Introduction
Chronic myeloid leukaemia (CML) is a clonal,
myeloproliferative disease (1).
The annual incidence of CML is 1–2 cases per 100,000 individuals
and the median age of presentation is 45–55 years, accounting for
15–20% of leukemia cases in adults (2). Based on clinical characteristics and
laboratory findings, CML is often divided into three phases,
chronic phase (CP), accelerated phase (AP) and blast phase (BP).
Without intervention, CML typically begins in CP and over the
course of several years progresses to AP, followed by BP. If
administered early, drug treatment prevents this progression to a
certain degree. A significant driver of progression from CP through
AP and BP is the acquisition of additional chromosomal
abnormalities to the Philadelphia (Ph) chromosome (2).
As the first consistent chromosomal abnormality
associated with a specific type of leukemia, the identification of
the Ph chromosome in 1960 by Nowell and Hungerford was a
breakthrough in cancer biology (3). The Ph chromosome is the result of a
t(9;22) reciprocal chromosomal translocation (4), involving the V-abl Abelson murine
leukemia viral oncogene homolog 1 (ABL) proto-oncogene normally
located on chromosome 9 and the breakpoint cluster region (BCR)
gene on chromosome 22 (5). The
translocation results in deregulation of ABL tyrosine kinase
activity and is defined as the pathogenetic principle (6). In addition to the classic BCR-ABL
translocation in CML (7), a number
of other gene fusions that contribute to oncogenesis have been
reported in previous studies (8).
Gene fusions are the result of aberrant chromosomal translocations
that join together the exons of two unrelated genes, producing a
chimeric mRNA transcript and protein (9). Previously, massively parallel RNA
sequencing (RNA-Seq) was performed to identify gene fusions
(10). RNA-Seq enables
identification of gene fusions in individual cancer samples and
facilitates comprehensive understanding of cellular transcriptomes
(11,12) through profiling the entire
transcriptome at a level of detail unattainable by microarray
(13).
In the present study, we performed RNA-Seq to detect
gene fusions in a patient who did not present with common types of
BCR-ABL gene fusions. Using an improved pipeline based on two tools
for the detection of gene fusion, we identified several candidates
of gene fusion in CP, including BCR-ABL1 and a novel gene fusion
ring finger protein 213 (RNF213)-solute carrier family 26, member
11 (SLC26A11). Expression levels and functional domain change of
the novel gene fusion were analyzed using a bioinformatic method
and compared with genes involved in normal tissue funtion.
Materials and methods
Sample information
A single CML case from a patient of the Jiang Yin
People’s Hospital forms the basis of this report. The sample for
gene fusion study was obtained in March 2011 and the patient was a
Chinese male aged 78 years old. Clinical information was obtained
from patient charts in the Jiang Yin People’s Hospital. CML in CP
was diagnosed in 2005 on the basis of a morphological study of bone
marrow and peripheral blood specimens (total of 8.1% myeloblast and
progranulocyte in blood smear with WBC=34.8×109/l, Hb=62
g/l and PLT=529×109/l). In addition to increased
myeloblast and progranulocyte levels, additional symptoms were
identified, including blood dilution and splenomegaly demonstrated
using ultrasonography. However, b3a2 b2a2 and ela2 fusion junctions
in the BCR-ABL gene were not detected in 2008. One year later,
detection of the classic BCR-ABL breakpoint remained negative.
Written informed consent from the patient was
obtained and the present study was reviewed and approved by the
Clinical Laboratories Department of the People’s Hospital in Jiang
Yin City (Jiang Yin, China).
Library preparation
Total RNA was extracted from the peripheral blood
using TRIzol according to the manufacturer’s instructions
(Invitrogen, Carlsbad, CA, USA). Following the TruSeq RNA
SamplePrep Guide (Illumina, Inc., San Diego, CA, USA), the Illumina
standard kit was used for mRNA-Seq sample preparation. Briefly, 10
μg of total RNA extracted from the sample was used for polyA mRNA
selection with polyT oligo-conjugated magnetic beads by two rounds
of purification, followed by thermal mRNA fragmentation. Using
reverse transcriptase (SuperScript II) and random primers, the
fragmented mRNA was subjected to cDNA synthesis. Following this,
cDNA was converted into double-stranded cDNA and after end repair
[Klenow fragment, T4 polynucleotide kinase, T4 polymerase and 3-‘A’
add process (Klenow exo-fragment)]the product was ligated to
Illumina Truseq adaptors. A 2% agarose gel was used to perform size
selection, which generated 380-bp cDNA libraries. Finally, the
libraries were enriched through 15 cycles of PCR and purified with
the QIAquick PCR purification kit (Qiagen, Hilden, Germany).
Elution buffer was used to dilute the enriched libraries to a final
concentration of 10 nM.
Primary analysis
Libraries from CML blood in CP were analyzed at a
concentration of 11 pM on a single Genome Analyzer IIx (GAIIx) lane
using 115-bp sequencing. Raw RNA-Seq data were filtered using
FASTx-tools (http://hannonlab.cshl.edu/fastx_toolkit/) according to
the following criteria: i) reads containing sequencing adaptors
were removed; ii) nucleotides with a quality score <20 were
trimmed from the end of the sequence; iii) reads <50 were
discarded; and iv) artificial reads were removed. Following the
filtering pipeline, all clean reads were trimmed to 90 bp long due
to 3′ low quality base. A total of 3.2 Gbp (3,193,208,820) of
cleaned, paired-end reads was produced. Raw sequence data were
submitted to the NCBI Short Read Archive (accession number,
SRP011486).
Reads mapping and expression
estimation
Clean and trimmed reads were aligned with the
Ensembl H. sapiens reference genome (build GRCh37) using
TopHat v1.3.3 (14).The program
initially removes a portion of the reads based on quality
information accompanying each read and then aligns the reads to the
reference genome. TopHat allows multiple alignments per read (up to
20 by default) and a maximum of two mismatches when mapping the
reads to the reference genome. The default parameters were used.
Aligned read files were then processed by Cufflinks v1.2.1
(15), which measures the relative
expression of the genes with the normalized RNA-Seq fragment
counts. Fragments per kilobase of exon per million fragments mapped
(FPKM) is used as the unit of measurement. A Bayesian inference
method (16) was used to calculate
confidence intervals for FPKM estimates. The reference GTF
annotation file used in Cufflinks was downloaded from the Ensembl
database (Homo_sapiens.GRCh37.63.gtf) (17). The gene expression data were
submitted to the GEO database (accession ID, GSE36522).
Gene fusion prediction
Using TopHat, all filtered RNA-Seq reads were mapped
to the reference transcriptome, which was downloaded from the
Ensembl database (Homo_sapiens.GRCh37.63.cdna.all.fa). Read pairs
mapping to the same transcripts were removed. Remaining reads were
analyzed by two software packages, deFuse (deFuse-0.4.2) (18) and TopHat-Fusion
(TopHat-Fusion-0.1.0) (19), to
identify candidate gene fusions. The bowtie-index used in the
TopHat-Fusion was downloaded from the TopHat homepage (H.
sapiens UCSC hg19). The parameters of the TopHat-Fusion used
were obtained from the ‘Getting Started’ (http://tophat-fusion.sourceforge.net/tutorial.html)
tutorial. The deFuse parameters used were default.
Candidate gene fusion filtering
deFuse results were filtered according to McPherson
et al(18) and Steidl et
al(20). Two candidate gene
fusions were obtained following the filtering pipeline. The
TopHat-Fusion results were parsed by in-house Perl scripts and
filtered according to the following pipeline: i) span reads were
>8 reads; ii) ratio of against reads vs. span reads was <0.5;
and iii) gene fusions involving ribosomal proteins or small nuclear
ribosomal proteins were excluded. Following parsing, only one
filtered candidate fusion remained. Filtered candidates detected
simultaneously by deFuse and TopHat-Fusion were considered reliable
candidate gene fusions. Following this filtering pipeline, a single
reliable candidate, RNF213-SLC26A11, was obtained.
Improvement of filtering method
To reduce the false-positive rate and remove
read-throughs, we improved the filtering standard by comparing the
conditions of our filtering method with those of the final results.
Two filtering standards of deFuse were modified: i) the number of
splitr_count was revised according to the average coverage of the
sequencing depth and ii) the events, which happened between 2
adjacent genes without any eversion or inversion, were considered
to be produced by read-through. The detailed conditions of the 14
candidates are listed in Table
I.
 | Table IFiltering improvement of deFuse by
manual checks. |
Table I
Filtering improvement of deFuse by
manual checks.
| Gene fusion | splitr_count |
splitr_span_p-value |
splitr_pos_p-value |
splitr_min_p-value | Adjacent | Eversion | Inversion | span_count | span_coverage2 | span_coverage1 | chr1/chr2a | pos1b | pos2b |
|---|
| RNF213/SLC26A11 | 57 | 1 | 0.99 | 0.39 | Y | Y | N | 115 | 1.29 | 1.29 | 17/17 | 78237577 | 78210727 |
| BCR/ABL1 | 1 | 1 | 0.31 | 0.26 | N | N | N | 5 | 0.51 | 0.62 | 22/9 | 23524426 | 133729451 |
| HRH2/U6.171 | 23 | 1 | 0.96 | 0.11 | Y | N | N | 27 | 0.93 | 0.95 | 5/5 | 175111312 | 175134782 |
| PMVK/PBXIP1 | 6 | 0.69 | 0.52 | 0.26 | Y | N | N | 10 | 0.71 | 0.71 | 1/1 | 154912826 | 154918038 |
|
SP110/AC093171.1 | 6 | 1 | 0.51 | 0.51 | Y | N | N | 13 | 1.23 | 1.16 | 2/2 | 231033762 | 231031005 |
|
AC068580.6/AC068580.7 | 9 | 0.21 | 0.84 | 0.95 | Y | N | N | 6 | 0.96 | 0.93 | 11/11 | 1782698 | 1792621 |
| OR10AA1P/MNDA | 16 | 0.89 | 0.79 | 0.6 | Y | N | N | 18 | 1.31 | 1.02 | 1/1 | 158772327 | 158811922 |
|
ACTBP8/RP11-459O1.2 | 6 | 1 | 0.91 | 0.58 | Y | N | N | 9 | 0.65 | 0.67 | 6/6 | 88990671 | 89008583 |
| GPR77/DHX34 | 8 | 0.76 | 0.9 | 0.25 | Y | N | N | 6 | 1.07 | 0.84 | 19/19 | 47845088 | 47856011 |
| UTY/DPPA2P1 | 9 | 0.41 | 0.49 | 0.53 | Y | N | N | 8 | 0.98 | 0.83 | Y/Y | 15362897 | 15346707 |
| EPX/MKS1 | 2 | 0.9 | 0.24 | 0.26 | Y | N | N | 6 | 0.75 | 1.02 | 17/17 | 56281795 | 56284250 |
| LRP10/REM2 | 1 | 0.9 | 0.89 | 0.88 | Y | N | N | 5 | 0.67 | 0.66 | 14/14 | 23350226 | 23353881 |
| LRRC6/TMEM71 | 6 | 0.9 | 0.47 | 0.35 | N | N | N | 7 | 0.79 | 0.89 | 8/8 | 133673873 | 133697188 |
Gene fusion validation
To detect fusion transcripts, we designed a forward
primer targeting the 5′ partner gene and reverse primer targeting
the 3′ partner. Primer pairs (Table
II) for the coding exons of the fusion genes were generated
using Primer 5 software (Premier Biosoft International, Palo Alto,
CA, USA) and the PCR volume comprised 20 μl sample, 2 μl 10X PCR
buffer, 2 μl cDNA template, 0.4 μl dNTP, 0.4 μl Taq enzyme
(Genscript, Piscataway, NJ, USA) and 0.2 pmol/μl each
oligonucleotide. PCR was performed using the following procedure:
95°C for 2 min, 35 cycles of 95°C for 15 sec, 62°C for 20 sec and
72°C for 20 sec, followed by 72°C for 2 min. The presence of the
fusion gene in the CML blood sample was confirmed. PCR products of
the fusion gene were cloned in the pGEM®-T Easy Vector
(Promega Corp., Madison, WI, USA) and then sequenced with the T7
primer using an Applied Biosystems 3730 DNA Analyzer (Life
Technologies Corporation, USA).
| Table IIPrimer pairs designed for detecting
gene fusion. |
Table II
Primer pairs designed for detecting
gene fusion.
| Gene name | Primers
(5′-3′) |
|---|
| RNF213 |
GACTCCTGCTCTTGCTTCTGG |
| SLC26A11 |
ATCGTCCCGTTGGCTGTG |
Gene fusion visualization
Views of the candidate gene fusions were generated
by running CIRCOS (available at http://circos.ca/). Reads coverage of the candidate
gene fusions in the sample were observed in the Integrative
Genomics Viewer (IGV).
Results
Characterization of sequencing and
mapping
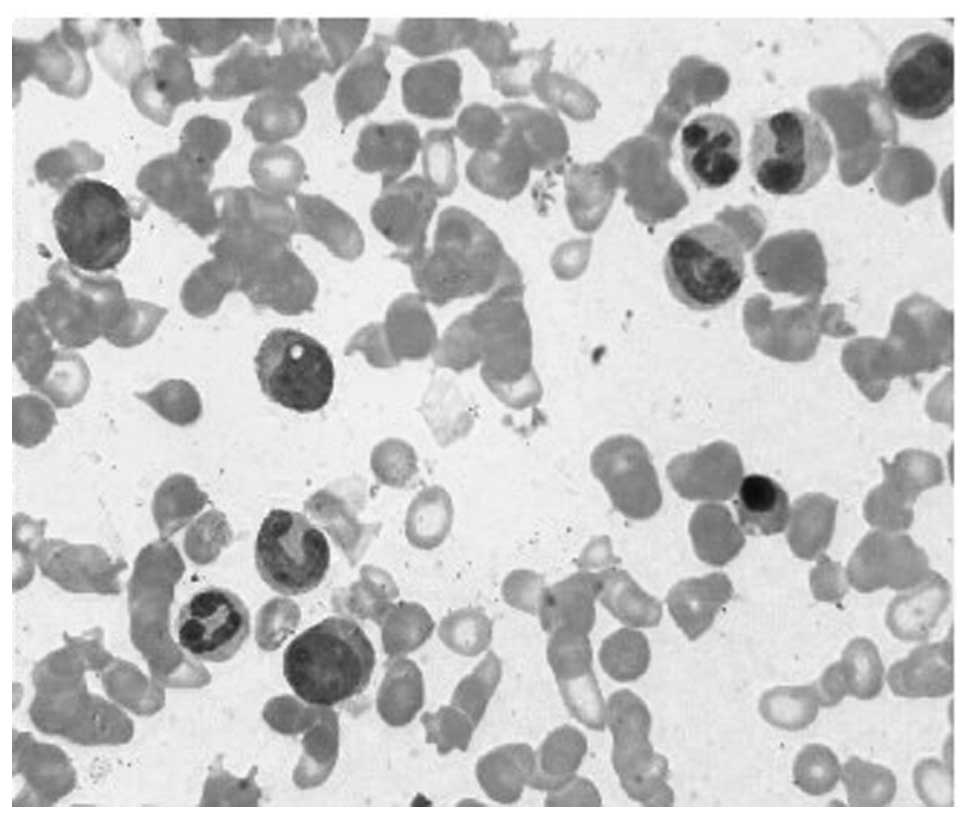
CML blood in CP was obtained from a male patient of
78 years old. The peripheral blood smear is demonstrated in
Fig. 1. Massively parallel paired
end cDNA sequencing was performed using the Illumina Genome
Analyzer IIx. In total, 43.6 million raw reads were produced.
Following filtering and trimming, 35.5 million clean reads were
obtained. All reads were aligned to the reference genome by TopHat.
In summary, 15.3 million read pairs were uniquely aligned. Reads
(74%) were mapped to Ensembl reference genes. The average coverage
of the sequencing depth was >30-fold of the human transcriptome.
Only 4% reads were mapped to the H. sapiens mitochondrial
genome. Raw 115-bp long reads from a single lane on the platform of
Genome Analyzer IIx (GAIIx) was analysed. In addition, 20.89%
(7,413,014) reads were splice junction reads. All observations were
indicative of a correctly constructed library.
Analysis of gene expression
To measure gene expression in the sample, we
utilized Cufflinks to estimate gene expression. Normalized
expression levels of each gene were measured in FPKM. By selecting
genes with an FPKM value >1, 22,607 expressed genes were
detected in the sample, including the majority of annotated
reference genes in H. sapiens.
Prediction of gene fusion events
Two algorithms, including deFuse and TopHat-Fusion,
were used to detect gene fusion events in the sample. Following
removal of read pairs mapping to the same genes, 5.4 million
(~30.9%, 5.4/17.7) reads pairs were obtained for gene fusion event
prediction. Several gene fusion candidates were discovered, the
majority of which were located within the chromosome. Following a
manual check of the raw results produced by deFuse, unreliable
results were removed. The filtered results of TopHat-Fusion and
deFuse were combined to produce 14 gene fusion candidates (Table I). Among the candidates, one gene
fusion event RNF213-SLC26A11 was detected by deFuse and
TopHat-Fusion with a 3-bp breakpoint shift.
Improvement of filtering gene fusion
The majority of candidate gene fusions were
identified within the same chromosome (Table I). Using manual checks and
selection, we identified that the majority of fusion events
occurred between adjacent genes on the same chromosome, which were
usually regarded as read-through (21), while only two candidates, including
RNF213-SLC26A11 and BCR-ABL1, were the most reliable gene fusion
pairs. BCR-ABL is a well-documented gene fusion in CML and is used
as a standard method for CML diagnosis (22). The RNF213-SLC26A11 fusion has not
been previously identified in CML. In the present study, this novel
fusion was further validated by reverse transcription polymerase
chain reaction (RT-PCR) and the function of RNF213-SLC26A11 was
predicted using bioinformatic methods.
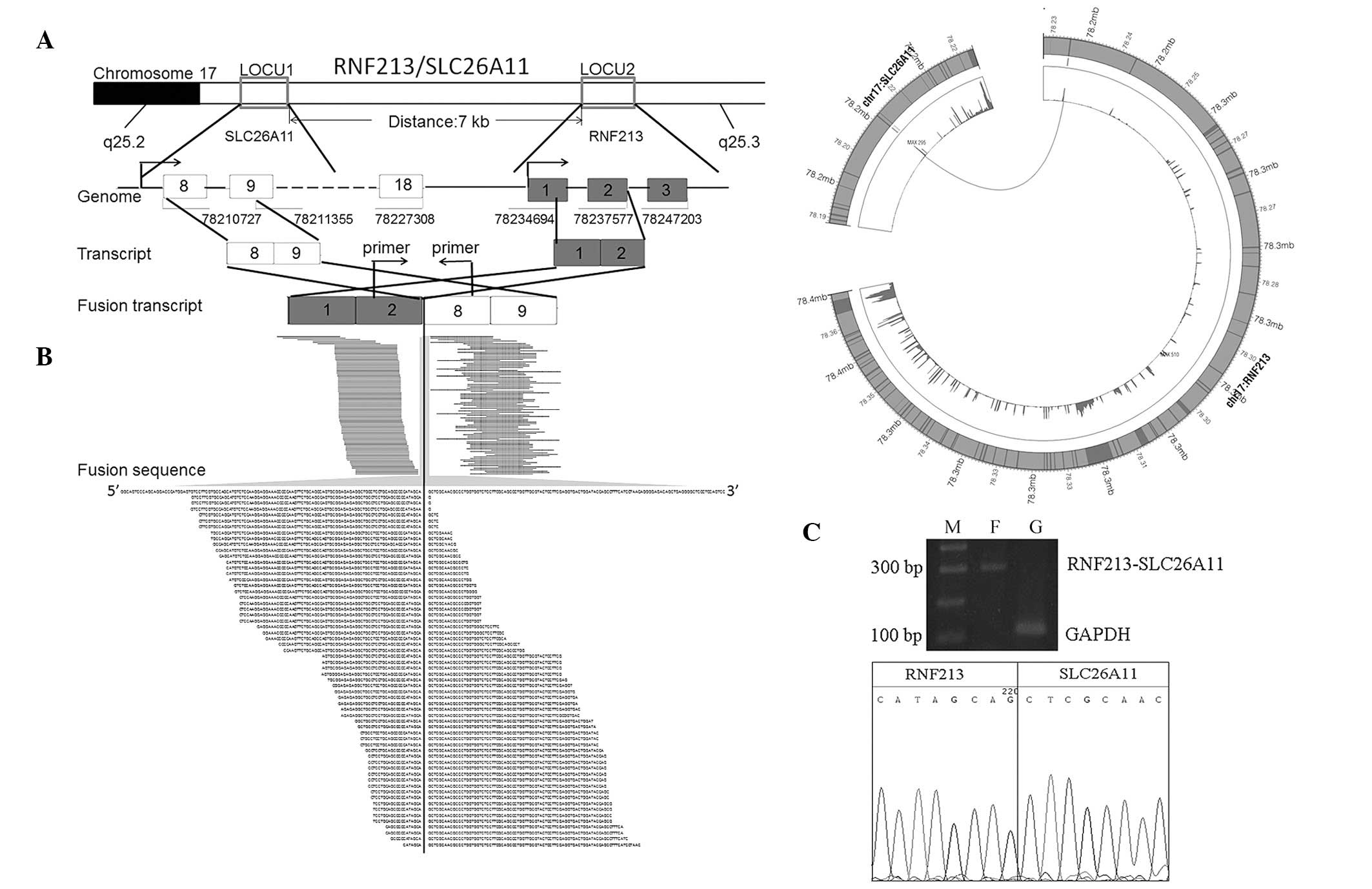
RT-PCR validation of RNF213-SLC26A11
To experimentally confirm the novel gene fusion
identified by RNA-Seq, the expression level of RNF213-SLC26A11 in
the sample was validated by RT-PCR. A primer pair, located at the
second intron of RNF213 (5′-GACTCCTGCTCTTGCTTC TGG-3′) and the 8th
exon region of SLC26A11 (5′-ATCGTC CCGTTGGCTGTG-3′) was designed.
The results confirmed the existence of the fusion event in the
sample (Fig. 2C), consistent with
conclusions based on RNA-Seq analysis.
Identification of novel gene fusion
RNF213-SLC26A11
As demonstrated in Fig.
2A, RNF213 and SLC26A11 are adjacent gene pairs located in
chromosome 17 separated by 7 kbp. The genes are transcribed in the
same direction in wild-type individuals. In the present sample, we
observed a fusion between the second intron of RNF213 and the
eighth exon of SLC26A11, generating a chimeric RNF213-SLC26A11
transcript, which has not previously been identified.
Analysis of reads coverage for RNF213 and
SLC26A11
Analysis of reads coverage of RNF213 and SLC26A11
demonstrated that each exon of RNF213 was expressed at normal
levels and the first 7 exons of SLC26A11 were expressed at
extremely low levels (Fig. 2B),
indicating that i) the RNF213-SLC26A11 fusion may occur in the
majority of cells or ii) the fusion event altered expression of
SLC26A11 using the promoter of RNF213, largely caused by the
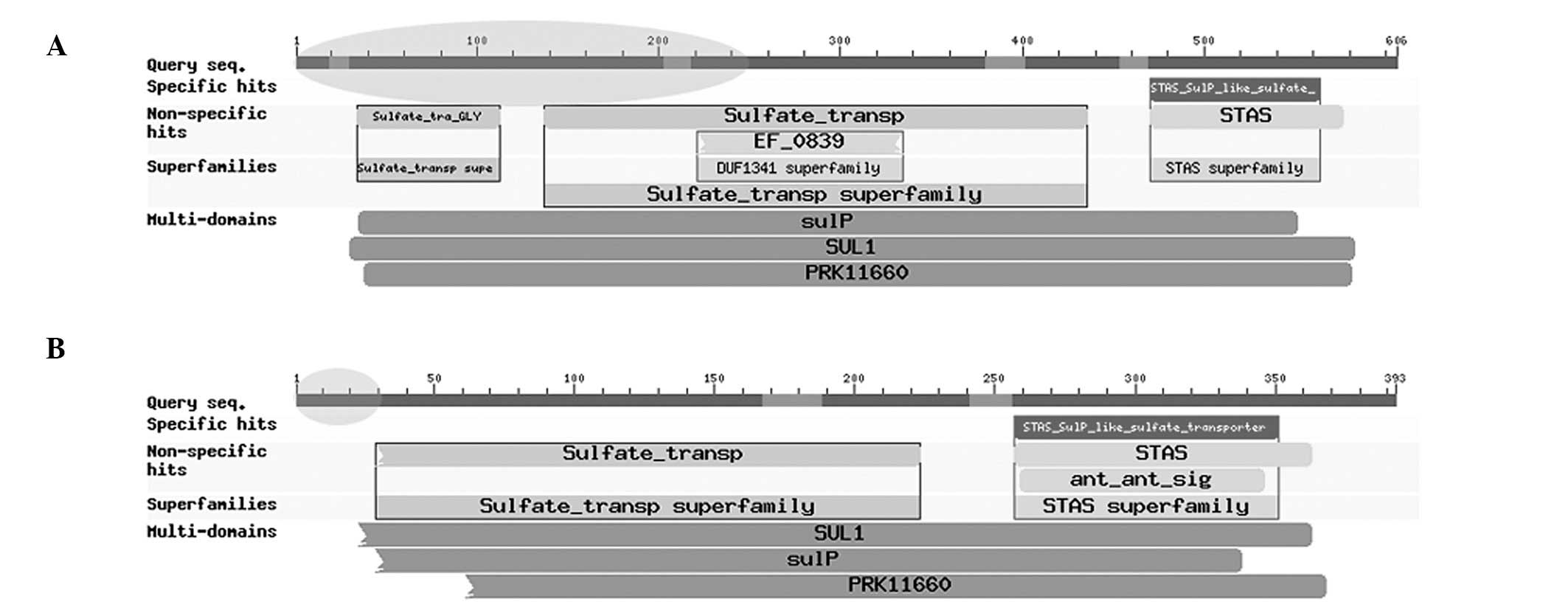
duplication of the first 2 exons of RNF213. RNF213 was normally
expressed. In addition, compared with domains of the whole SLC26A11
gene under normal conditions (Fig.
3A), specific domains of the gene SLC26A11 in the chimeric
transcript, including Sulfate_tra_GLY (pfam13792), EF_0839
(TIGR03581), sulfate_transp (pfam00916), sulP (TIGR00815), SUL1
(COG0659) and PRK11660 (PRK11660), were partially damaged (Fig. 3B). These observations indicate that
the normal function of RNF213 did not appear to be affected, while
loss of function in SLC26A11 was identified. However, further
investigation is required to understand the specific mechanism and
functional consequence of this fusion event.
Identification of recurrent gene
fusion
In addition to the novel gene fusion, we identified
another gene fusion candidate, which had been previously identified
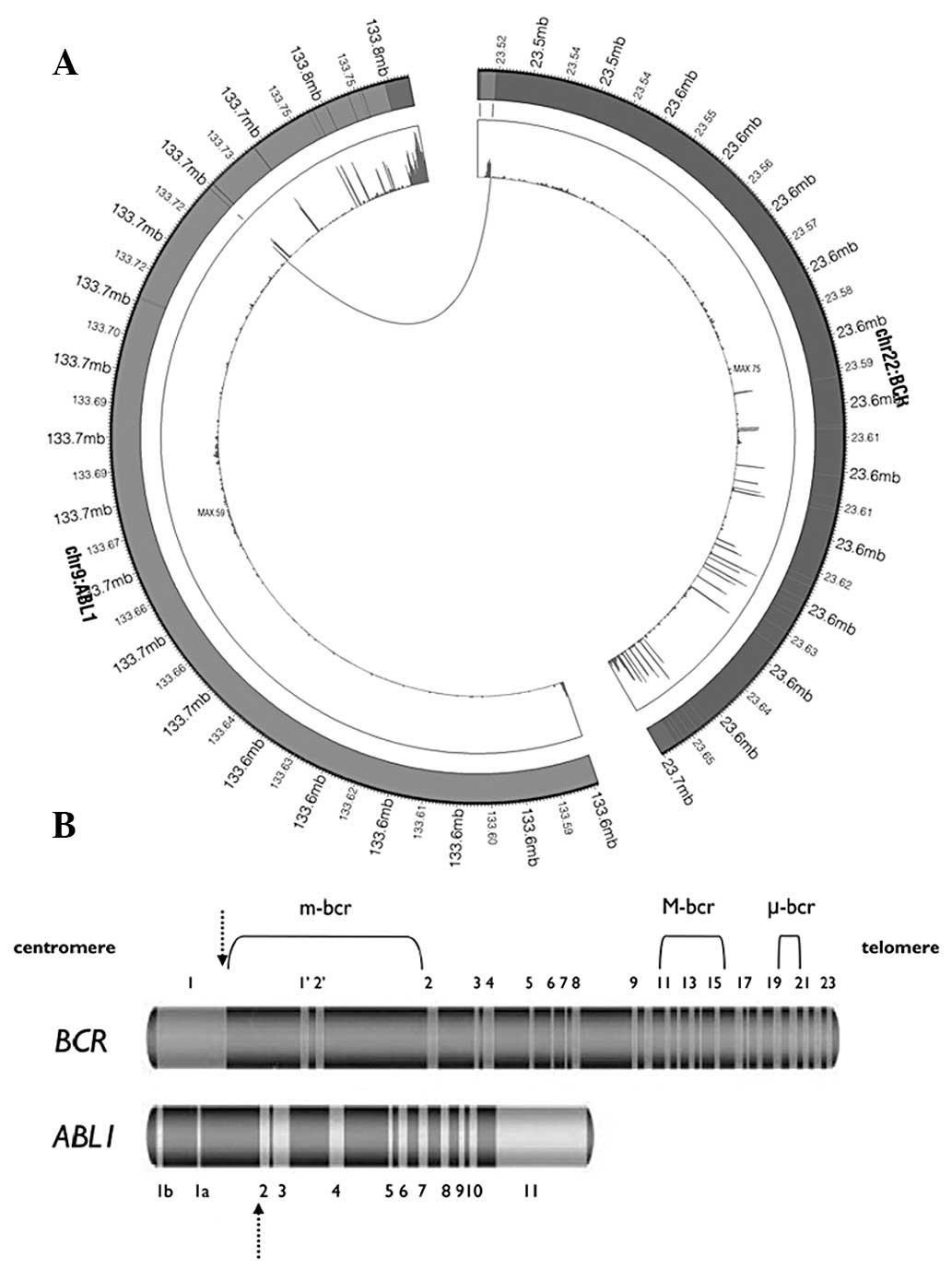
in CML (23). As demonstrated in
Fig. 4B, BCR is located in
chromosome 22 while ABL1 is on chromosome 9. In the present sample,
a fusion was identified between the first intron of BCR and the
second exon of ABL1, generating a chimeric transcript BCR-ABL1
(Fig. 4). Deininger et
al(24) previously reported
that this fusion is a e1a2 fusion junction and is extremely rare
(25).
Discussion
A number of methods are currently available for the
detection of gene fusions, including chromosome banding analysis
(karyotyping) followed by fluorescence in situ hybridization
studies and molecular analysis based on RT-PCR (26). The RNA-Seq method currently enables
genome-wide identification of novel fusion transcripts at the
highest resolution level (8,27,28).
The BCR-ABL fusion transcript is considered to be a major
consequence of the Ph translocation in CML (29). In the present study, whole
transcriptome high-throughput sequencing was performed to detect
additional fusion events to BCR-ABL, consistent with clinical
diagnosis of CML. Using an improved bioinformatics method and
RT-PCR, we identified extremely low levels of BCR-ABL1 in an
ela2-type sample (FPKM: BCR=20.28, ABL=9.80). An additional novel
gene fusion was also discovered.
To obtain the most reliable gene fusion candidates
in the blood sample obtained from a CML patient in CP, the
filtering method of deFuse was improved. Sample analysis using
deFuse and TopHat-Fusion methods identified several gene fusion
candidates. TopHat-Fusion results were filtered, mapping reads were
checked manually and ribosomes were removed from the raw results
produced by deFuse to obtain 14 gene fusion candidates. However,
the majority of candidates were identified within the chromosome.
Following detailed observation in IGV and improvement of the
filtering method of deFuse, two reliable gene fusions were
identified, including the novel gene fusion RNF213-SLC26A11 and the
recurrent gene fusion BCR-ABL1.
From analysis of the structure of the novel chimeric
transcript RNF213-SLC26A11, we identified that SLC26A11 expression
is largely mediated by promoter displacement of RNF213. SLC26A11
was initially characterized as a putative sulfate transporter
predominately expressed in high endothelial venules. SLC26A11 is
widely expressed and mRNA expression has been identified in the
kidney, placenta and brain (30).
The blood sample analyzed in the present study was obtained from a
CML patient in CP. Expression of the first 7 exons of SLC26A11 in
this sample was observed at extremely low levels (Fig. 2B). In addition, SLC26A11 has been
demonstrated to be expressed in the normal blood tissue in acute
myoleid leukemia (http://www.genecards.org/cgi-bin/carddisp.pl?gene=SLC26A11;
http://www.genecards.org/info.shtml#expression_images).
Furthermore, a number of domains identified in normal human samples
of SLC26A11 were also identified to be damaged by the formation of
the novel chimeric transcript RNF213-SLC26A11 (Fig. 3B), including Sulfate_tra_GLY,
EF_0839, sulfate_transp, sulP, SUL1 and PRK11660.
STAS_SulP_like_sulfate_transporter (cd07042) and STAS (pfam01740)
were observed to be unaffected. The present results indicate that
the sulfate transport function of SLC26A11 was weakened, while its
function in transport activity regulation and general NTP binding
were maintained following fusion.
To explore the correlation between the
RNF213-SLC26A11 gene fusion and CML, the invidual functions of
RNF213 and SLC26A11 under normal conditions were identified. RNF213
encodes a protein containing a C3HC4-type RING finger domain, a
specialized type of Zn-finger that binds two atoms of zinc and is
thought to be involved in mediating protein-protein interactions.
The protein also contains an AAA domain, which is associated with
ATPase activity. RNF213 is a susceptibility gene for Moyamoya
disease, a vascular disorder of intracranial arteries (31). In addition, the protein functions
as a translocation partner in anaplastic large cell lymphoma and
inflammatory myofibroblastic tumor cases. A t(2;17)(p23;q25)
translocation has been identified with the anaplastic lymphoma
kinase gene located on chromosome 2 and a t(8;17)(q24;q25)
translocation has been identified with the MYC gene on chromosome 8
(31). SLC26A11 encodes a member
of the solute linked carrier 26 family of anion exchangers. Members
of this family of proteins are essential for a number of cellular
functions, including homeostasis and intracellular electrolyte
balance. The encoded protein is a sodium-independent sulfate
transporter that is sensitive to the anion exchanger inhibitor
4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (RefSeq, Oct
2009). These functions indicate that the novel gene fusion
candidate RNF213-SLC26A11 commonly occurs and affects homeostasis
to a limited degree only. At present, the impact of this fusion on
CML remains unclear. The mechanism of this functional change and
its affect on the blood during CML require further
investigations.
In addition to identification of the novel gene
fusion RNF213-SLC26A11, we identified the recurrent gene fusion
BCR-ABL1 at extremely low levels of expression. Analysis of the
structure of this gene fusion demonstrated that the transcript
discovered in our sample exhibits a e1a2 fusion junction, producing
a 190-kDa protein (P190BCR-ABL). P190BCR-ABL CML has been
previously identified in only 1% of CML patients and this junction
is correlated with poor response to therapy with tyrosine kinase
inhibitors (TKIs), with few, usually short-lived responses.
Individuals with this gene fusion must be identified as high-risk
patients, monitored closely for efficacy during therapy with TKI
and offered stem cell transplantation early if eligible for this
procedure (25).
Following improvement of the filtering method, we
analyzed RNA-Seq data and discovered two gene fusions in a blood
sample obtained from a CML patient in CP, including a novel gene
fusion RNF213-SLC26A11 and the recurrent BCR-ABL1 fusion. In
addition, the present study has demonstrated that the current
filtering method of deFuse must be revised according to the average
coverage of the sequencing depth. This is likely to enable the
improved filtering method to be applied to gene fusion detection in
other tumor tissues.
References
|
1
|
Frazer R, Irvine AE and McMullin MF:
Chronic myeloid leukaemia in the 21st century. Ulster Med J.
76:8–17. 2007.PubMed/NCBI
|
|
2
|
Faderl S, Talpaz M, Estrov Z and
Kantarjian HM: Chronic myelogenous leukemia: biology and therapy.
Ann Intern Med. 131:207–219. 1999. View Article : Google Scholar
|
|
3
|
Nowefl PC and Hungerford DA: A minute
chromosome in human chronic granulocytic leukemia. Science.
142:14971960.
|
|
4
|
Rowley JD: Letter: A new consistent
chromosomal abnormality in chronic myelogenous leukaemia identified
by quinacrine fluorescence and Giemsa staining. Nature.
243:290–293. 1973. View
Article : Google Scholar
|
|
5
|
Tefferi A, Bren GD, Wagner KV, Schaid DJ,
Ash RC and Thibodeau SN: The location of the Philadelphia
chromosomal breakpoint site and prognosis in chronic granulocytic
leukemia. Leukemia. 4:839–842. 1990.PubMed/NCBI
|
|
6
|
Lugo TG, Pendergast AM, Muller AJ and
Witte ON: Tyrosine kinase activity and transformation potency of
BCR-ABL oncogene products. Science. 247:1079–1082. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shtivelman E, Lifshitz B, Gale RP and
Canaani E: Fused transcript of abl and bcr genes in chronic
myelogenous leukaemia. Nature. 315:550–554. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mitelman F, Johansson B and Mertens F: The
impact of translocations and gene fusions on cancer causation. Nat
Rev Cancer. 7:233–245. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ha KC, Lalonde E, Li L, et al:
Identification of gene fusion transcripts by transcriptome
sequencing in BRCA1-mutated breast cancers and cell lines. BMC Med
Genomics. 4:752011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Maher CA, Kumar-Sinha C, Cao X, et al:
Transcriptome sequencing to detect gene fusions in cancer. Nature.
458:97–101. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tang F, Barbacioru C, Wang Y, et al:
mRNA-Seq whole-transcriptome analysis of a single cell. Nat
Methods. 6:377–382. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mortazavi A, Williams BA, McCue K,
Schaeffer L and Wold B: Mapping and quantifying mammalian
transcriptomes by RNA-Seq. Nat Methods. 5:621–628. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Z, Gerstein M and Snyder M: RNA-Seq:
a revolutionary tool for transcriptomics. Nat Rev Genet. 10:57–63.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Trapnell C, Pachter L and Salzberg SL:
TopHat: discovering splice junctions with RNA-Seq. Bioinformatics.
25:1105–1111. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Roberts A, Trapnell C, Donaghey J, Rinn JL
and Pachter L: Improving RNA-Seq expression estimates by correcting
for fragment bias. Genome Biol. 12:R222011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jiang H and Wong WH: Statistical
inferences for isoform expression in RNA-Seq. Bioinformatics.
25:1026–1032. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hubbard TJ, Aken BL, Ayling S, et al:
Ensembl 2009. Nucleic Acids Res. 37:D690–D697. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McPherson A, Hormozdiari F, Zayed A, et
al: deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq
data. PLoS Comput Biol. 7:e10011382011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim D and Salzberg SL: TopHat-Fusion: an
algorithm for discovery of novel fusion transcripts. Genome Biol.
12:R722011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Steidl C, Shah SP, Woolcock BW, et al: MHC
class II transactivator CIITA is a recurrent gene fusion partner in
lymphoid cancers. Nature. 471:377–381. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nacu S, Yuan W, Kan Z, et al: Deep RNA
sequencing analysis of readthrough gene fusions in human prostate
adenocarcinoma and reference samples. BMC Med Genomics. 4:112011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Melo JV, Gordon DE, Cross NC and Goldman
JM: The ABL-BCR fusion gene is expressed in chronic myeloid
leukemia. Blood. 81:158–165. 1993.PubMed/NCBI
|
|
23
|
Quintas-Cardama A and Cortes J: Molecular
biology of BCR-ABL1-positive chronic myeloid leukemia. Blood.
113:1619–1630. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Deininger MW, Goldman JM and Melo JV: The
molecular biology of chronic myeloid leukemia. Blood. 96:3343–3356.
2000.PubMed/NCBI
|
|
25
|
Verma D, Kantarjian HM, Jones D, et al:
Chronic myeloid leukemia (CML) with P190 BCR-ABL: analysis of
characteristics, outcomes and prognostic significance. Blood.
114:2232–2235. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Skotheim RI, Thomassen GO, Eken M, et al:
A universal assay for detection of oncogenic fusion transcripts by
oligo microarray analysis. Mol Cancer. 8:52009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen W, Kalscheuer V, Tzschach A, et al:
Mapping translocation breakpoints by next-generation sequencing.
Genome Res. 18:1143–1149. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Campbell PJ, Stephens PJ, Pleasance ED, et
al: Identification of somatically acquired rearrangements in cancer
using genome-wide massively parallel paired-end sequencing. Nat
Genet. 40:722–729. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hermans A, Selleri L, Gow J and Grosveld
GC: Absence of alternative splicing in BCR-ABL mRNA in chronic
myeloid leukemia cell lines. Blood. 72:2066–2069. 1988.PubMed/NCBI
|
|
30
|
Vincourt JB, Jullien D, Amalric F and
Girard JP: Molecular and functional characterization of SLC26A11, a
sodium-independent sulfate transporter from high endothelial
venules. FASEB J. 17:890–892. 2003.PubMed/NCBI
|
|
31
|
Pruitt KD, Tatusova T and Maglott DR: NCBI
Reference Sequence (RefSeq): a curated non-redundant sequence
database of genomes, transcripts and proteins. Nucleic Acids Res.
33:D501–D504. 2005. View Article : Google Scholar
|