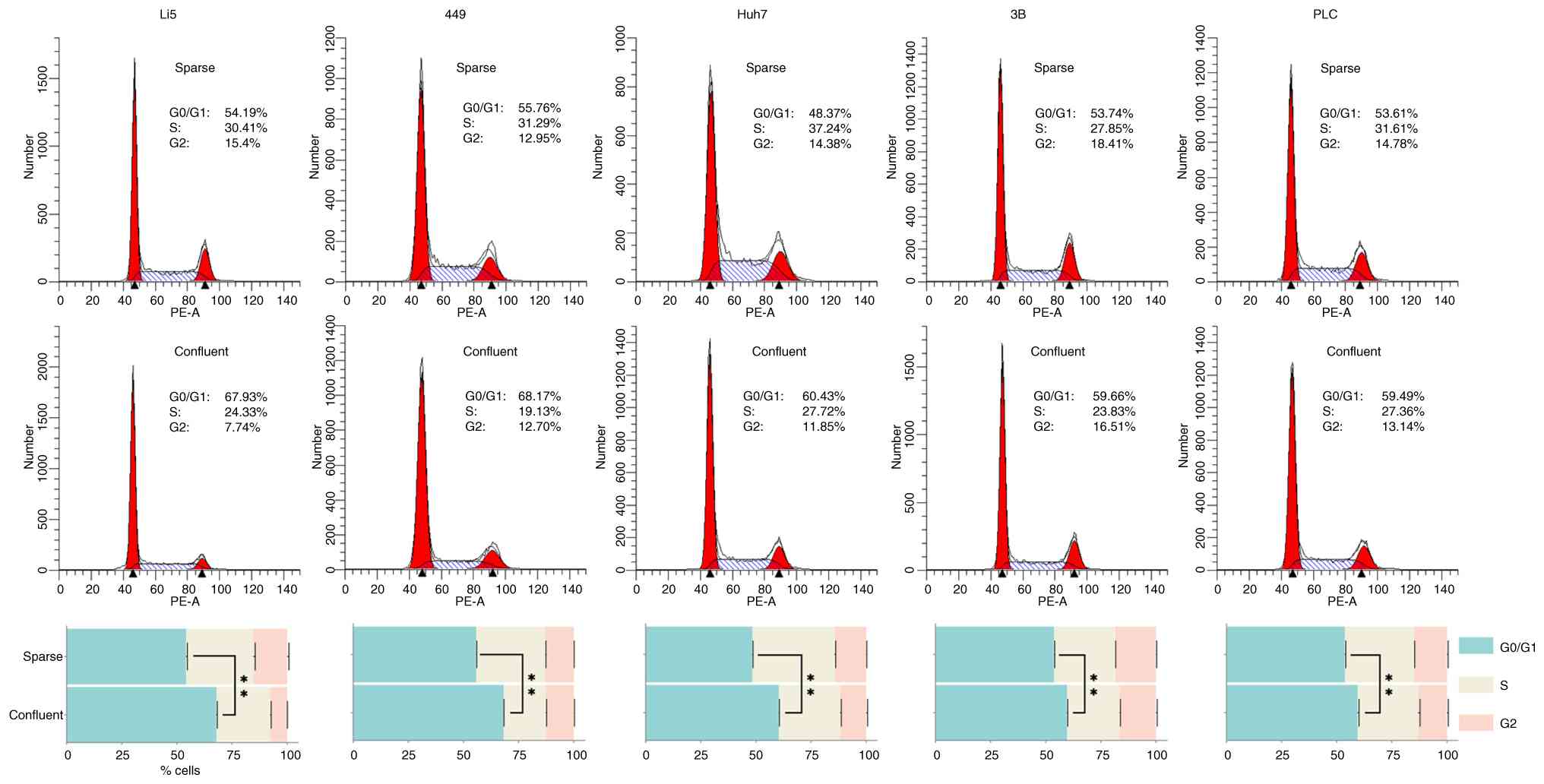
The magnitude of G0/G1 phase arrest
induced by cell-cell contact exhibits variability among HCC
lines
Cancer cells are known to be insensitive to CIP;
initially, the responses of four HCC lines, SNU-449, HuH-7, Hep
3B2.1-7 and PLC/PRF/5, and an immortalized human hepatocyte cell
line (HepLi5) to high-density cell-cell contact were investigated.
As expected, confluent HepLi5 exhibited G0/G1 phase arrest with a
proportion of cells in G0/G1 phase increased from 54.19 to 67.93%,
compared with sparse cultures (Fig.
1). Nonetheless, all 4 HCC cell lines exhibited varying
magnitudes of G0/G1 phase arrest as well; specifically, SNU-449 and
HuH-7 suffered more severe inhibition (increased from 55.76 to
68.17%, and 48.37 to 60.43% respectively), contrasting with milder
extents observed in Hep 3B2.1-7 and PLC/PRF/5 (increased from 53.74
to 59.66%, and 53.61 to 59.49% respectively) (Fig. 1).
CD276 expression coordinates with
E-Cad and expression of cell cycle markers
CD276 has been confirmed to promote the
proliferation of cancer cells, but whether it plays a role in CIP
is currently unclear. Therefore, to investigate this, the
alterations of CD276 expression within these cells under confluent
culture conditions were examined. The expression of CD276 was
increased in the four HCC cell lines, whereas no such elevation was
found in HepLi5 (Fig. 2).
Moreover, CD276 expression demonstrated a more substantial
upregulation in Hep 3B2.1-7 and PLC/PRF/5 relative to SNU-449 and
Huh-7 (Fig. 2). Intriguingly,
cells exhibiting prominent CIP, namely SNU-449, Huh-7 and HepLi5,
concurrently displayed notable elevations in E-Cad expression
(Fig. 2). Subsequently,
alterations of cell cycle-associated markers, such as p27 and
cyclin D1, were also found (Fig.
2). These findings suggest that elevated CD276 expression might
help cells overcome CIP, possibly through the modulation of E-Cad
expression.
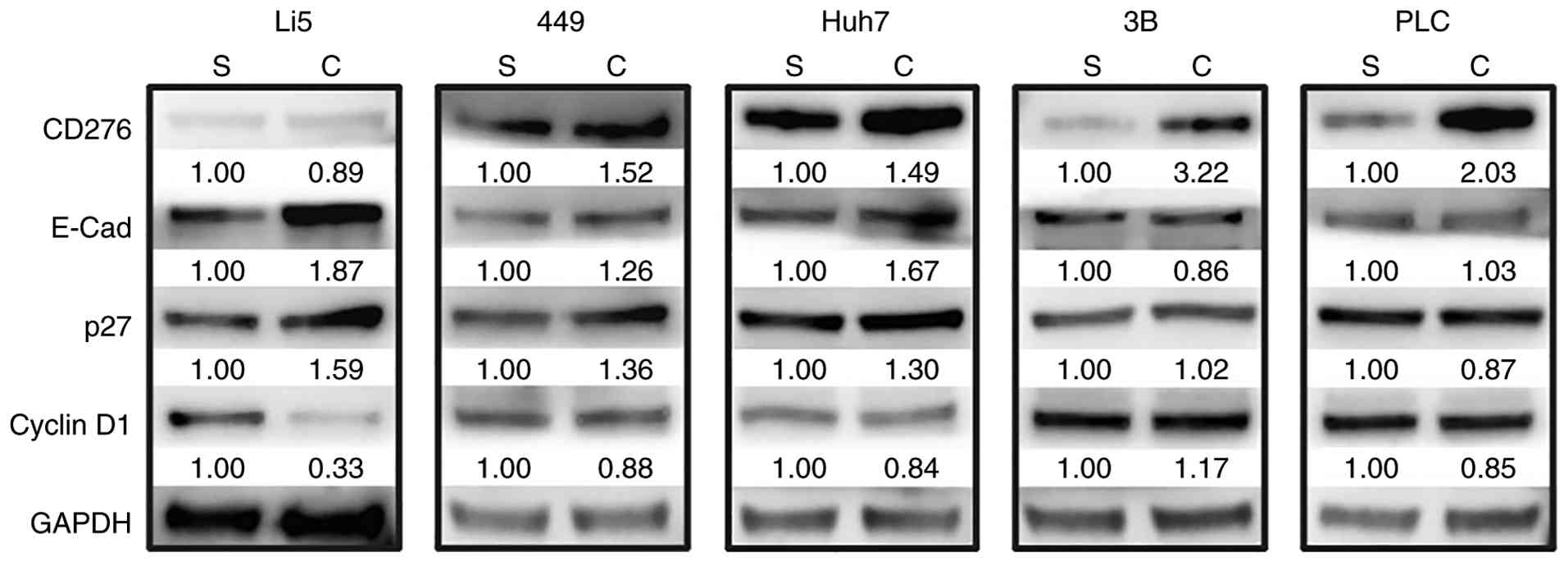 | Figure 2Alterations in the expression of CD276
and cell cycle-related proteins were observed after the occurrence
of CIP. The expression of CD276, E-Cad, p27 and cyclin D1 in HepLi5
and the four HCC cell lines (449, Huh-7, Hep 3B2.1-7 and PLC/PRF/5)
was analyzed by western blotting. GAPDH was used as a loading
control. CIP, contact inhibition of proliferation; 449, SNU-449;
3B, Hep 3B2.1-7; PLC, PLC/PRF/5; S, sparse cultures; C, confluent
cultures. |
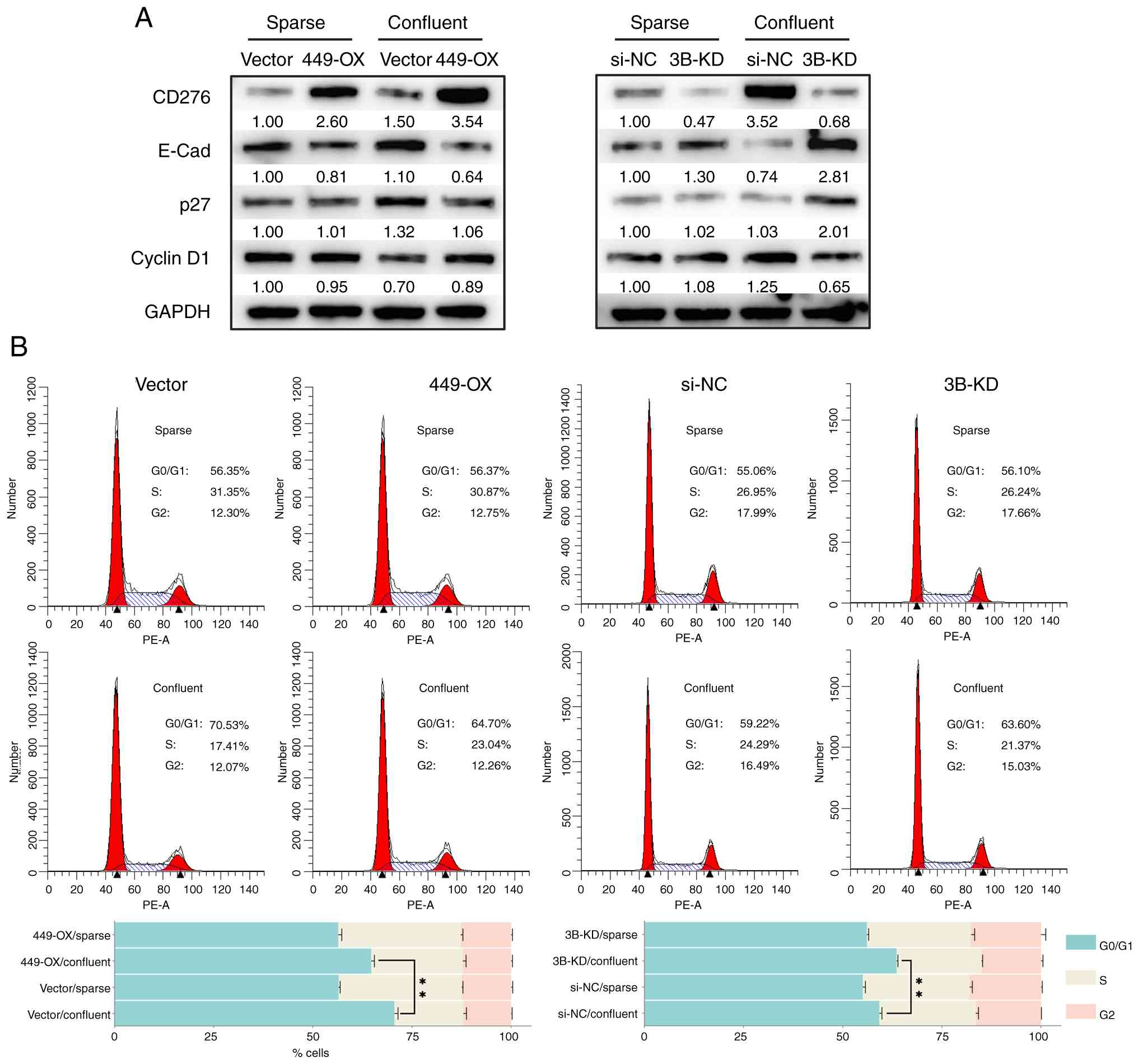
Depending on high-density cell-cell
contact, CD276 emerges as a critical determinant in regulating
E-Cad expression
To further investigate the impact of CD276 on E-Cad
expression, cell lines with either CD276 overexpression or
knockdown were established. For SNU-449 cells which were sensitive
to CIP, CD276 (abbreviated as 449-OX in following context) was
overexpressed. While for Hep 3B2.1-7 cells, which were less
sensitive to CIP and showed a marked increase in CD276 expression
responding to cell-cell contact, CD276 expression [abbreviated as
CD276-knockdown Hep 3B2.1-7 cells (3B-KD) in following context] was
suppressed. Under sparse culture conditions, CD276 expression was
markedly enhanced in 449-OX cells and suppressed in 3B-KD cells,
respectively. Under confluent culture conditions, CD276 expression
was further increased in 449-OX cells, while it remained markedly
suppressed in 3B-KD cells (Fig.
3A). Under sparse culture conditions, the difference in CD276
expression caused only slight changes in E-Cad expression (Fig. 3A). However, under confluent
conditions, as the difference in CD276 expression became more
pronounced, the difference in E-Cad expression also became more
evident, manifesting as suppressed E-Cad expression when CD276 was
highly expressed and enhanced E-Cad expression when CD276
expression was reduced (Fig. 3A).
These results indicate that CD276 plays a role in regulating E-Cad
expression. Similarly, under confluent culture conditions, more
pronounced differences in p27 and cyclin D1 expression were
observed in confluent 449-OX and 3B-KD cells (Fig. 3A). Consistent with the changes in
p27 and cyclin D1 expression, confluent 449-OX cells exhibited a
markedly reduced G0/G1 phase arrest compared with control cells;
meanwhile, G0/G1 phase arrest was more pronounced in the 3B-KD
cells (Fig. 3B). The cell cycle
profiles of 449-OX and 3B-KD cells were further studied, revealing
no discernible variations in the proportion of cells in G0/G1 phase
compared with control cells when cultured under sparse conditions
(Fig. 3B). Consequently, the
findings of the present study suggested that CD276's regulation of
E-Cad expression is highly dependent on high-density cell-cell
contact.
SIRT1 is a regulator in the cascade of
CD276 mediated E-Cad expression
To ascertain whether CD276 could regulate cell
proliferation via SIRT1, the expression of SIRT1 under confluent
culture conditions was examined and a downregulation of SIRT1 was
observed in HepLi5 cells, whereas no such decline was found in two
HCC cell lines, SNU-449 and Hep 3B2.1-7 (Fig. 4A). Subsequently, in 449-OX cells,
an elevation was found in SIRT1 expression when cultured under
confluent conditions (Fig. 4B); by
contrast, in 3B-KD cells, SIRT1 expression decreased (Fig. 4C), with its expression change being
consistent with that of CD276 (Figs.
4B and 3A). Moreover, when
LY294002, an inhibitor of PI3K pathway, was added, the increase in
SIRT1 expression in 449-OX cells was abolished, while LY294002 did
not significantly affect CD276 expression (Fig. 4B). These findings strongly
suggested that CD276 indeed exerts a regulatory effect on SIRT1,
with the modulation occurring via the PI3K/AKT pathway.
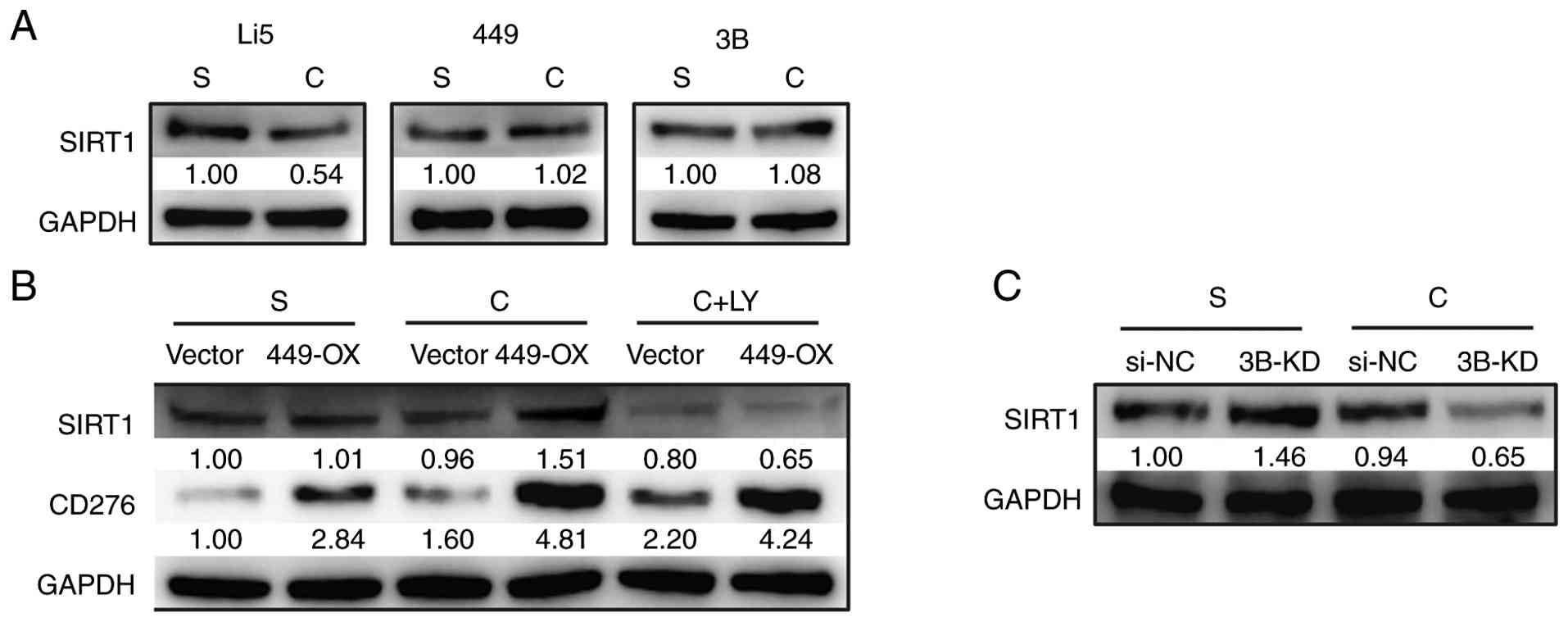 | Figure 4SIRT1 acts as a downstream regulator
of CD276 in mediating E-Cad. (A) The expression of SIRT1 was
analyzed by western blotting in HepLi5, SNU-449 and 3B cell lines
under sparse or confluent conditions. (B) The expression of SIRT1
in 449-OX cells was analyzed by western blotting under three
conditions: sparse, confluent, and confluent with additional
treatment of LY294002. (C) The expression of SIRT1 was analyzed by
western blotting in 3B-KD cells under sparse and confluent
conditions. 3B, Hep 3B2.1-7; 449-OX, CD276-ovexpressing SNU-449
cells; Li5, HepLi5; 449, SNU-449; 3B, Hep 3B2.1-7; S, sparse
cultures; C, confluent cultures; LY, LY294002; si-NC, small
interfering-negative control; 3B-KD, CD276-knockdown Hep 3B2.1-7
cells. |
Discussion
The present study investigated the role of CD276 in
proliferation inhibition induced by high-density cell-cell contact,
elucidating that CD276 modulates SIRT1 via the PI3K/AKT
pathway, leading to suppressed E-Cad expression and thereby
partially alleviating CIP. Previous studies have established
CD276's capability to enhance cellular proliferation through the
PI3K/AKT, Jak2/Stat3, and MEK pathways (9-11),
while the present study represented the first documentation of
CD276's function in enabling cancer cells to maintain their
proliferative potential by suppressing E-Cad during CIP.
Initially recognized as a member of the immune
checkpoint proteins, CD276 plays a pivotal role in regulating the
immune system, crucial for both immune tolerance and recognition
(18). Its ubiquitous expression
across various cancer cell types facilitates the evasion of these
cells from cytotoxic T-cell and natural killer cell surveillance
(24). Over time, accumulating
evidence has shed light on CD276's involvement in tumor
proliferation, metastasis, treatment resistance and poor prognosis
(17). More recently, studies have
shown the existence of a CD276/SIRT1 axis in cancer (22,25),
which either promotes EMT or suppresses growth arrest. Both the
investigations consistently revealed that CD276 modulates SIRT1
through AKT phosphorylation. Previously, Cho and Dai (21) confirmed that SIRT1 regulates E-Cad
expression, thereby influencing CIP. Given these precedents and
upon discovering CD276's potential involvement in CIP and its
capacity to regulate E-Cad, the authors were enlightened to
consider SIRT1's role, as a downstream factor of CD276, in this
process. Consequently, the investigation of the present study
successfully delineated a regulatory pathway involving
CD276/PI3K/SIRT1/E-Cad.
Considering the pivotal roles of CD276 in inducing
immune tolerance, facilitating immune evasion, and promoting tumor
proliferation and migration, therapeutic interventions targeting
CD276 have garnered significant interest. To date, numerous
clinical programs investigating CD276 inhibitors are currently
underway, including enoblituzumab, omburtamab, HS-20093, ifinatamab
deruxtecan and vobramitamab, all of which have consistently
demonstrated varying degrees of antitumor activity (17,19).
The therapeutic mechanisms of these agents are primarily based on
either the immunosuppressive function of CD276 or its pan-cancer
expression pattern. However, the present research on the role of
CD276 in CIP suggests that CD276 likely functions within the core
or central region of tumor masses, areas characterized by limited
blood supply. This implies that monoclonal antibodies and
antibody-drug conjugates currently in Phase 1/2 clinical trials may
not efficiently penetrate and act on the interior of such tumor
cell clusters. Nevertheless, developing potent small-molecule
inhibitors targeting CD276, especially when combined with targeted
nanoparticle delivery systems, could overcome this limitation. Such
an approach may inhibit tumor proliferation at an earlier stage and
holds significant promise for application in neoadjuvant
therapy.
The cascade that was validated in the present study,
involving CD276/PI3K/SIRT1/E-Cad, not only enriches the author's
comprehension of CD276's multifaceted functions but also expands
the potential applications of CD276-targeted therapies in cancer
treatment, suggesting new avenues for intervention in tumor
progression and potentially augmenting the effectiveness of
immunotherapies. In solid tumors, the proliferation of tumor cells
is accompanied by their ability to breach CIP imposed by cell-cell
interactions, ultimately leading to insensitivity to contact
inhibition in their proliferation. Tumor cells not only overcome
CIP but also evade contact inhibition of locomotion (CIL), enabling
them to leave the primary site and migrate (26). Given the ubiquitous nature of this
phenomenon across solid tumors, its regulatory mechanisms are
likely highly conserved. Therefore, the signaling axis mediated by
CD276 may also operate in other solid tumor types. The present
study is the first to identify the role of the CD276/E-Cad axis and
its downstream pathways in this process, providing new insights
into the pathogenesis and treatment of solid tumors.
In vitro tumor research based on
two-dimensional (2D) cell culture offers advantages such as
operational simplicity and low cost. Due to the absence of nutrient
heterogeneity, this system often provides high experimental
reproducibility. In the present study, a 2D, anchorage-dependent
culture system, was utilized. However, such a system might have
inherent limitations in the present work, notably the inadequate
cell junction formation and insufficient response to mechanical
cues (27,28), leading to physiological and
cellular response that differ from those observed in vivo.
Extensive research has underscored the critical role of mechanical
transduction in CIL and CIP (26).
Moreover, it poorly mimics tumor growth in 3D settings, as it
assumes spatial uniformity among cells, making it difficult to
accurately study internal tumor cell distribution and growth
dynamics. Consequently, numerous studies have shifted towards 3D
culture models to better investigate cell-cell contact, yielding
substantial insights (21,29,30).
These 3D systems partially mimic key aspects of solid tumors more
closely, such as structural complexity, central hypoxia and
gradients of oxygen and nutrients. Notably, 3D cultures enable
cells to receive contact stimuli from multiple orientations,
thereby providing a more accurate reflection of in vivo
conditions than 2D monolayer cultures and serving as a more
realistic model for studying contact inhibition (31). Despite inherent limitations, 2D
culture in the present study successfully unraveled CD276's role in
cell-cell contact inhibition. It was hypothesized that the efficacy
of the 2D culture system may stem from CD276's early engagement in
contact inhibition processes, which might be highly sensitive to
contact and occurs at the moment cells just make contact, thus
becoming observable within 36 h in the present study. Recently,
Sutton et al (32) reported
that membrane-localized 4Ig-CD276 enhances cancer cell
proliferation through a dimerization-dependent intrinsic signaling.
Insightfully, it is plausible that high-density cell-cell contact
fosters 4Ig-CD276 dimerization, triggering downstream activation,
although mere overexpression of CD276 alone may not elicit
substantial effects. Hence, while 3D cultures often provide a more
intricate simulation of in vivo conditions, the results of
the present study emphasized the utility of 2D models for capturing
early molecular events mediated by CD276 in contact inhibition,
potentially due to its swift response to initial cell contacts and
the need for specific structural configurations, such as
dimerization, to full regulatory functionality.
In vitro cell experiments often involve
prolonged culture durations, which can culminate in elevated cell
densities at the time of analysis, possibly activating contact
inhibition-related proteins and introducing bias into the outcomes.
A recent study on CD276's function in promoting EMT in esophageal
squamous cell carcinoma (33)
revealed enhanced E-Cad expression in CD276 knockdown cells;
conversely, suppression was observed in cells overexpressing CD276.
This observation differs slightly from the results of the present
study and might be attributed to a lower/higher cell density in the
culture system during protein harvest. Therefore, meticulous
management of cell density throughout experiments is crucial in
studies of cell cycle to ensure the validity of interpretations and
minimize artifacts attributable to contact inhibition.
In summary, employing a 2D culture system, a
regulatory pathway involving CD276/PI3K/SIRT1/E-Cad that governs
CIP was elucidated, thereby expanding the potential applications of
CD276-targeted therapies in cancer treatment. The findings of the
present study also indicated that CD276's involvement in cell cycle
regulation is contingent upon high-density cell-cell contact,
highlighting the importance of considering cellular context when
examining its function in proliferation control.
Supplementary Material
Second set of validated western blot
results. (A-C) The western blot results in Panels A, B, and C
represent an independent biological replicate or transfection
experiment corresponding to Figs.
2, 3A and 4 in the main text, respectively. Li5,
HepLi5; 449, SNU-449; 3B, Hep 3B2.1-7; S, sparse cultures; C,
confluent cultures; si-NC, small interfering-negative control;
3B-KD, CD276-knockdown Hep 3B2.1-7 cells; LY, LY294002.
Acknowledgements
The authors are grateful to Professor Lanjuan Li
(State Key Laboratory for Diagnosis and Treatment of Infectious
Diseases, The First Affiliated Hospital, Zhejiang University School
of Medicine, China.) for generously providing us with the
immortalized human hepatocyte cell line (HepLi5).
Funding
Funding: The present study was supported by the Non-profit
Central Research Institute Fund of Chinese Academy of Medical
Sciences (grant no. 20-PT320-02).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
XY performed experiments, validated data and wrote
the original draft. RS conducted statistical analysis. GC managed
and maintained the research data. HX supervised the study. LZ
conceived the study. SZ conceived and supervised the study. All
authors read and approved the final version of the manuscript. XY
and SZ confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ribatti D: A revisited concept: Contact
inhibition of growth. From cell biology to malignancy. Exp Cell
Res. 359:17–19. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Mendonsa AM, Na TY and Gumbiner BM:
E-cadherin in contact inhibition and cancer. Oncogene.
37:4769–4780. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
St Croix B, Sheehan C, Rak JW, Florenes
VA, Slingerland JM and Kerbel RS: E-Cadherin-dependent growth
suppression is mediated by the cyclin-dependent kinase inhibitor
p27(KIP1). J Cell Biol. 142:557–571. 1998.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Motti ML, Califano D, Baldassarre G,
Celetti A, Merolla F, Forzati F, Napolitano M, Tavernise B, Fusco A
and Viglietto G: Reduced E-cadherin expression contributes to the
loss of p27kip1-mediated mechanism of contact inhibition in thyroid
anaplastic carcinomas. Carcinogenesis. 26:1021–1034.
2005.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Lin WH, Cooper LM and Anastasiadis PZ:
Cadherins and catenins in cancer: Connecting cancer pathways and
tumor microenvironment. Front Cell Dev Biol.
11(1137013)2023.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Perrais M, Chen X, Perez-Moreno M and
Gumbiner BM: E-cadherin homophilic ligation inhibits cell growth
and epidermal growth factor receptor signaling independently of
other cell interactions. Mol Biol Cell. 18:2013–2025.
2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zeng Q and Hong W: The emerging role of
the hippo pathway in cell contact inhibition, organ size control,
and cancer development in mammals. Cancer Cell. 13:188–192.
2008.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Chapoval AI, Ni J, Lau JS, Wilcox RA,
Flies DB, Liu D, Dong H, Sica GL, Zhu G, Tamada K and Chen L:
B7-H3: a costimulatory molecule for T cell activation and IFN-gamma
production. Nat Immunol. 2:269–274. 2001.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Flem-Karlsen K, Tekle C, Andersson Y,
Flatmark K, Fodstad O and Nunes-Xavier CE: Immunoregulatory protein
B7-H3 promotes growth and decreases sensitivity to therapy in
metastatic melanoma cells. Pigment Cell Melanoma Res. 30:467–476.
2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Zhong C, Tao B, Chen Y, Guo Z, Yang X,
Peng L, Xia X and Chen L: B7-H3 regulates glioma growth and cell
invasion through a JAK2/STAT3/Slug-Dependent signaling pathway.
Onco Targets Ther. 13:2215–2224. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wu X, Ding C, Liu Y, Dong K and Zhang H:
B7-H3 promotes proliferation and migration of lung cancer cells by
modulating PI3K/AKT pathway via ENO1 activity. Transl Cancer Res.
13:833–846. 2024.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kang FB, Wang L, Jia HC, Li D, Li HJ,
Zhang YG and Sun DX: B7-H3 promotes aggression and invasion of
hepatocellular carcinoma by targeting epithelial-to-mesenchymal
transition via JAK2/STAT3/Slug signaling pathway. Cancer Cell Int.
15(45)2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Jiang B, Zhang T, Liu F, Sun Z, Shi H, Hua
D and Yang C: The co-stimulatory molecule B7-H3 promotes the
epithelial-mesenchymal transition in colorectal cancer. Oncotarget.
7:31755–31771. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Li Y, Zhang J, Han S, Qian Q, Chen Q, Liu
L and Zhang Y: B7-H3 promotes the proliferation, migration and
invasiveness of cervical cancer cells and is an indicator of poor
prognosis. Oncol Rep. 38:1043–1050. 2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hu X, Xu M, Hu Y, Li N and Zhou L: B7-H3,
negatively regulated by miR-128, promotes colorectal cancer cell
proliferation and migration. Cell Biochem Biophys. 79:397–405.
2021.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Feng R, Chen Y, Liu Y, Zhou Q and Zhang W:
The role of B7-H3 in tumors and its potential in clinical
application. Int Immunopharmacol. 101 (Pt B)(108153)2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Liu S, Liang J, Liu Z, Zhang C, Wang Y,
Watson AH, Zhou C, Zhang F, Wu K, Zhang F, et al: The role of CD276
in cancers. Front Oncol. 11(654684)2021.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Getu AA, Tigabu A, Zhou M, Lu J, Fodstad O
and Tan M: New frontiers in immune checkpoint B7-H3 (CD276)
research and drug development. Mol Cancer. 22(43)2023.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Feustel K, Martin J and Falchook GS: B7-H3
inhibitors in oncology clinical trials: A review. J Immunother
Precis Oncol. 7:53–66. 2024.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Garcia-Peterson LM and Li X: Trending
topics of SIRT1 in tumorigenicity. Biochim Biophys Acta Gen Subj.
1865(129952)2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Cho EH and Dai Y: SIRT1 controls cell
proliferation by regulating contact inhibition. Biochem Biophys Res
Commun. 478:868–872. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Liao H, Ding M, Zhou N, Yang Y and Chen L:
B7-H3 promotes the epithelial-mesenchymal transition of NSCLC by
targeting SIRT1 through the PI3K/AKT pathway. Mol Med Rep.
25(79)2022.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Pan X, Li J, Du W, Yu X, Zhu C, Yu C, Cao
H, Zhang Y, Chen Y and Li L: Establishment and characterization of
immortalized human hepatocyte cell line for applications in
bioartificial livers. Biotechnol Lett. 34:2183–2190.
2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Mortezaee K: B7-H3 immunoregulatory roles
in cancer. Biomed Pharmacother. 163(114890)2023.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wang R, Sun L, Xia S, Wu H, Ma Y, Zhan S,
Zhang G, Zhang X, Shi T and Chen W: B7-H3 suppresses
doxorubicin-induced senescence-like growth arrest in colorectal
cancer through the AKT/TM4SF1/SIRT1 pathway. Cell Death Dis.
12(453)2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Nakamura F: The role of
mechanotransduction in contact inhibition of locomotion and
proliferation. Int J Mol Sci. 25(2135)2024.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Pontes Soares C, Midlej V, de Oliveira ME,
Benchimol M, Costa ML and Mermelstein C: 2D and 3D-organized
cardiac cells shows differences in cellular morphology, adhesion
junctions, presence of myofibrils and protein expression. PLoS One.
7(e38147)2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Li Y, Huang G, Li M, Wang L, Elson EL, Lu
TJ, Genin GM and Xu F: An approach to quantifying 3D responses of
cells to extreme strain. Sci Rep. 6(19550)2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Martianov I, Cler E, Duluc I, Vicaire S,
Philipps M, Freund JN and Davidson I: TAF4 inactivation reveals the
3 dimensional growth promoting activities of collagen 6A3. PLoS
One. 9(e87365)2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Rogers S, McCloy RA, Parker BL,
Gallego-Ortega D, Law AMK, Chin VT, Conway JRW, Fey D, Millar EKA,
O'Toole S, et al: MASTL overexpression promotes chromosome
instability and metastasis in breast cancer. Oncogene.
37:4518–4533. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Grimes DR and Fletcher AG: Close
encounters of the cell kind: The impact of contact inhibition on
tumour growth and cancer models. Bull Math Biol.
82(20)2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Sutton MN, Glazer SE, Muzzioli R, Yang P,
Gammon ST and Piwnica-Worms D: Dimerization of the 4Ig isoform of
B7-H3 in tumor cells mediates enhanced proliferation and
tumorigenic signaling. Commun Biol. 7(21)2024.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhang X, Xu C, Wang C, Pei Y, He M, Wan Z,
Hou J and Wang L: CD276 promotes epithelial-mesenchymal transition
in esophageal squamous cell carcinoma through the TGF-β/SMAD
signaling. Clin Exp Metastasis. 41:81–90. 2024.PubMed/NCBI View Article : Google Scholar
|