Introduction
Creutzfeldt-Jakob disease (CJD) is a rare, fatal and
rapidly progressive neurodegenerative disorder caused by misfolded
prion proteins (PrPSc), which induce abnormal
conformational changes in normal cellular prions (PrPC),
leading to widespread neuronal loss, spongiform changes, and
gliosis (1,2). The sporadic form [sporadic CJD (sCJD)]
accounts for 85-90% of cases, with an estimated annual incidence of
1-1.5 per million in the population worldwide (3-5)
sCJD primarily affects individuals aged 50-70 years and is
characterized by rapidly progressive dementia, myoclonus, ataxia,
visual disturbances and pyramidal or extrapyramidal signs (1,5).
As sCJD progresses rapidly and shares features with
autoimmune, infectious, metabolic, toxic and neoplastic disorders,
timely recognition is critical for accurate diagnosis and
appropriate management. Probable sCJD is diagnosed through a
combination of clinical findings and supportive paraclinical
evidence, including periodic sharp wave complexes on an
electroencephalography (EEG), cortical ribboning or basal ganglia
hyperintensities on magnetic resonance imaging (MRI) (6-8),
and positive cerebrospinal fluid (CSF) biomarkers, such as 14-3-3
protein or real-time quaking-induced conversion (RT-QuIC) assays
(9).
The present study describes the case of a
55-year-old female patient whose initial psychiatric symptoms
rapidly progressed to severe cognitive and motor decline,
ultimately meeting criteria for probable sCJD. By highlighting the
integration of clinical assessment, neuroimaging, EEG and CSF
biomarkers, the present case report aimed to emphasize the
diagnostic challenges and the importance of early multidisciplinary
evaluation in rapidly progressive neurocognitive syndromes.
Case report
Presentation of the case
A 55-year-old woman with a history of
well-controlled systemic arterial hypertension under regular
treatment, and no prior neurological or psychiatric conditions,
presented to the Department of Internal Medicine, Hospital General
Dr. Belisario Domínguez ISSSTE, Tuxtla Gutiérrez, Chiapas, Mexico,
in February 4, 2025, with an 8-week history of gradually
progressive cognitive decline. The initial symptoms included
short-term memory loss, impaired executive functions and behavioral
alterations, such as irritability, mild disinhibition and apathy,
leading to an initial psychiatric evaluation under the suspicion of
major depressive disorder.
Her condition rapidly deteriorated, progressing to
gait instability with frequent falls, wide-based ataxia, dysarthria
of the scanning type and spontaneous myoclonus, which became
multifocal and stimulus-sensitive, predominantly affecting the left
side. The cognitive deterioration advanced to overt dementia with
temporal-spatial disorientation, partial mutism, and severe
functional impairment.
Upon admission, her Glasgow coma scale score was
13/15. She was alert but minimally verbal, with axial rigidity,
bradykinesia, bradyphrenia and hypokinetic dysarthria. Global motor
responses to painful stimuli were preserved, without
lateralization. Cerebellar signs, including dysmetria and limb
ataxia, were evident, as well as multifocal myoclonus triggered by
external stimuli. No focal motor or sensory deficits were
observed.
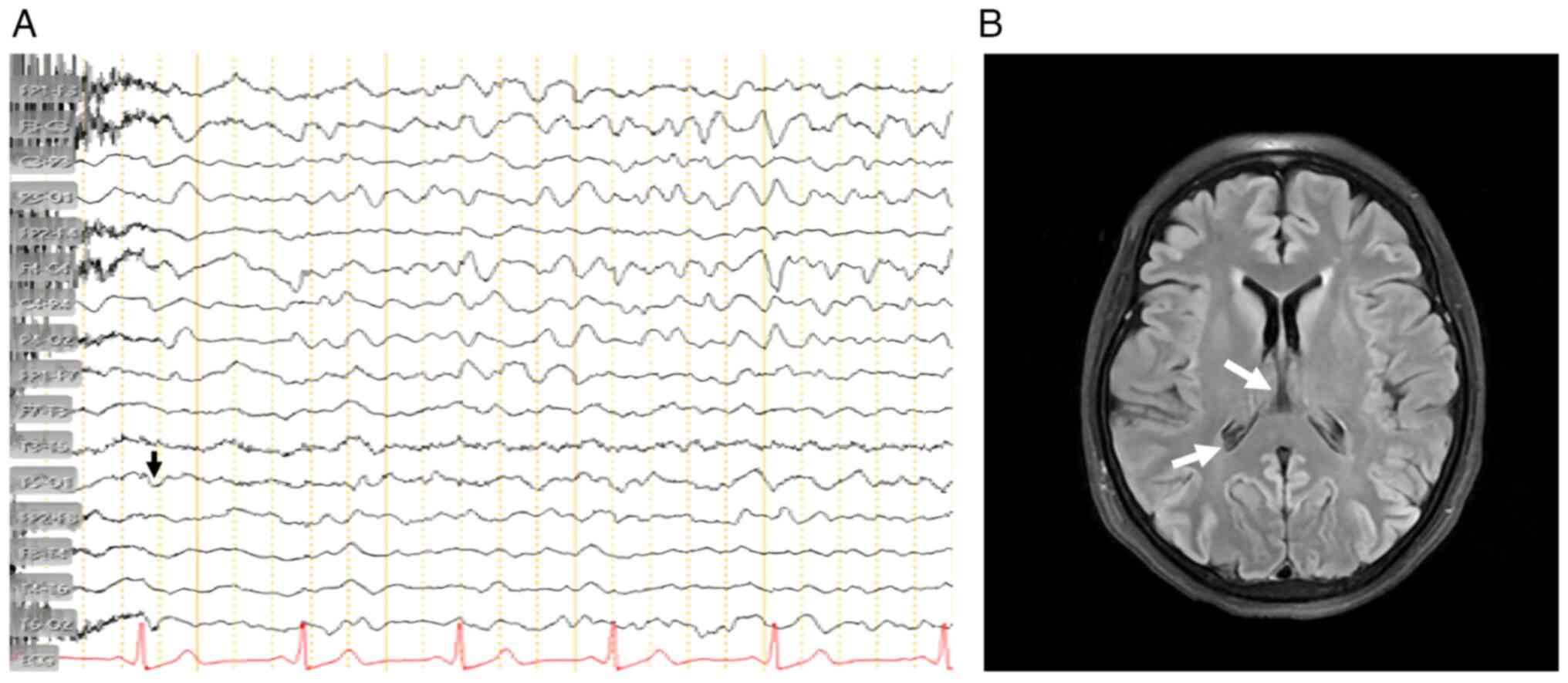
An EEG demonstrated generalized periodic discharges
with triphasic complexes at 1-2 Hz, a pattern typical of prion
encephalopathies. The EEG findings are illustrated in Fig. 1A. A brain MRI in FLAIR and DWI
sequences revealed cortical ribboning in the parieto-occipital
cortex and basal ganglia, characteristic of CJD. Representative MRI
images are shown in Fig. 1B.
CSF analysis was positive for 14-3-3 protein (50,026
AU/ml; reference, <20,000 AU/ml=negative), determined using an
automated enzyme immunoassay (ELISA-based) method that reports
results in arbitrary units (AU/ml) and are interpreted
qualitatively (positive/negative) according to the manufacturer’s
reference range. The test was performed using a monoclonal antibody
specific for the 14-3-3 γ-isoform, analyzed with an automated
chemiluminescence imaging system (Bio-Rad ChemiDoc MP; Bio-Rad
Laboratories, Inc.). Routine CSF analyses revealed normal cell
counts and biochemical parameters, and no evidence of infection or
pleocytosis. A broad panel of paraneoplastic (anti-Hu, anti-Yo,
anti-Ri, anti-Ma2, anti-CV2, amphiphysin), autoimmune (NMDA-R,
AMPA-R, LGI1, CASPR2, GABA-B-R) and infectious (HSV, VZV, CMV, EBV,
HIV, VDRL) panels were negative. Metabolic, including thyroid
function, vitamin B12, folate, and ammonia, were within normal
limits (10).
This clinical and diagnostic profile supported a
probable diagnosis of sCJD. The case is notable for its initial
psychiatric presentation and rapid neurological decline,
highlighting the diagnostic challenges that may delay recognition
of prion diseases in early stages.
Diagnostic workup and imaging
techniques
The diagnosis of probable sCJD was established
according to the updated 2018 Centers for Disease Control and
Prevention (CDC) diagnostic criteria and the UCSF Prion Center
guidelines (2023), integrating the clinical course, characteristic
EEG findings, MRI features and positive CSF biomarker (14-3-3
protein) (10).
Diagnostic criteria applied
The diagnosis of probable sCJD was established based
on the updated diagnostic criteria from the Centers for Disease
Control and Prevention (CDC) and the UCSF Prion Center, which
incorporate clinical features, EEG patterns, MRI findings, and CSF
biomarkers (10,11). During hospitalization, the patient
experienced progressive neurological deterioration, characterized
by worsening cognitive decline, generalized myoclonus, and
decreased level of consciousness. Subsequently, she developed
healthcare-associated pneumonia complicated by acute respiratory
distress syndrome (ARDS). Despite receiving supportive and
palliative management, her condition continued to deteriorate, and
she passed away on June 22, 2025.
Ethical considerations
The present case report was conducted in accordance
with the ethical standards of the institutional and national
research committees, and with the 1964 Declaration of Helsinki and
its later amendments (12). Written
informed consent was obtained from the patient's legal
representative for the publication of this case and any
accompanying images. All data were anonymized to protect patient
confidentiality. The case report was approved by the Health
Research Coordination of the General Hospital ‘Dr. Belisario
Domínguez’, ISSSTE, under official authorization no. CIS/0100/2025.
The case did not involve experimental procedures and was based
solely on routine clinical care.
Discussion
sCJD is a subacute, fatal prion encephalopathy.
Despite its low incidence rates, it requires a high index of
suspicion in cases of rapidly progressive dementia, particularly in
previously functional patients without a relevant neuropsychiatric
history (2,13,14). The
present case report exemplifies the diagnostic challenges posed by
an early psychiatric presentation in a previously independent,
middle-aged woman, in whom the rapid evolution of symptoms
underscored the importance of close collaboration between
psychiatry and neurology for timely recognition of
neurodegenerative etiologies (3,15-19).
The progression of the condition of the patient from
anterograde memory loss and executive dysfunction to ataxia,
scanning dysarthria and multifocal myoclonus aligns with the
characteristic triad of cognitive decline, myoclonus and cerebellar
dysfunction described in cases of sCJD in the literature (6,7,20-22).
In the literature, an EEG demonstrated periodic
triphasic complexes (observed in ~65-75% of cases), while an MRI
revealed cortical ribboning in the parieto-occipital cortex and
basal ganglia, a highly sensitive and specific imaging marker for
prion disease. These findings, combined with a positive 14-3-3
protein assay in CSF, provided strong diagnostic support in the
absence of histopathological confirmation (8,9,23,24)
(Fig. 1B). The combination of EEG
and MRI findings improved diagnostic confidence in the absence of
histopathology (14,25,26).
The 14-3-3 protein result (50,026 AU/ml; reference,
<20,000 AU/ml=negative) reflects a semi-quantitative positive
reading from an enzyme immunoassay system. When interpreted
alongside typical clinical and neuroimaging features, this
biomarker significantly increases diagnostic confidence.
Nevertheless, false positives may occur in acute neurological
conditions, such as stroke or encephalitis; thus, the results
should be interpreted within the clinical context (27-30).
The 14-3-3 protein assay was performed using an automated enzyme
immunoassay (ELISA-based) method that reports results in arbitrary
units (AU/ml), which are interpreted qualitatively
(positive/negative) according to the manufacturer's reference
range. In the event that diagnostic uncertainty persists, it is
recommended to repeat the test within a time frame of 2 to 3 weeks.
Conversely, a negative result does not rule out, but makes it less
likely; repeated testing may also be indicated if doubts remain
(29,31). Differential diagnoses included
autoimmune encephalitis (e.g., anti-NMDAR), metabolic and toxic
encephalopathies, infections (HIV, syphilis) and neoplastic
processes. These were excluded through comprehensive evaluation,
and the clinical course and imaging were inconsistent with
Alzheimer's disease, corticobasal degeneration, or other
tauopathies (7,32).
The present case report highlights the importance of
early neurological assessment when behavioral or cognitive decline
progresses rapidly, particularly when initial symptoms mimic
psychiatric illness. Early recognition enabled appropriate
supportive and palliative management while avoiding unnecessary or
invasive interventions (14).
In the context of the current literature, the
present case report reinforces the need for vigilance when
evaluating rapidly progressive neurocognitive syndromes. Despite
diagnostic advances, establishing a definitive diagnosis without
neuropathological confirmation remains challenging. Emerging
biomarkers, such as the real-time quaking-induced conversion
(RT-QuIC) assay, tau proteins and neurofilament light chain show
promise in complementing clinical and imaging criteria, potentially
reducing reliance on postmortem confirmation (31,33-35).
The case presented herein is distinctive for its
initial psychiatric misdiagnosis, rapid clinical decline and clear
demonstration of classical sCJD markers, allowing a robust
diagnosis based on combined clinical, imaging and laboratory
evidence. Some limitations include the absence of neuropathological
confirmation, which remains the gold standard for definitive
diagnosis, and the single-patient design, which limits
generalizability. Nevertheless, the combination of clinical,
imaging and biomarker findings strongly supports a probable
diagnosis of sCJD. Future research is required to focus on
validating integrated biomarker panels and advanced neuroimaging
techniques to improve early, non-invasive diagnosis and patient
management.
Acknowledgements
The authors would like to thank Mr. Julio V. Barrios
Nuñez from the University of Colima (Colima, Mexico) for providing
assistance with English language editing.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
VRJR was involved in the clinical management of the
patient, in the conceptualization of the case report, and in the
drafting of the initial version of the manuscript. WSRG supervised
the overall study, provided institutional resources, contributed to
project administration, and performed the critical review of the
manuscript for important intellectual content. In addition, WSRG
actively participated in the conception of the case report,
contributed to the clinical analysis and interpretation of
patient's data, and was involved in the interpretation of key
diagnostic studies relevant to the case. EDS participated in data
collection, organization of clinical and laboratory records,
literature review, and manuscript editing. ATSV coordinated the
acquisition and interpretation of diagnostic studies, including EEG
and MRI, and contributed to validation of clinical data and final
revision of the manuscript. DAFG processed and prepared the
diagnostic imaging and figures for publication using medical image
visualization software and contributed to data interpretation and
manuscript editing. All authors contributed to data interpretation,
reviewed the manuscript critically for accuracy and intellectual
content, and approved the final version of the manuscript. VRJR and
WSRG confirm the authenticity of all the raw data. All authors have
read and approved the final version of the manuscript and that all
are accountable for all aspects of the work.
Ethics approval and consent to
participate
The present case report was reviewed and approved by
the Research Ethics Committee and the Health Research Committee of
the General Hospital ‘Dr. Belisario Domínguez’, Coordination of
Teaching and Health Research, Institute for Social Security and
Services for State Workers (ISSSTE), Tuxtla Gutiérrez, Chiapas,
Mexico. The case was conducted in accordance with the ethical
standards of the institutional and national research committees and
with the 1964 Declaration of Helsinki and its later amendments.
Written informed consent was obtained from the patient's legal
representative for her participation in the present study. All data
were anonymized to protect patient confidentiality. The case did
not involve experimental procedures and was based solely on routine
clinical care.
Patient consent for publication
Written informed consent was obtained from the
patient's legal representative for the publication of the present
case report and any accompanying images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Noor H, Baqai MH, Naveed H, Naveed T,
Rehman SS, Aslam MS, Lakdawala FM, Memon WA, Rani S, Khan H, et al:
Creutzfeldt-Jakob disease: A comprehensive review of current
understanding and research. J Neurol Sci.
467(123293)2024.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ladogana A, Puopolo M, Croes EA, Budka H,
Jarius C, Collins S, Klug GM, Sutcliffe T, Giulivi A, Alperovitch
A, et al: Mortality from Creutzfeldt-Jakob disease and related
disorders in Europe, Australia, and Canada. Neurology.
64:1586–1591. 2005.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ritchie DL and Smith C: Pathological
spectrum of sporadic Creutzfeldt-Jakob disease. Pathology.
57:196–206. 2025.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Jurcau MC, Jurcau A, Diaconu RG, Hogea VO
and Nunkoo VS: A systematic review of sporadic Creutzfeldt-Jakob
disease: Pathogenesis, diagnosis, and therapeutic attempts. Neurol
Int. 16:1039–1065. 2024.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Hall WA and Masood W: Creutzfeldt-Jakob
disease. In: StatPearls [Internet]. StatPearls Publishing, Treasure
Island, FL, 2025.
|
|
6
|
Appleby BS, Appleby KK and Rabins PV: Does
the presentation of Creutzfeldt-Jakob disease vary by age or
presumed etiology? A meta-analysis of the past 10 years. J
Neuropsychiatry Clin Neurosci. 19:428–435. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Geschwind MD: Rapidly progressive
dementia. Continuum (Minneap Minn). 22:510–537. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Steinhoff BJ, Zerr I, Glatting M,
Schulz-Schaeffer W, Poser S and Kretzschmar HA: Diagnostic value of
periodic complexes in Creutzfeldt-Jakob disease. Ann Neurol.
56:702–708. 2004.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Vitali P, MacCagnano E, Caverzasi E, Henry
RG, Haman A, Torres-Chae C, Johnson DY, Miller BL and Geschwind MD:
Diffusion-weighted MRI hyperintensity patterns differentiate CJD
from other rapid dementias. Neurology. 76:1711–1719.
2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Centers for Disease Control and
Prevention: CJD Diagnostic Criteria. Centers for Disease Control
and Prevention, Atlanta, GA, 2024. https://www.cdc.gov/creutzfeldt-jakob/hcp/clinical-overview/diagnosis.html?CDC_AAref_Val=https://www.cdc.gov/prions/cjd/diagnostic-criteria.html.
Accessed July 5, 2025.
|
|
11
|
University of California, San Francisco
(UCSF): Familial Prion Disease. https://memory.ucsf.edu/genetics/familial-prion-disease.
Accessed July 2025.
|
|
12
|
World Medical Association. World Medical
Association Declaration of Helsinki: Ethical principles for medical
research involving human subjects. JAMA. 310:2191–2194.
2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Puoti G, Bizzi A, Forloni G, Safar JG,
Tagliavini F and Gambetti P: Sporadic human prion diseases:
Molecular insights and diagnosis. Lancet Neurol. 11:618–628.
2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zerr I, Kallenberg K, Summers DM, Romero
C, Taratuto A, Heinemann U, Breithaupt M, Varges D, Meissner B,
Ladogana A, et al: Updated clinical diagnostic criteria for
sporadic Creutzfeldt-Jakob disease. Brain. 132:2659–2668.
2009.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Kaur J, Lam MT, Singh S and Somal NK: Slow
to respond: A rapidly progressive case of sporadic
Creutzfeldt-Jakob disease. Cureus. 16(e53381)2024.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Bahrami SN, Karimi AS, Khosravi S and
Almasi-Dooghaee M: Parkinsonism as an initial presentation of
Creutzfeldt-Jakob disease: A case report and review of literature.
Clin Case Rep. 12(e9278)2024.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zitser J, Forner S, Wong K, Chengshi J,
Neuhaus J, Martindale J, Raudabaugh BJ, Benisano K, Goodman-O'Leary
K, Metcalf S, et al: Neuropsychiatric symptoms in sporadic
Creutzfeldt-Jakob disease. Brain: Mar 28, 2025 (Epub ahead of
print).
|
|
18
|
Mader EC, El-Abassi R,
Villemarette-Pittman NR, Santana-Gould L, Olejniczak PW and England
JD: Sporadic Creutzfeldt-Jakob disease with focal findings: Caveats
to current diagnostic criteria. Neurol Int. 5(e1)2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Hermann P, Appleby B, Brandel JP, Caughey
B, Collins S, Geschwind MD, Green A, Haïk S, Kovacs GG, Ladogana A,
et al: Biomarkers and diagnostic guidelines for sporadic
Creutzfeldt-Jakob disease. Lancet Neurol. 20:235–246.
2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Frank A, Bendig J, Schnalke N,
Klingelhoefer L, Reichmann H, Akgün K, Ziemssen T and Falkenburger
BH: Serum neurofilament indicates accelerated neurodegeneration and
predicts mortality in late-stage Parkinson's disease. NPJ
Parkinsons Dis. 10(14)2024.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Baiardi S, Rossi M, Giannini G, Mammana A,
Polischi B, Sambati L, Mastrangelo A, Magliocchetti F, Cortelli P,
Capellari S, et al: Head-to-head comparison of four cerebrospinal
fluid and three plasma neurofilament light chain assays in
Parkinsonism. NPJ Parkinsons Dis. 11(98)2025.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Xiang C and Cong S, Tan X, Ma S, Liu Y,
Wang H and Cong S: A meta-analysis of the diagnostic utility of
biomarkers in cerebrospinal fluid in Parkinson's disease. NPJ
Parkinsons Dis. 8(165)2022.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Gilman S, Wenning GK, Low PA, Brooks DJ,
Mathias CJ, Trojanowski JQ, Wood NW, Colosimo C, Dürr A, Fowler CJ,
et al: Second consensus statement on the diagnosis of multiple
system atrophy. Neurology. 71:670–676. 2008.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Postuma RB, Poewe W, Litvan I, Lewis S,
Lang AE, Halliday G, Goetz CG, Chan P, Slow E, Seppi K, et al:
Validation of the MDS clinical diagnostic criteria for Parkinson's
disease. Mov Disord. 33:1601–1608. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Centeno M, Tierney TM, Perani S, Shamshiri
EA, St Pier K, Wilkinson C, Konn D, Vulliemoz S, Grouiller F,
Lemieux L, et al: Combined electroencephalography-functional
magnetic resonance imaging and electrical source imaging improves
localization of pediatric focal epilepsy. Ann Neurol. 82:278–287.
2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Vulliemoz S, Lemieux L, Daunizeau J,
Michel CM and Duncan JS: The combination of EEG source imaging and
EEG-correlated functional MRI to map epileptic networks. Epilepsia.
51:491–505. 2010.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Collins S, Boyd A, Fletcher A, Gonzales M,
McLean CA, Byron K and Masters CL: Creutzfeldt-Jakob disease:
Diagnostic utility of 14-3-3 protein immunodetection in
cerebrospinal fluid. J Clin Neurosci. 7:203–208. 2000.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Van Everbroeck B, Boons J and Cras P:
Cerebrospinal fluid biomarkers in Creutzfeldt-Jakob disease. Clin
Neurol Neurosurg. 107:355–360. 2005.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Dorey A, Tholance Y, Vighetto A,
Perret-Liaudet A, Lachman I, Krolak-Salmon P, Wagner U, Struyfs H,
De Deyn PP, El-Moualij B, et al: Association of cerebrospinal fluid
prion protein levels and the distinction between Alzheimer disease
and Creutzfeldt-Jakob disease. JAMA Neurol. 72:267–275.
2015.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Otto M, Wiltfang J, Tumani H, Zerr I,
Lantsch M, Kornhuber J, Weber T, Kretzschmar HA and Poser S:
Elevated levels of tau-protein in cerebrospinal fluid of patients
with Creutzfeldt-Jakob disease. Neurosci Lett. 225:210–212.
1997.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Foutz A, Appleby BS, Hamlin C, Liu X, Yang
S, Cohen Y, Chen W, Blevins J, Fausett C, Wang H, et al: Diagnostic
and prognostic value of human prion detection in cerebrospinal
fluid. Ann Neurol. 81:79–92. 2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Dalmau J, Lancaster E, Martinez-Hernandez
E, Rosenfeld MR and Balice-Gordon R: Clinical experience and
laboratory investigations in patients with anti-NMDAR encephalitis.
Lancet Neurol. 10:63–74. 2011.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Atarashi R, Satoh K, Sano K, Fuse T,
Yamaguchi N, Ishibashi D, Matsubara T, Nakagaki T, Yamanaka H,
Shirabe S, et al: Ultrasensitive human prion detection in
cerebrospinal fluid by real-time quaking-induced conversion. Nat
Med. 17:175–178. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
34
|
Kuzkina A, Bargar C, Schmitt D, Rößle J,
Wang W, Schubert AL, Tatsuoka C, Gunzler SA, Zou WQ, Volkmann J, et
al: Diagnostic value of skin RT-QuIC in Parkinson's disease: A
two-laboratory study. NPJ Parkinsons Dis. 7(99)2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Senesi M, Lewis V, Varghese S, Stehmann C,
McGlade A, Doecke JD, Ellett L, Sarros S, Fowler CJ, Masters CL, et
al: Diagnostic performance of CSF biomarkers in a
well-characterized Australian cohort of sporadic Creutzfeldt-Jakob
disease. Front Neurol. 14(1072952)2023.PubMed/NCBI View Article : Google Scholar
|