Introduction
Hemolytic uremic syndrome (HUS) is a thrombotic
microangiopathy (TMA) characterized by the triad of
microangiopathic hemolytic anemia, thrombocytopenia and acute
kidney injury (1). HUS is classified
into two main types: i) Typical HUS, most commonly caused by Shiga
toxin-producing Escherichia coli infections; and ii)
atypical HUS (aHUS), which is primarily related to the
dysregulation of the complement system due to genetic mutations.
Typical HUS mainly affects children, whereas aHUS is more frequent
among adults, particularly women during pregnancy or postpartum.
Distinguishing between these forms is essential for an accurate
diagnosis and management (2,3). First documented by Gasser et al
(4) in 1955, HUS primarily affects
the renal vasculature, resulting in arteriolar fibrinoid necrosis
(5). Its global incidence is ~2 to 3
cases per 100,000 individuals annually (6). While typical HUS affects children of
both sexes equally, aHUS is more frequently diagnosed in adult
women, potentially linked to pregnancy-related triggers (7). As aforementioned, the syndrome is
categorized into two forms: Typical HUS, commonly associated with
infections such as Shiga toxin-producing Escherichia coli
(STEC), and aHUS, often driven by genetic mutations affecting the
complement system (8). Both variants
arise from the dysregulation of the alternative complement pathway,
leading to the formation of the membrane attack complex (C5b-9)
(9). This process damages
endothelial cells and promotes a procoagulant state, inducing
platelet deposition and resulting in microthrombi within the renal
microvasculature that determines disease severity (10).
Clinically, HUS presents with fatigue, pallor due to
anemia and lethargy, progressing to renal manifestations, including
hematuria, proteinuria, and, in severe cases, anuria (11). Extrarenal complications, such as
cardiac and neurological involvement, occur in up to 20% of
patients (12,13). Diagnosis is established through
laboratory findings, such as elevated levels of lactate
dehydrogenase, schistocytes, thrombocytopenia and renal impairment,
supported by imaging to assess kidney involvement (14). A histological evaluation may be
required to differentiate HUS from other TMAs, such as thrombotic
thrombocytopenic purpura (15,16).
Treatment is tailored to disease severity and recurrence risk
(17), encompassing supportive care
(e.g., hydration and transfusions), immunosuppressive therapy
(18), and, in refractory cases,
renal replacement therapy or kidney transplantation. Genetic
testing is recommended, as ~50% of aHUS cases are associated with
mutations in complement-regulating genes, notably complement factor
H (CFH) (19-21).
Additional risk factors include certain medications, autoimmune
disorders and chronic illnesses (22,23).
Pregnancy is a recognized risk factor for aHUS,
likely due to the decreased production of complement regulatory
proteins and altered maternal immunity (24). However, diagnosis in the postpartum
period is challenging, as its features overlap with obstetric
conditions such as preeclampsia and hemolysis, elevated liver
enzymes and low platelets (HELLP) syndrome, often delaying
appropriate management (25,26). Given the rarity of aHUS and its
potential for severe morbidity and mortality, the present study
describes the case of a 29-year-old female patient who developed
aHUS 4 days following a cesarean delivery.
Case report
Initial presentation
A 29-year-old female patient (G2P1C1) presented to
the Emergency Department at the Gynecology and Obstetrics Hospital
No. 15 of the Mexican Social Security Institute (Chihuahua, Mexico)
on the 4th day postpartum following a cesarean section, reporting
purulent discharge from the vaginal and surgical sites, along with
urinary symptoms. The patient described a 4-day history of fever
(38.9˚C), asthenia, fatigue, dyspnea and pallor. Her medical
history was notable for a cesarean section at 39 weeks of
gestation, complicated by an estimated 3-liter obstetric
hemorrhage.
Upon admission, laboratory tests revealed severe
normocytic normochromic anemia (hemoglobin level, 4.6 g/dl) and
acute kidney injury (creatinine, 10.6 mg/dl; urea, 286 mg/dl; blood
urea nitrogen, 133.47 mg/dl); urinalysis revealed leukocyturia
(20-22 leukocytes per field) with granular casts. The patient
received empirical intravenous antibiotics and 3 units of packed
red blood cells before being transferred to a secondary care
facility the Regional General Hospital No. 1 of the Mexican Social
Security Institute, Morelos, Mexico for further management.
Diagnostic workup
The nephrology service evaluation identified anuria
(urine output 0.08 ml/kg/h), metabolic acidosis, azotemia and
hyperkalemia, necessitating transfer to the intensive care unit
(ICU) for intermittent hemodialysis. Initially, acute tubular
necrosis secondary to ischemic injury from obstetric hemorrhage was
suspected. A wound culture was obtained and dual antibiotic therapy
was initiated. The general surgery team drained 20 ml of purulent
fluid from the surgical site, resulting in transient clinical
improvement.
After 3 days, the patient developed lower
respiratory symptoms requiring supplemental oxygen. A chest
computed tomography scan revealed bilateral hilar reticular
opacities. Given these findings, and considering it was peak
influenza season, empirical treatment with oseltamivir (75 mg,
administered orally twice daily) was initiated following the
institutional protocol (27).
However, subsequent testing ruled out influenza infection.
Treatment course
The patient subsequently exhibited signs of fluid
overload and acute pulmonary edema, which was refractory to
non-invasive ventilation, leading to 4 days of mechanical
ventilation.
Due to persistent severe anemia, thrombocytopenia
and laboratory evidence of hemolysis (schistocytes on blood smear,
elevated lactate dehydrogenase without hyperbilirubinemia), a
peripheral blood smear was performed, revealing >10 schistocytes
per high-power field. She received fresh frozen plasma and
intravenous corticosteroids. Thrombotic microangiopathy was
investigated; however, complement levels were normal and
autoantibody tests yielded negative results: ANCA, 0.8 ng/m;
anti-dsDNA, 9.3 IU/ml; C3, 107 mg/dl; C4, 24.8 mg/dl; ADAMTS13
activity, 70%; and anticardiolipin, 1.2; these findings ruled out
thrombotic thrombocytopenic purpura.
Given the ongoing hemolytic anemia, renal impairment
and the absence of positive autoantibodies, a percutaneous kidney
biopsy was performed. Renal tissue samples were fixed in 10%
neutral buffered formalin for 24 h at room temperature, embedded in
paraffin, and cut into 4-µm-thick sections. Hematoxylin and eosin
(H&E), periodic acid-Schiff (PAS), Masson's trichrome and Jones
silver staining were performed following standard histopathological
protocols using reagents supplied by Sigma-Aldrich®
(Merck KGaA). The sections were stained at room temperature
according to the manufacturer's recommendations (H&E,
hematoxylin 10 min, eosin 3 min; PAS, periodic acid 5 min, Schiff's
reagent 15 min; Masson's trichrome: Sequential staining was
performed with Weigert's iron hematoxylin, Biebrich scarlet-acid
fuchsin, phosphotungstic/phosphomolybdic acid and aniline blue;
Jones silver: Methenamine silver impregnation with periodic acid
oxidation). Microscopic examination and image acquisition were
performed using a Nikon Eclipse E200 microscope® (Nikon
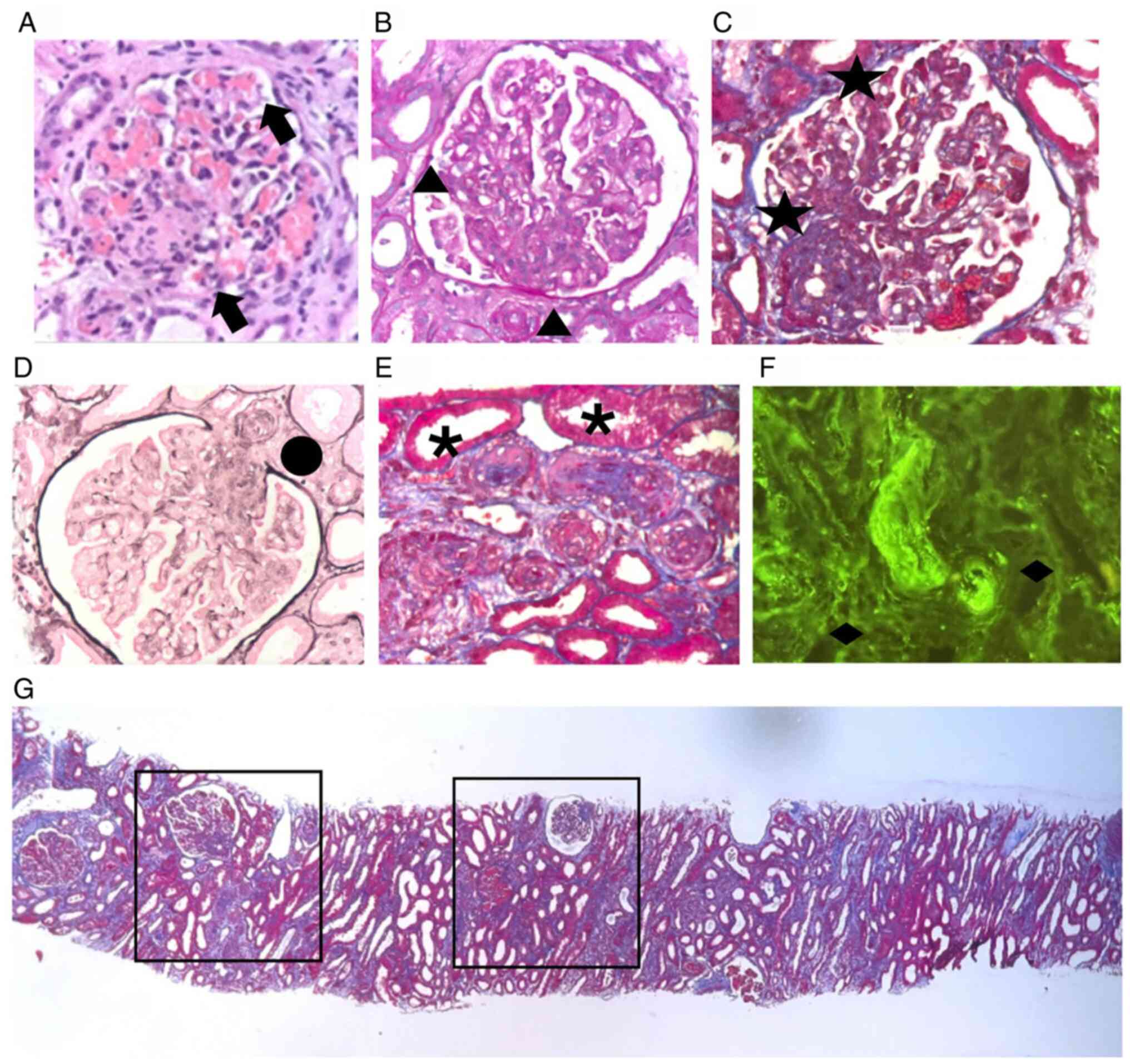
Corporation). A histopathological analysis demonstrated thrombotic
microangiopathy in both acute and chronic phases, with glomerular
and interlobular artery thrombosis, membranoproliferative
glomerulonephritis, diffuse endothelialitis without immune complex
deposition and severe arteriolonephrosclerosis with onion-skin
elastosis. Later, the sections were incubated overnight at 4˚C with
the following primary antibodies: IgA (1:100; cat. no. A0262), IgG
(1:200; cat. no. A0423), IgM (1:100; cat. no. A0425), C1q (1:100;
cat. no. A0136), C3c (1:200; cat. no. A0062), fibrinogen (1:100;
cat. no. A0080), kappa light chain (1:200; cat. no. A0191), lambda
light chain (1:200; cat. no. A0193) and albumin (1:200; cat. no.
A0001) (all from Dako, Agilent Technologies, Inc.). After washing,
the slides were incubated with horseradish peroxidase
(HRP)-conjugated secondary antibodies [goat anti-rabbit IgG (cat.
no. P0448) or goat anti-mouse IgG (cat. no. P0447) (both from Dako,
Agilent Technologies, Inc.), depending on the primary antibody] for
1 h at room temperature. Visualization was achieved using
3,3'-diaminobenzidine (DAB) substrate solution (cat. no. K3468;
Dako, Agilent Technologies, Inc.). Immunohistochemical staining for
immunoglobulins (IgA, IgG and IgM), complement components (C1q and
C3c), fibrinogen, kappa, lambda and albumin was uniformly negative,
confirming the diagnosis of HUS (Fig.
1).
Clinical outcomes
Several days later, the patient developed
exsanguinating hemoptysis. Bronchoscopy revealed diffuse alveolar
hemorrhage without specific lesions, leading to a diagnosis of
diffuse alveolar hemorrhage. However, no biopsy was performed, as
no specific lesions were visualized. The patient required
reintubation on hospital day 15 and was successfully extubated
after 10 days. However, she developed ventilator-associated
pneumonia, necessitating broad-spectrum antibiotics, which delayed
the initiation of immunosuppressive therapy. The rapid postpartum
onset of anemia, thrombocytopenia, and acute kidney injury in the
patient described herein closely mirrors the presentation described
in previously reported cases of pregnancy-associated aHUS, in which
early complement blockade has often been linked to improved
hematologic recovery, although renal outcomes remain variable. A
distinctive feature in the present case was the presence of
multiple severe complications, including diffuse alveolar
hemorrhage and ventilator-associated pneumonia, which are less
frequently described in the literature and may have contributed to
delays in targeted treatment. Following improvement and resolution
of the infection, she was discharged with ongoing intermittent
hemodialysis and remains under nephrology follow-up for renal
replacement therapy. Renal transplantation from a first-degree
relative (sister) was performed following renal replacement
therapy, with preserved graft function on follow-up. A concise
summary of key laboratory parameters at major clinical milestones
is presented in Table I. The
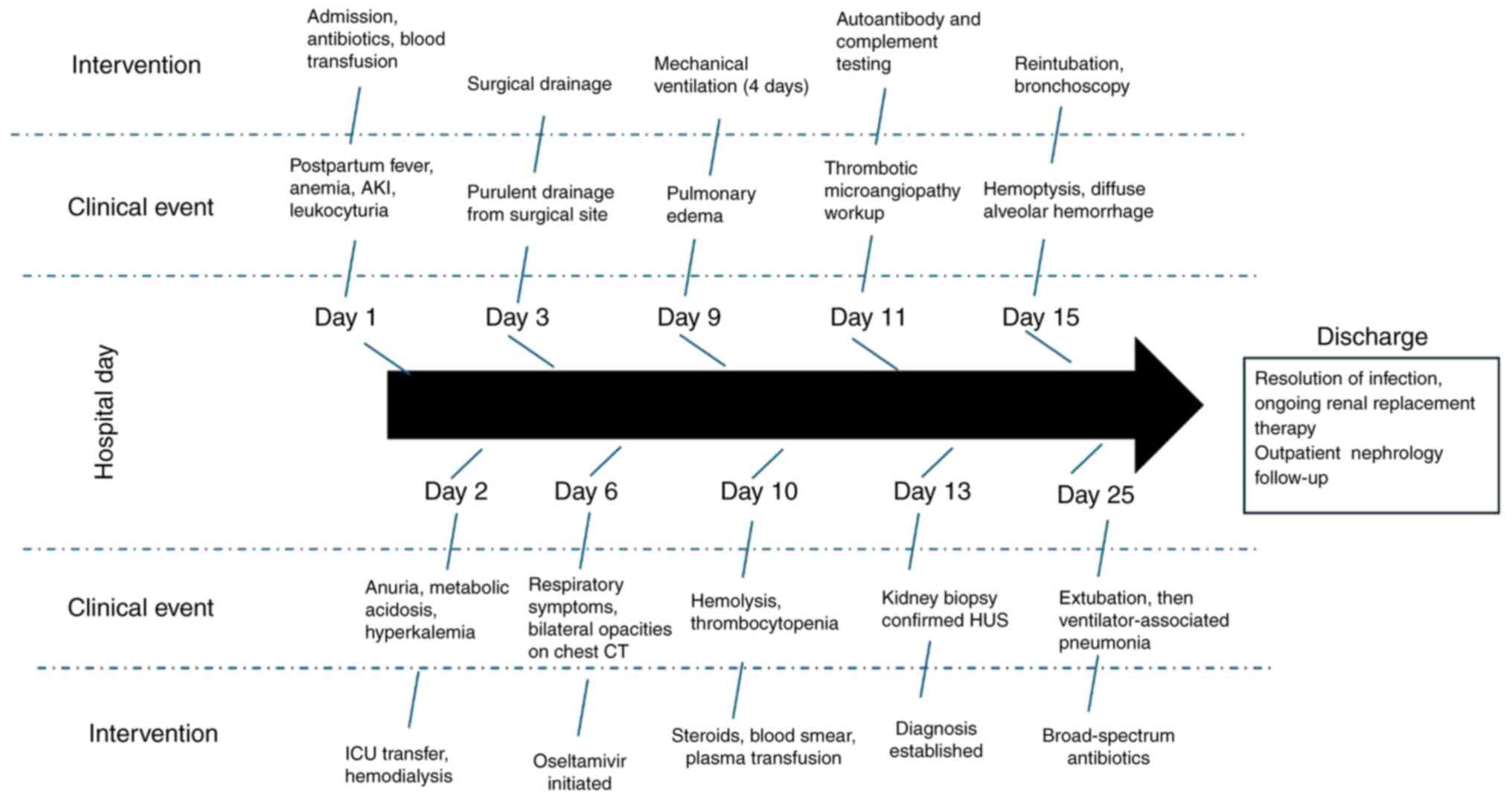
chronological sequence of clinical events and interventions is
depicted in Fig. 2.
 | Table IKey laboratory values and clinical
milestones following kidney transplantation. |
Table I
Key laboratory values and clinical
milestones following kidney transplantation.
| Hospital
day/follow-up day | Clinical
event/intervention | Hemoglobin
(g/dl) | Hematocrit (%) | Platelets
(x10³/µl) | Creatinine
(mg/dl) | LDH (U/l) | BUN (mg/dl) | Notes |
|---|
| Pre-Tx | Pre-transplant
laboratory tests | 12.0 | 37 | 241 | 16.2 | - | 46 | Stable baseline |
| Day 0 | Living donor kidney
transplant | - | - | - | - | - | - | Warm ischemia 5
min; cold ischemia 48 min |
| POD 2 | Routine laboratory
tests | 8.4 | 26 | 69 | 1.0 | 212 | 10 | Albumin 2.2 g/d;;
tacrolimus 2.6 ng/ml |
| POD 4 | Routine
follow-up | 8.6 | 25 | 94 | 0.9 | 191 | 9 | Tacrolimus 3.4
ng/m; |
| POD 12 | Outpatient
control | 10.7 | 34 | 401 | 1.0 | - | - | Improving
counts |
| 2nd visit | Stable | 12.0 | 36 | 333 | 1.0 | - | 13 | - |
| 3rd visit | Ultrasound: mild
collection | 12.7 | 38 | 276 | 1.0 | - | 14 | Upper pole
collection 19 cc |
| Latest visit | Staples and double
J removal | - | - | - | 1.0 | - | - | Asymptomatic |
Discussion
The present case report details a young female
patient who, on the 4th day postpartum following a cesarean
section, experienced an acute onset of hemolytic anemia,
thrombocytopenia, significant hemorrhage and acute kidney injury,
culminating in a diagnosis of pregnancy-associated HUS. HUS, a rare
TMA. It has an estimated global incidence of 2 to 3 cases per
100,000 individuals annually, and it predominantly affects children
and young adults, with incidence peaks at ages 21 and 25 uears for
males and females, respectively (28).
TMAs are categorized by their underlying mechanisms:
Primary TMAs, such as thrombotic thrombocytopenic purpura (TTP) and
typical HUS (HUS-Tx), stem from intrinsic genetic defects, whereas
secondary TMAs, including aHUS, are precipitated by infections,
autoimmune conditions, or other triggers such as aHUS or
microangiopathic HUS (29). Although
rare, HUS can manifest during pregnancy or the immediate postpartum
period, particularly in genetically susceptible young women
(30). Typically emerging within the
first few days postpartum, it affects ~1 in 25,000 pregnancies.
Despite its rarity, HUS carries severe implications, with up to 70%
of affected women requiring long-term renal replacement therapy,
underscoring the need for prompt recognition to improve prognosis
(31).
The identification of HUS is challenging, as its
differential diagnosis includes all forms of TMAs, (both those
directly and indirectly mediated by the complement system), as well
as common pathologies that present with similar clinical
manifestations (5). The patient in
the case described herein presented a clinical picture consistent
with various hemolytic disorders associated with pregnancy and the
puerperium. Additionally, the history of significant bleeding
during the obstetric event is particularly noteworthy, as previous
reports have suggested that blood loss >1,500 ml in women
without a prior pathological history may indicate a diagnosis of
HUS (32).
Acute anemia is a frequent finding in postpartum HUS
and presents a broad differential etiology, often delaying initial
diagnostic suspicion (33). Renal
manifestations, including azotemia and electrolyte imbalances,
aligned with prior descriptions, yet necessitated the exclusion of
more common entities such as preeclampsia, HELLP syndrome and
hypovolemic acute kidney injury through systematic evaluation
(34).
This protocol included, for example, the
identification of abundant schistocytes on the blood smear, which
suggests microangiopathy, and an elevated level of lactate
dehydrogenase, indicative of prolonged cellular lysis (35). These findings, together with other
laboratory results, clinical signs of cellular ischemia, refractory
anemia and worsening symptoms, strongly supported the suspicion of
microangiopathy.
For this reason, the analyses of antibodies and
complement fractions were performed (36). To determine the triggering cause, the
use of the ADAMTS13 test via fluorometric assay was also warranted
(37). After evaluating the results,
the diagnosis of the patient was ultimately determined.
Additionally, it was found that complement levels were within
normal ranges (C3, 107 mg/dl; C4, 24.8 mg/dl) and ADAMTS13 activity
was 70%, effectively ruling out thrombotic thrombocytopenic
purpura. aHUS has a strong genetic basis, with mutations commonly
identified in complement regulatory genes, such as CFH, CFI, MCP,
C3 and deletions in CFHR1/3. Genetic testing is critical for
confirming diagnosis, assessing prognosis and guiding family
counseling. However, genetic analysis was not performed in the
present study due to limited availability at the Gynecology and
Obstetrics Hospital No. 15 of the Mexican Social Security
Institute. Expanding access to such testing in the future is vital
for optimizing patient management and understanding disease
pathogenesis. Given the overlapping clinical features,
distinguishing aHUS from other pregnancy-associated thrombotic
microangiopathies is crucial. HELLP syndrome typically presents
with hemolysis, elevated liver enzymes and low platelets before or
shortly after delivery, often associated with hypertension and
proteinuria, both of which were absent in the patient described
herein. Although severe preeclampsia is characterized by
hypertension and signs of organ dysfunction, the patient in the
present study had no hypertensive episodes or proteinuria
throughout her pregnancy or postpartum course. TTP is usually
marked by severe ADAMTS13 deficiency (<10%), neurological
symptoms and minor renal involvement; by contrast, the ADAMTS13
activity of the patient in the present study was within normal
limits (70%) and acute kidney injury was a prominent feature.
The constellation of findings, postpartum anemia,
thrombocytopenia, acute kidney injury, normal ADAMTS13 activity and
confirmation of thrombotic microangiopathy on kidney biopsy,
strongly supports the diagnosis of pregnancy-associated aHUS,
effectively distinguishing it from other differential
diagnoses.
However, multiple complications arose at that time,
extending her treatment and duration of hospitalization and
necessitating subsequent outpatient follow-up while she continued
renal replacement therapy. Eculizumab was considered as a
therapeutic option; however, it was not administered due to the
presence of active infectious complications, including sepsis and
ventilator-associated pneumonia, which contraindicated its use in
this setting. Current treatment options for aHUS include plasma
exchange or infusion, immunosuppressive therapies, such as
corticosteroids and complement inhibitors such as eculizumab
(38). Eculizumab has exhibited
notable efficacy by inhibiting terminal complement activation,
reducing disease progression, and improving survival rates
(39). However, its use may be
contraindicated in patients with active infections, as was the case
here, and it is also limited by availability and cost. Plasma
exchange remains a mainstay therapy when eculizumab is unavailable
or unsuitable, although critical illness may restrict its
feasibility (40,41). Additionally, there were institutional
and resource limitations regarding its availability. As regards
plasma therapy, the patient received fresh frozen plasma infusions
and intravenous corticosteroids, which led to temporary
stabilization. Plasmapheresis (plasma exchange) was considered but
ultimately not initiated, primarily due to the patient's critical
hemodynamic status and the decision of the multidisciplinary team
prioritizing infection control and respiratory management.
The present case report highlights the rapid
clinical progression of a rare condition and emphasizes the
importance of maintaining a high index of suspicion in women
presenting with similar characteristics to facilitate prompt
treatment. This case shares a number of clinical and laboratory
features with previously reported cases of pregnancy-associated
aHUS, including rapid postpartum onset of anemia, thrombocytopenia
and acute kidney injury (30). To
further address the similarities and differences between the
present case and those previously reported, a comparative summary
is presented in Table II. This
table contrasts key aspects of presentation, laboratory findings,
complications, treatment strategies, and outcomes between our
patient and selected cases from the literature (42-45).
As demonstrated, the present case shares core features with other
pregnancy-associated aHUS presentations, such as rapid postpartum
onset of anemia, thrombocytopenia and acute kidney injury, but is
distinguished by the presence of severe complications such as
diffuse alveolar hemorrhage and ventilator-associated pneumonia,
which are rarely documented. These additional complications likely
delayed the initiation of targeted therapy, particularly complement
blockade, and may have contributed to the persistence of
dialysis-dependent renal failure, in contrast to some published
cases where earlier intervention was associated with improved
hematologic recovery and, in certain instances, partial renal
recovery.
| Table IIComparative features of the present
case and selected previously reported cases of pregnancy-associated
atypical hemolytic uremic syndrome. |
Table II
Comparative features of the present
case and selected previously reported cases of pregnancy-associated
atypical hemolytic uremic syndrome.
| | Author(s) (Refs.),
year of publication |
|---|
| Feature | Present case | Saad et al
(30), 2016 | Frimat et al
(42), 2024 | Fakhouri (44), 2016 | Fakhouri et
al (45), 2021 |
|---|
| Onset | 4 Days
postpartum | Immediate
postpartum | Late pregnancy or
postpartum | Postpartum | Postpartum |
| Main clinical
features | Severe anemia,
thrombocytopenia, acute kidney injury, diffuse alveolar hemorrhage,
ventilator-associated pneumonia | Severe anemia,
thrombocytopenia, AKI | TMA signs with
anemia, thrombocytopenia, AKI | Anemia,
thrombocytopenia, AKI | Anemia,
thrombocytopenia, AKI |
| Laboratory
findings | Hb 4.6 g/dl,
schistocytes >10/HPF, creatinine 10.6 mg/dl, normal complement
levels | Similar hematologic
profile, elevated LDH, normal C3/C4 | Variable complement
abnormalities | Complement
abnormalities in subset | Complement
abnormalities in subset |
| Histopathology | TMA in acute and
chronic phases, MPGN pattern, severe arteriolonephrosclerosis | Not always
performed; when done, TMA lesions | Not always
performed; when done, TMA lesions | TMA lesions | TMA lesions |
| Complications | Diffuse alveolar
hemorrhage, ventilator-associated pneumonia | Not reported | Not reported | Not reported | Not reported |
| Treatment | Broad-spectrum
antibiotics, plasma, corticosteroids; delayed immunosuppression; no
Eculizumab due to complications and timing | Plasma exchange,
supportive care; some cases Eculizumab | Variable:
supportive care, plasma exchange, complement blockade | Eculizumab early in
course | Eculizumab, some
discontinuation |
| Outcome | Discharged on
dialysis, ongoing renal replacement therapy | Variable renal
recovery; some dialysis dependence | Variable renal
recovery | Hematologic
remission; variable renal recovery | Hematologic
remission; variable renal recovery |
Variability in treatment decisions reflects
differing clinical scenarios and resource availability,
highlighting the importance of individualized, multidisciplinary
care (43). In this regard, it is
worth noting that the use of eculizumab and other C5 inhibitors, (a
humanized monoclonal antibody that inhibits terminal complement
activity) has been shown to promote symptom resolution and improve
survival, although their impact on renal function is more complex
to analyze (44,45). In the case described herein, the
complexity of the complications and associated pathologies made it
difficult to determine their precise impact; however, a prompter
diagnosis may have reduced the morbidity of the patient. Although
the present case report highlights key aspects of aHUS management,
its findings are limited to a single patient experience and may not
be generalizable. Larger case series and systematic studies are
required to establish definitive clinical guidelines. Other
limitations of the present case report include the absence of
genetic testing results and limited long-term follow-up data beyond
initial discharge. Future studies are thus required to focus on
improving access to genetic diagnostics and on long-term outcomes
in pregnancy-associated aHUS. Heightened awareness and timely
diagnosis remain crucial to improve patient prognosis and guide
therapy. Finally, an important limitation of the present report is
the absence of peripheral blood smear images, kidney biopsy
histopathology annotations and laboratory trend graphs due to
institutional constraints, which would have further supported the
diagnosis.
In conclusion, TMAs such as HUS, although rare,
confer high morbidity and mortality, often precipitated by
pregnancy or puerperal sepsis. The present case report describes a
case of postpartum HUS confirmed by renal biopsy following a
comprehensive workup including negative antibody and complement
tests and ADAMTS13 analysis, which excluded alternative diagnoses.
Initial management with plasmapheresis and intravenous steroids
yielded favorable outcomes despite in-hospital complications. Given
the diagnostic complexity, ruling out differential diagnoses and
securing histopathological confirmation remain essential for
effective management.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
KPQB, HARL, LFMB, AVU, RAAM and JASO participated
equally in the preparation of this manuscript, both in the medical
care process of the patient and during data collection, literature
search, information synthesis, and writing of this manuscript. KPQB
and HARL confirm the authenticity of all the raw data. All authors
have read and approved the final manuscript.
Ethics approval and consent for
participation
The present study was performed in accordance with
the ethical standards of the Declaration of Helsinki, 1964.
Informed consent was obtained from the patient for inclusion in the
study. Ethics approval was waived by the local committee as no
personal data were used.
Patient consent for publication
Written informed consent was obtained from the
patient for the publication of the present case report and any
related images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Afshar-Kharghan V: Atypical hemolytic
uremic syndrome. Hematol Am Soc Hematol Educ Prog. 2016:217–225.
2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Salvadori M and Bertoni E: Update on
hemolytic uremic syndrome: Diagnostic and therapeutic
recommendations. World J Nephrol. 2:56–76. 2013.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Jokiranta TS: HUS and atypical HUS. Blood.
129:2847–2856. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Gasser C, Gautier E, Steck A, Siebenmann
RE and Oechslin R: Hemolytic-uremic syndrome: Bilateral necrosis of
the renal cortex in acute acquired hemolytic anemia. Schweiz Med
Wochenschr. 85:905–909. 1955.PubMed/NCBI
|
|
5
|
Lee H, Kang E, Kang HG, Kim YH, Kim JS,
Kim HJ, Moon KC, Ban TH, Oh SW, Jo SK, et al: Consensus regarding
diagnosis and management of atypical hemolytic uremic syndrome.
Korean J Intern Med. 35:25–40. 2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Aldharman SS, Almutairi SM, Alharbi AA,
Alyousef MA, Alzankrany KH, Althagafi MK, Alshalahi EE, Al-Jabr KH,
Alghamdi A and Jamil SF: The Prevalence and incidence of hemolytic
uremic syndrome: A systematic review. Cureus.
15(e39347)2023.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Roumenina LT, Loirat C, Dragon-Durey MA,
Halbwachs-Mecarelli L, Sautes-Fridman C and Fremeaux-Bacchi V:
Alternative complement pathway assessment in patients with atypical
HUS. J Immunol Methods. 365:8–26. 2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Rondeau E, Ardissino G, Caby-Tosi MP,
Al-Dakkak I, Fakhouri F, Miller B and Scully M: Global aHUS
Registry. Pregnancy in women with atypical hemolytic uremic
syndrome. Nephron. 146:1–10. 2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ávila A, Cao M, Espinosa M, Manrique J and
Morales E: Recommendations for the individualised management of
atypical hemolytic uremic syndrome in adults. Front Med (Lausanne).
10(1264310)2023.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Yan K, Desai K, Gullapalli L, Druyts E and
Balijepalli C: Epidemiology of atypical hemolytic uremic syndrome:
A systematic literature review. Clin Epidemiol. 12:295–305.
2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Yerigeri K, Kadatane S, Mongan K, Boyer O,
Burke LLG, Sethi SK, Licht C and Raina R: Atypical Hemolytic-uremic
syndrome: Genetic basis, clinical manifestations, and a
multidisciplinary approach to management. J Multidiscip Healthc.
16:2233–2249. 2023.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhang K, Lu Y, Harley KT and Tran MH:
Atypical hemolytic uremic syndrome: A brief review. Hematol Rep.
9(7053)2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Bayer G, von Tokarski F, Thoreau B,
Bauvois A, Barbet C, Cloarec S, Mérieau E, Lachot S, Garot D,
Bernard L, et al: Etiology and outcomes of thrombotic
microangiopathies. Clin J Am Soc Nephrol. 14:557–566.
2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Sheerin NS and Glover E: Haemolytic uremic
syndrome: Diagnosis and management. F1000Res 8: F1000 Faculty
Rev-1690, 2019.
|
|
15
|
Scully M and Goodship T: How I treat
thrombotic thrombocytopenic purpura and atypical haemolytic uraemic
syndrome. Br J Haematol. 164:759–766. 2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Domínguez-Vargas A, Ariño F, Silva D,
González-Tórres HJ, Aroca-Martinez G, Egea E and Musso CG:
Pregnancy-associated atypical hemolytic uremic syndrome: A case
report with MCP gene mutation and successful eculizumab treatment.
AJP Rep. 14:e96–e100. 2024.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Tseng MH, Lin SH, Tsai JD, Wu MS, Tsai IJ,
Chen YC, Chang MC, Chou WC, Chiou YH and Huang CC: Atypical
hemolytic uremic syndrome: Consensus of diagnosis and treatment in
Taiwan. J Formos Med Assoc. 122:366–375. 2023.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Ardissino G, F, Possenti I, Testa S,
Consonni D, Paglialonga F, Salardi S, Borsa-Ghiringhelli N, Salice
P, Tedeschi S, et al: Early volume expansion and outcomes of
hemolytic uremic syndrome. Pediatrics: Dec 7, 2016 (Epub ahead of
print). doi: 10.1542/peds.2015-2153.
|
|
19
|
Leon J, LeStang MB, Sberro-Soussan R,
Servais A, Anglicheau D, Frémeaux-Bacchi V and Zuber J:
Complement-driven hemolytic uremic syndrome. Am J Hematol. 98
(Suppl 4):S44–S56. 2023.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Valoti E, Alberti M, Iatropoulos P, Piras
R, Mele C, Breno M, Cremaschi A, Bresin E, Donadelli R, Alizzi S,
et al: Rare functional variants in complement genes and Anti-FH
Autoantibodies-associated aHUS. Front Immunol.
10(853)2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Piras R, Valoti E, Alberti M, Bresin E,
Mele C, Breno M, Liguori L, Donadelli R, Rigoldi M, Benigni A, et
al: CFH and CFHR structural variants in atypical Hemolytic Uremic
Syndrome: Prevalence, genomic characterization and impact on
outcome. Front Immunol. 13(1011580)2023.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Valério P, Barreto JP, Ferreira H, Chuva
T, Paiva A and Costa JM: Thrombotic microangiopathy in oncology-a
review. Transl Oncol. 14(101081)2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Schwotzer N, Frémeaux-Bacchi V and
Fakhouri F: Hemolytic uremic syndrome: A question of terminology.
Clin J Am Soc Nephrol. 18:831–833. 2023.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wildes DM, Harvey S, Costigan CS, Sweeney
C, Twomey É, Awan A and Gorman KM: Eculizumab in STEC-HUS: A
paradigm shift in the management of pediatric patients with
neurological involvement. Pediatr Nephrol. 39:315–324.
2024.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Stefanovic V: The Extended use of
eculizumab in pregnancy and complement activation(-)associated
diseases affecting maternal, fetal and neonatal kidneys-the future
is now? J Clin Med. 8(407)2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Timmermans SAMEG, Abdul-Hamid MA,
Potjewijd J, Theunissen ROMFIH, Damoiseaux JGMC, Reutelingsperger
CP and van Paassen P: Limburg Renal Registry. C5b9 formation on
endothelial cells reflects complement defects among patients with
renal thrombotic microangiopathy and severe hypertension. J Am Soc
Nephrol. 29:2234–2243. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Secretaría de Salud México: Frequently
asked questions: Seasonal influenza [Internet]. Mexico City:
Government of Mexico. In Spanish. Available from: https://www.gob.mx/salud/acciones-y-programas/preguntas-frecuentes-influenzaestacional.
|
|
28
|
Kottke-Marchant K: Diagnostic approach to
microangiopathic hemolytic disorders. Int J Lab Hematol. 39 (Suppl
1):S69–S75. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Avila Bernabeu AI, Cavero Escribano T and
Cao Vilarino M: Atypical hemolytic uremic syndrome: New challenges
in the complement blockage era. Nephron. 144:537–549.
2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Saad AF, Roman J, Wyble A and Pacheco LD:
Pregnancy-associated atypical hemolytic-uremic syndrome. AJP Rep.
6:e125–e128. 2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Konopásek P and Zieg J: Eculizumab use in
patients with pneumococcal-associated hemolytic uremic syndrome and
kidney outcomes. Pediatr Nephrol. 38:4209–4215. 2023.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Miyazaki Y, Fukuda M, Hirayu N, Nabeta M
and Takasu O: Pregnancy-associated atypical hemolytic uremic
syndrome successfully treated with ravulizumab: A case report.
Cureus. 16(e54207)2024.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Mirza M, Sadiq N and Aye C:
Pregnancy-associated atypical hemolytic uremic syndrome and
Life-long kidney failure. Cureus. 14(e25655)2022.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Che M, Moran SM, Smith RJ, Ren KYM, Smith
GN, Shamseddin MK, Avila-Casado C and Garland JS: A case-based
narrative review of pregnancy-associated atypical hemolytic uremic
syndrome/complement-mediated thrombotic microangiopathy. Kidney
Int. 105:960–970. 2024.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Java A: Atypical hemolytic uremic
syndrome: Diagnosis, management, and discontinuation of therapy.
Hematology Am Soc Hematol Educ Program. 2024:200–205.
2024.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Gaire S, Shrestha M, Bhattarai CD,
Dhungana S, Gyawali S and Bajgain A: Hemolytic uremic syndrome: A
case report. JNMA J Nepal Med Assoc. 61:472–474. 2023.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Raina R, Krishnappa V, Blaha T, Kann T,
Hein W, Burke L and Bagga A: Atypical Hemolytic-uremic syndrome: An
update on pathophysiology, diagnosis, and treatment. Ther Apher
Dial. 23:4–21. 2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Kaplan BS, Ruebner RL, Spinale JM and
Copelovitch L: Current treatment of atypical hemolytic uremic
syndrome. Intractable Rare Dis Res. 3:34–45. 2024.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Cofiell R, Kukreja A, Bedard K, Yan Y,
Mickle AP, Ogawa M, Bedrosian CL and Faas SJ: Eculizumab reduces
complement activation, inflammation, endothelial damage,
thrombosis, and renal injury markers in aHUS. Blood. 125:3253–3262.
2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Yang L, Liu F, Li X, He L, Gu Y, Sun M and
Liu Z and Liu Z: Plasma exchange combined with eculizumab in the
management of atypical hemolytic uremic in pediatric patients: A
case report. Medicine (Baltimore). 104(e42090)2025.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Loirat C, Garnier A, Sellier-Leclerc AL
and Kwon T: Plasmatherapy in atypical hemolytic uremic syndrome.
Semin Thromb Hemost. 36:673–81. 2010.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Frimat M, Gnemmi V, Stichelbout M, Provôt
F and Fakhouri F: Pregnancy as a susceptible state for thrombotic
microangiopathies. Front Med (Lausanne). 11(1343060)2024.PubMed/NCBI View Article : Google Scholar
|
|
43
|
De Meyer F, Chambaere K, Van de Velde S,
Van Assche K, Beernaert K and Sterckx S: Factors influencing
obstetricians' acceptance of termination of pregnancy beyond the
first trimester: A qualitative study. BMC Med Ethics.
26(32)2025.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Fakhouri F: Pregnancy-related thrombotic
microangiopathies: Clues from complement biology. Transfus Apher
Sci. 54:199–202. 2016.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Fakhouri F, Fila M, Hummel A, Ribes D,
Sellier-Leclerc AL, Ville S, Pouteil-Noble C, Coindre JP, Le
Quintrec M, Rondeau E, et al: Eculizumab discontinuation in
children and adults with atypical hemolytic-uremic syndrome: A
prospective multicenter study. Blood. 137:2438–2449.
2021.PubMed/NCBI View Article : Google Scholar
|