Introduction
Alveolar macrophages, continuously exposed to
various microbial pathogens and their products, are crucial for
pulmonary defense. Lipopolysaccharide (LPS) is a major component of
the cell wall in gram-negative bacteria and may lead to activation
of inflammatory cells, including alveolar macrophages, through
toll-like receptor 4 (TLR-4). TLR-4 is a member of the pattern
recognition receptors, which are important in the clearance of
microbial pathogens. Binding of TLR-4 results in the activation of
both the myeloid differentiation factor 88 (MyD88)-dependent and
MyD88-independent pathway, leading to the activation of nuclear
factor-κB (NF-κB), production of various inflammatory mediators and
ultimately cellular injury (1).
These mediators include tumor necrosis factor-α (TNF-α),
interleukin (IL)-6, IL-2 and IL-10 (2,3). The
LPS-induced TLR-4 signaling pathway has been well studied, but its
association with other receptors, such as triggering receptor
expressed on myeloid cells (TREMs), and its molecular mechanism are
not well defined. In this study, we investigated the correlation
between TREM-2 and TLR-4 in alveolar macrophages in response to
LPS, which may provide a new therapeutic strategy in the prevention
and treatment of endotoxin-induced inflammatory response and
cellular injury.
TREMs are also members of the pattern recognition
receptors, playing important roles in regulating the immune
response. TREM receptors include various members expressed mainly
on immune cells of myeloid lineage (4). Of these members, TREM-1 and TREM-2
are the most studied to date. TREM-1 is mainly expressed on
neutrophils and monocytes, amplifying the inflammatory responses
associated with sepsis (5,6). Silencing of TREM-1 by siRNA
significantly attenuates TLR-4 signaling by reducing the production
of MyD88, CD14, IκBα, IL-1β, MCP-1 and IL-10 in macrophages treated
with LPS (7). By contrast, TREM-2
is mainly expressed on macrophages, microglia, osteoclasts and
dendritic cells and has been reported to negatively regulate immune
responses (4). In
TREM-2−/− mice, cytokine production by macrophages in
response to LPS is upregulated; however, overexpression of TREM-2
in microglia leads to reduced production of TNF-α and inducible
nitric oxide synthase (iNOS) (8,9).
This evidence indicates that TREM-1 enhances inflammatory responses
and that TREM-2 plays an anti-inflammatory role in the regulation
of the immune response. TREM-1 and TREM-2 signal through a
transmembrane adaptor molecule, DAP12, which contains an
immunoreceptor tyrosine-based activation motif (ITAM). After
initiation by the tyrosine phosphorylation of ITAM, DAP-12 signals
to activate or suppress inflammatory responses, depending on which
receptor it binds to (4,10).
RNA interference (RNAi)-mediated gene silencing is
now widely used for clinical and experimental purposes. RNAi was
first recognized as a natural antiviral mechanism by which long
double-stranded RNA (dsRNA) molecules produced during viral
replication are cleaved by the endonuclease Dicer into short 21-23
nucleotide fragments known as small interfering RNAs (siRNAs).
siRNAs are associated with a multiprotein nuclease complex known as
the RNA-induced silencing complex (RISC), that selects and degrades
mRNAs homologous to siRNAs, resulting in target gene silencing.
siRNA can be delivered endogenously as a form of short-hairpin RNA
(shRNA), which is generated by reverse complement of the siRNA
sequence, and carried by plasmid or viral vectors into target cells
and subsequently processed to siRNA. Compared with synthetic siRNA,
the effects of shRNA are long-lasting, since it is continuously
produced within the cells. Lentivirus vector is a newly developed
shRNA vehicle, which is capable of transfecting various cell lines
in both dividing and non-dividing phases with low immunogenicity,
and integrates transgenes into the host genome, leading to
long-term expression of the transgene and stable achievement of
target gene silencing (11,12).
In this study, we constructed a lentivirus-mediated
shRNA system targeting TREM-2 in murine alveolar macrophages and
investigated the effects of TREM-2 silencing on the expression of
TLR-4, TNF-α and IL-10 in response to LPS treatment.
Materials and methods
Cell culture
Alveolar macrophages were harvested by
bronchoalveolar lavage from healthy male C57BL/6 mice (8-12 weeks
old, 20-25 g, provided by the Experimental Animal Research
Institutes of Chinese Academy of Medical Sciences, China). Lungs
were subjected to lavage 10 times with phosphate-buffered saline
(PBS) (37°C, 1 ml/time). Subsequently, cells were obtained from the
lavage fluid and >95% of these cells were confirmed to be
alveolar macrophages. More than 10 mice were used to obtain
6×106 alveolar macrophages for each group. These cells
were divided into 6 wells per group. The isolated alveolar
macrophages were cultured in RPMI-1640 medium (Gibco, Carlsbad, CA,
USA) containing 10% fetal bovine serum, 100 U/ml penicillin and 100
μg/ml streptomycin (13). The
study was approved by the ethics committee of Shanxi Medical
University, Taiyuan, China.
Selection of TREM-2 shRNA
Taking into account the murine TREM-2 sequence from
GenBank (accession no.: NM_031254), we designed two siRNA sequences
and a scrambled siRNA using Ambion software (Ambion Life
Technologies, Beijing, China): siRNA 573, 5′-GAU GCUGGA
GAUCUCUGGG-3′; siRNA 737, 5′-GGAGGUACG UGAG AGAAUU-3′; scrambled
siRNA, 5′-GGCACAAGC UGGAG UACAA-3′. A BLAST search was performed to
prevent interference with non-specific genes. For shRNA
construction, the sense and antisense sequences of siRNA were
combined by a 9-base hairpin of the following sequence:
5′-TTCAAGAGA-3′, and an HI promoter and RNA polymerase III
termination sequence (TTTTT) were added. A double-stranded DNA
oligonucleotide of these shRNA was annealed from two
single-stranded ones and connected to the eukaryotic pGCsi plasmid
(TransGen Biotech, China), which had been double digested by
HindIII and BamHI (Takara, Japan). The recombinant
plasmid was transfected into DH5α-competent E. coli cells
(TransGen Biotech). A colony was selected for real-time polymerase
chain reaction (RT-PCR) and the recombinant plasmids were extracted
for sequence detection to ensure the accuracy of our design.
Moreover, alveolar macrophages were transfected with
recombinant shRNA plasmids in 12-well plates for 48 h using
TransFectin Lipid at the concentration indicated by the
manufacturer’s instructions (Bio-Rad, Hercules, CA, USA). The
transfection efficiency was measured by flow cytometric analysis of
green fluorescent protein (GFP), which was expressed by the pGCsi
plasmid. mRNA of TREM-2 was extracted from transfected alveolar
macrophages and detected by RT-PCR to select the optimal shRNA
sequence.
Lentivirus-mediated shRNA expression
system
shRNAs targeting TREM-2 were cloned into the
lentiviral expression vector pLenti-EGFP-U6 (VGTC, Beijing, China),
which was transfected into 293T cells together with pLP1, pLP2 and
pLP/VSVG using a lentivirus packaging system and polybrene-gene
companion II according to the manufacturer’s instructions (VGTC).
ELISA was used to test the virus titer. At 48 h, packaged
lentivirus particles were harvested, purified and extracted for
sequence detection. As a negative control, a lentivirus vector
containing scrambled shRNA was constructed and an empty vector
without shRNA was used as a blank control.
Silencing of TREM-2 and LPS treatment of
alveolar macrophages
The recombinant lentivirus vectors were delivered
into alveolar macrophages in 24-well plates in the presence of
polybrene (Sigma, St. Louis, MO, USA) at various concentrations.
Each well contained 1×106 cells and these cells were
cultured with LPS in RPMI-1640 medium in different conditions prior
to or following recombinant lentivirus transfection. Alveolar
macrophages were divided into 5 groups with triplicates per group:
the TREM-2 shRNA+LPS group, the non-sense shRNA+LPS group, the
empty vector+LPS group, the non-treatment group which underwent no
interference and the LPS group which received LPS treatment alone.
Cell viability was determined by MTT assay after transfection. RNA
and protein samples were harvested at the indicated time points
following treatment.
Real-time fluorescence quantitative
PCR
We used real-time fluorescence quantitative PCR to
evaluate mRNA expression of TREM-2 and TLR-4. Total RNA was
extracted from alveolar macrophages using TRIzol reagent
(Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s
instructions. RNA (5 μg) was reverse transcribed to complementary
DNA using a reverse transcription PCR Purification kit (Promega,
Madison, WI, USA). The real-time PCR reaction was conducted in a
total volume of 50 μl with 10 μl of cDNA templates. The
housekeeping gene β-actin was used to normalize target gene
expression. Primer sequences were as follows:
5′-TGGTCAGAGGGCTGGACTGT-3′ (sense), 5′-TCCTGG CTGGACTTAAGCTGTAG-3′
(antisense) for TREM-2 and 5′-TCAGAACTTCAGTGGCTGGATTTAT-3′ (sense),
5′-TAGGGTTTCCTGTCAGTATCAAGTTTG-3′ (antisense) for TLR-4. The
primers for β-actin were 5′-AGCGGTTCC GATGCCCT-3′ (sense) and
5′-AGAGGTCTTTACGGAT GTCAACG-3′ (antisense). The reaction conditions
were set as follows: an initial 2 min at 94°C followed by 40 cycles
(15 sec at 94°C, 30 sec at 60°C, 30 sec at 72°C) and a final 10 min
at 72°C. Quantification was performed by measuring the threshold
cycle (CT value) and using the standard curve as a reference.
Flow cytometry
Flow cytometry was applied to detect protein levels
of TREM-2 and TLR-4. Rat anti-mouse polyclonal antibody (R&D
Systems, Minneapolis, MN, USA) and goat anti-rat IgG conjugated to
PE (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were used as
primary and secondary antibodies, respectively. The expression of
TREM-2 and TLR-4 by alveolar macrophages was measured by FACScan
flow cytometry (BD Biosciences) and data were analyzed with
CellQuest software.
ELISA
Levels of TNF-α and IL-10 in cell-free supernatant
were determined by enzyme-linked immunosorbent assay (ELISA)
(R&D Co.) according to the manufacturer’s instructions.
Statistical analysis
Data were analyzed using Statistical Package for
Social Sciences version 14.0 for Windows (SPSS) software. Results
were presented as the means ± SEM. Analysis of variance (ANOVA) was
applied to experiments with three or more groups. We used the SNK-q
test to compare means between two groups. P<0.05 was considered
to indicate a statistically significant difference.
Results
Optimal sequence of TREM-2 shRNA
In this study, a eukaryotic pGCsi plasmid carrying
TREM-2 shRNA was constructed after selection from GenBank in order
to determine the optimal target sequence for TREM-2 silencing.
RT-PCR showed that the sequence of the recombinant plasmid was in
accordance with our design. Alveolar macrophages were transfected
by the recombinant plasmid with a transfection efficiency of
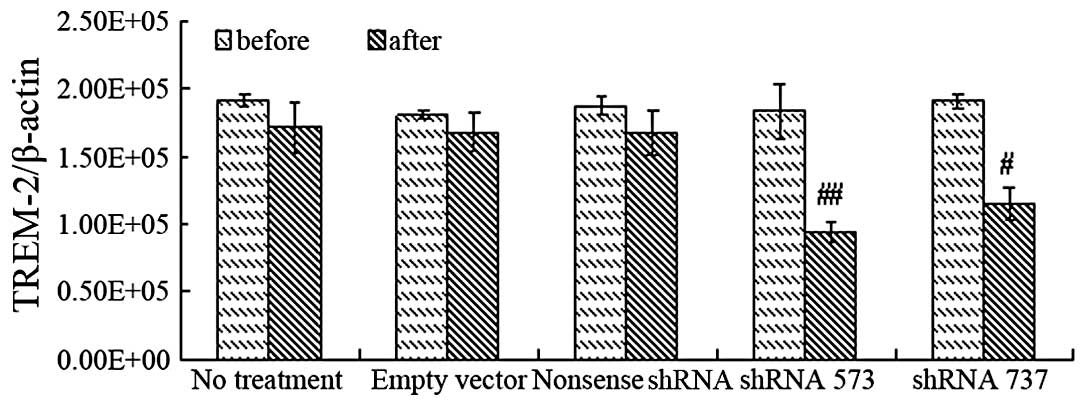
60.75±4.16%, as measured by flow cytometry. Analysis of TREM-2 mRNA
by RT-PCR suggested a higher inhibition effect of shRNA 573 than
that of shRNA 737, while the scrambled shRNA and empty vector had
no inhibitory effects on TREM-2 expression (Fig. 1). Therefore, shRNA 573 was selected
as the target sequence for TREM-2 silencing.
LPS-induced inflammatory response
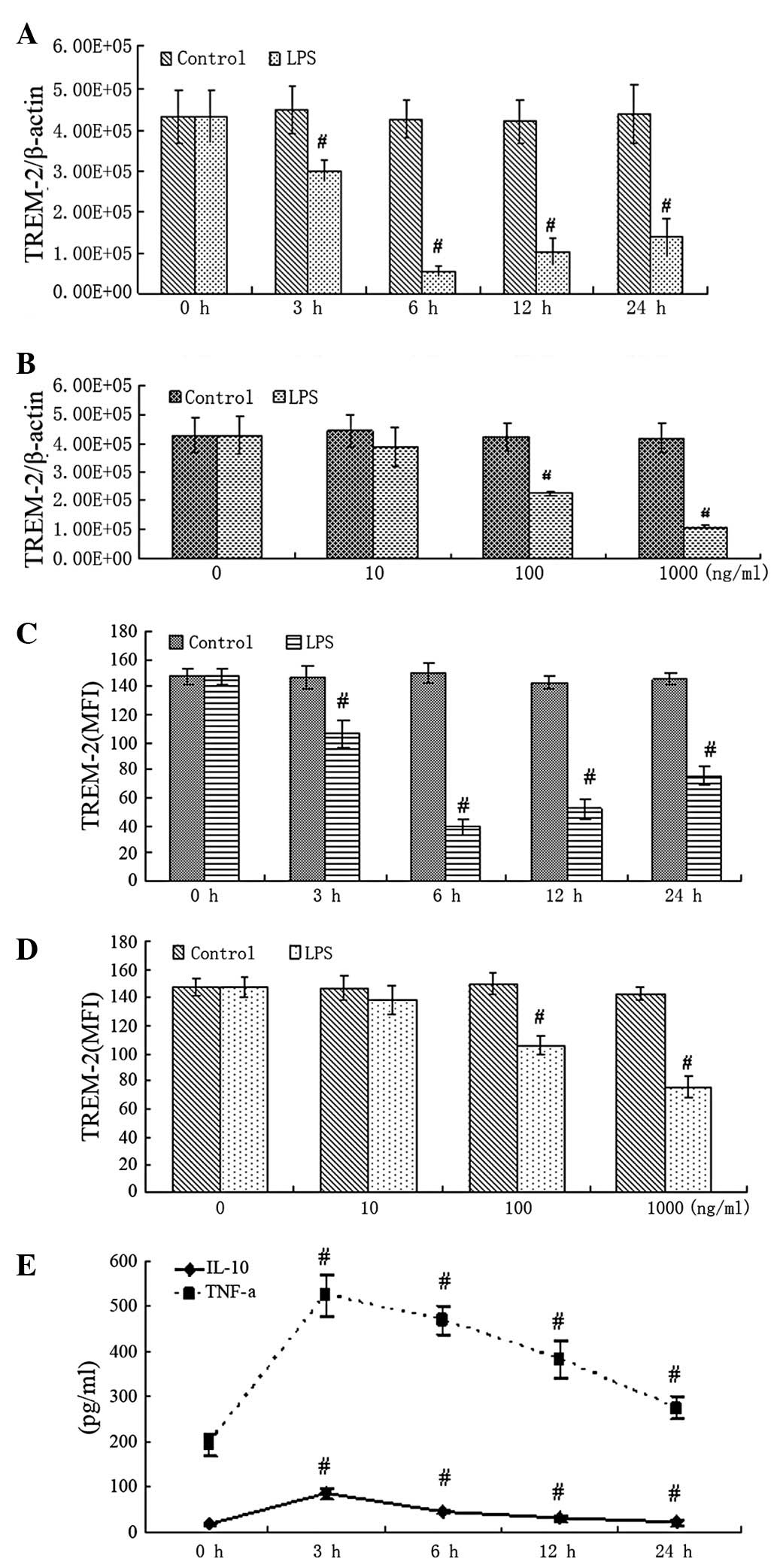
To evaluate how LPS affected TREM-2 expression in
alveolar macrophages, mRNA and protein levels of TREM-2 were
studied at different concentrations of LPS at various time points.
RT-PCR showed that mRNA of TREM-2 was significantly downregulated 3
h after LPS treatment at a concentration of 100 ng/ml. Increasing
doses of LPS showed more significant reduction effects (Fig. 2A and B). Flow cytometry revealed
similar changes in TREM-2 protein (Fig. 2C and D). Inflammatory mediators
(TNF-α and IL-10) in the supernatants were highly upregulated 3 h
after LPS treatment, as shown by ELISA (Fig. 2E). These results indicate that
TREM-2 is downregulated, but TNF-α and IL-10 are upregulated
significantly in response to LPS treatment.
TREM-2 expression
ELISA results demonstrated that the virus titer was
2.87×109 TU/ml in 293T cells transfected with
recombinant lentivirus-mediated shRNA. In the negative and blank
control groups, virus titers were both 2.0×109 TU/ml.
Our study used flow cytometry to evaluate the transfection
efficiency of lentivirus-mediated TREM-2 shRNA treatment in
alveolar macrophages, and a time- and dose-dependent effect of
transfection was observed. When the concentration of lentivirus was
2×107 TU/well and polybrene was 6 μg/ml at 72 h, the
transfection efficiency reached the highest level (Fig. 3A and B) with cell viability of
89.2±3.2% as measured by the MTT method (Table I). RT-PCR showed a significant
reduction of TREM-2 mRNA in alveolar macrophages treated with
lentivirus-mediated shRNA, but not in the cells treated with
non-sense shRNA or empty lentivirus vector (Fig. 3C).
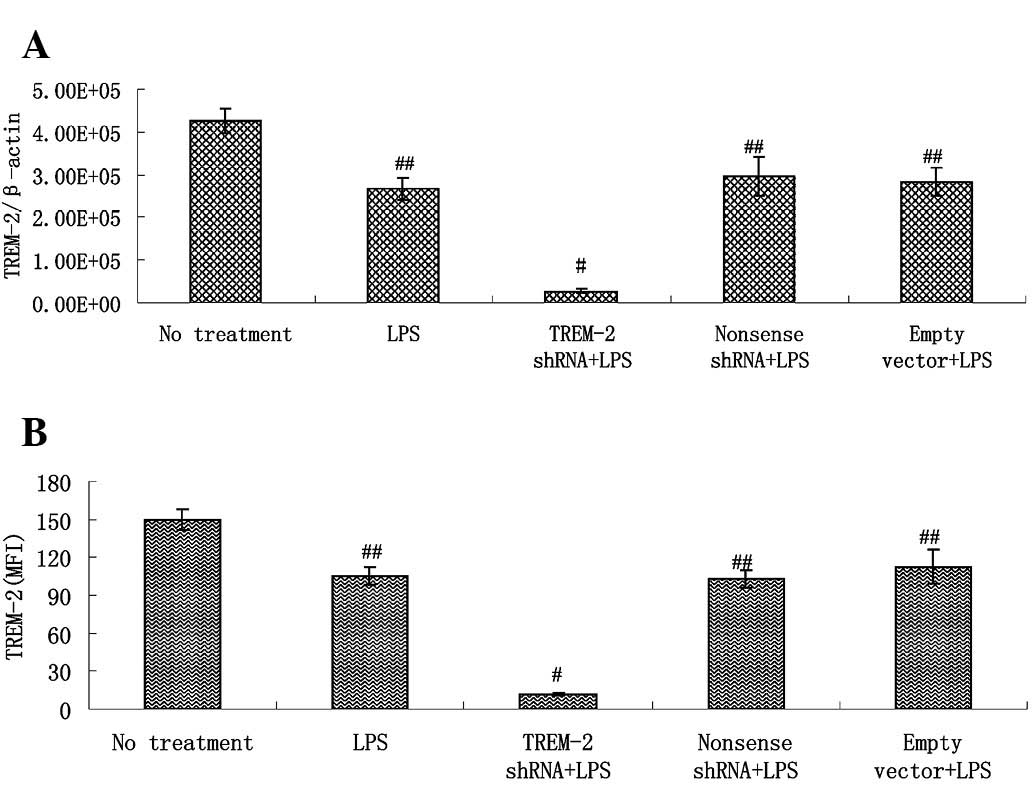
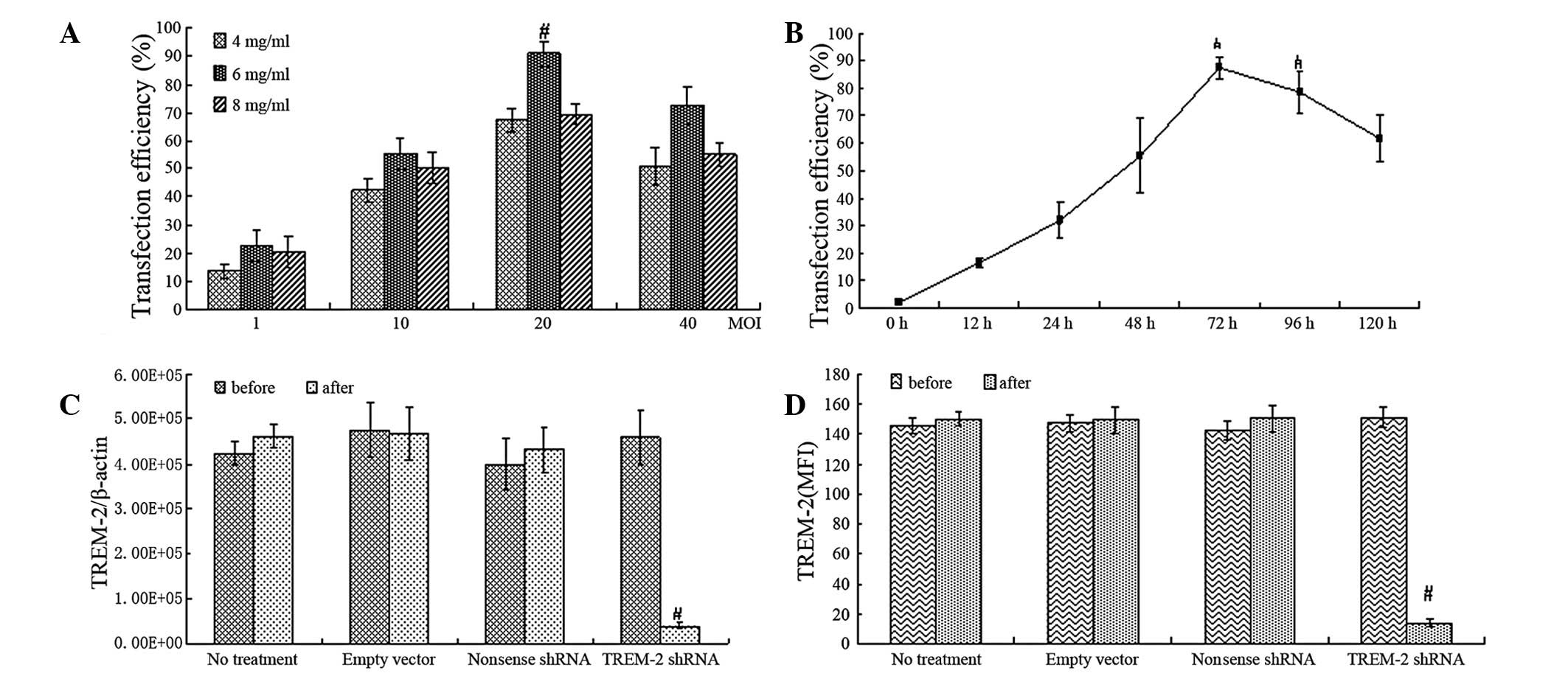 | Figure 3Suppression of TREM-2 gene and protein
expression in alveolar macrophages by delivery of
lentivirus-mediated TREM-2 shRNA. (A and B) Flow cytometry analysis
of transfection efficiency with different multiplicity of infection
(MOI, the number of lentivirus/well, the density of cells were
1×106/well, MOI=1, 10, 20, 40) and various doses of
polybrene (4, 6 and 8 mg/ml) at different time points (0-120 h). (C
and D) RT-PCR and flow cytometry showed levels of TREM-2 mRNA and
protein in alveolar macrophages transfected with
lentivirus-mediated shRNA or non-sense shRNA or empty lentivirus
vector. Data were presented as the means ± SEM (n=6),
#P<0.05 compared with control. TREM-2, triggering
receptor expressed on myeloid cells-2; shRNA, short hairpin
RNA. |
 | Table ICell viability (%) 72 h after
lentivirus-mediated TREM-2 shRNA treatment with different
multiplicity of infection and doses of polybrene. |
Table I
Cell viability (%) 72 h after
lentivirus-mediated TREM-2 shRNA treatment with different
multiplicity of infection and doses of polybrene.
| Concentration of
polybrene |
|---|
|
|
|---|
| MOI | 4 g/ml | 6 μg/ml | 8 μg/ml |
|---|
| 1 | 80.1±1.9 | 75.5±2.4 | 70.8±3.5 |
| 10 | 87.2±2.1 | 88.5±2.1 | 75.1±3.7 |
| 20 | 90.3±1.7 | 89.2±3.2 | 79.1±4.7 |
| 40 | 90.5±2.4 | 91.2±4.1 | 80.2±4.5 |
Alveolar macrophages, cultured with LPS after
delivery of lentivirus-mediated shRNA for 3 h, showed a more
significant reduction of TREM-2 gene expression than those in the
LPS control group. By contrast, delivery of non-sense shRNA or
empty vectors had no such effect (Fig.
4A). TREM-2 protein levels in transfected alveolar macrophages
showed similar changes to the TREM-2 mRNA (Fig. 4B).
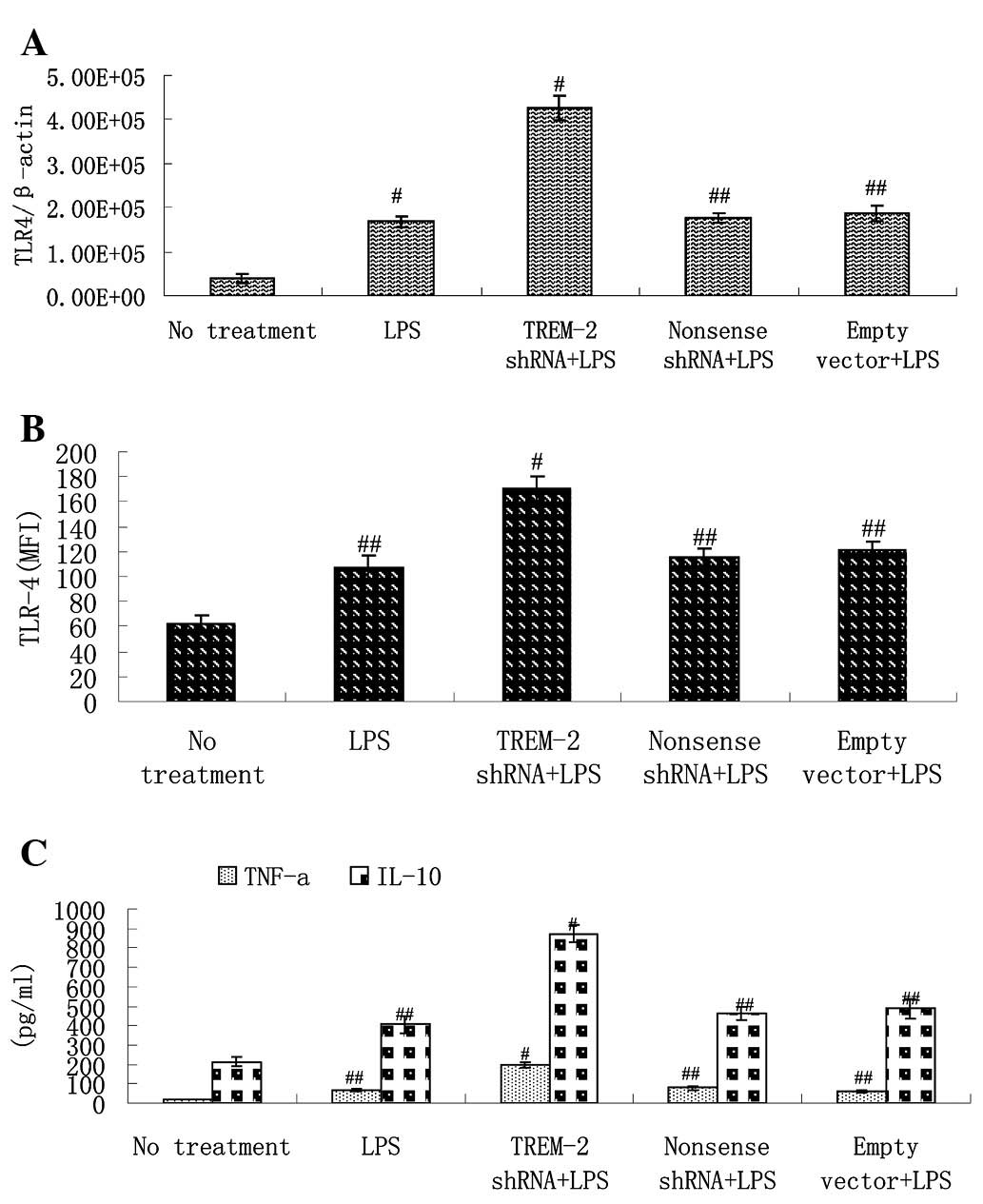
Silencing of TREM-2 upregulates TLR-4
expression
We investigated the effects of TREM-2 shRNA on
levels of TLR-4 in response to LPS in murine alveolar macrophages.
Levels of TLR-4 mRNA and protein were evaluated by RT-PCR and flow
cytometry, respectively. We observed that TLR-4 gene expression was
upregulated by LPS treatment. However, delivery of TREM-2 shRNA
increased levels of TLR-4 mRNA more significantly at 3 h after LPS
stimulation and delivery of non-sense shRNA or empty lentivirus
vector exhibited no such effect (Fig.
5A). Flow cytometry indicated similar changes in TLR-4 protein
(Fig. 5B).
Effects of TREM-2 silencing on
inflammatory mediators in response to LPS treatment
LPS stimulation increased levels of TNF-α and IL-10
in murine alveolar macrophages. Delivery of TREM-2 shRNA augmented
expression of both TNF-α and IL-10 by >1-fold compared with the
LPS control group, which indicates that silencing of TREM-2
significantly amplifies the production of TNF-α and IL-10 in
response to LPS in murine alveolar macrophages (Fig. 5C).
Discussion
In this study, we have elucidated the contribution
of TREM-2 to the inflammatory response induced by LPS and its
correlation with TLR-4 in alveolar macrophages using
lentivirus-mediated shRNA. The optimal sequence was selected for
TREM-2 silencing and a recombinant lentivirus vector was
constructed to deliver the selected shRNA into alveolar
macrophages. Real-time fluorescence quantitative PCR and flow
cytometry showed that TREM-2 was highly inhibited at the
transcriptional and translational levels by use of
lentivirus-mediated shRNA. Silencing of TREM-2 enhanced the
expression of TLR-4 and the inflammatory mediators TNF-α and IL-10,
which are recognized as downstream cytokines in the TLR-4 pathway.
These results suggest that TREM-2 may suppress the inflammatory
response through regulation of the TLR-4 pathway and this needs to
be confirmed by further studies.
Lentivirus vector-mediated shRNA is a highly
efficient RNAi technology with lower off-target effects and less
endogenous interference with host genes than synthetic siRNA
oligonucleotides (11,12). For several years, lentivirus
vector-mediated shRNA technology has been widely used for its
therapeutic potential in neurological disorders such as Alzheimer’s
disease (AD) (12,14). Lentiviral vectors are capable of
delivering long-term transgenes in various cell types in
vivo and in vitro and suppress target genes in a
specific manner, which provides broad applications in the study of
genome functions and for clinical purposes (11,12).
However, there have been few studies applying this technology to
TREM-2 in alveolar macrophages. In this study, we observed that
lentivirus-mediated shRNA treatment significantly suppressed TREM-2
expression in alveolar macrophages.
The TREM family members, as innate immune receptors,
play critical roles in modulating inflammatory responses, either by
amplifying or dampening TLR-4-induced signaling (4). TREM-1 was established by initial
findings as an amplifier of inflammatory responses and was also
found to be associated with inflammatory bowel disease (IBD)
(4,15). On the contrary, TREM-2 is
considered to be an important negative modulator of immune
response. Previous studies have shown TREM-2 ligands on the surface
of peritoneal and bone-marrow derived macrophages (4,8).
Others have reported that TREM-2 binds to anionic ligands on the
surface of bacteria through Phexin-A and functions as a phagocytic
receptor for bacteria (4,16,17).
Upon the binding of pathogens, TREM-2 is able to positively enhance
phagocytosis and negatively suppress production of TNF-α and IL-6
in microglia in order to maintain the local immunosuppressive
microenvironment in the central neural system (CNS) (9). Hamerman et al reported that
TREM-2, associated with its counterpart DAP-12, inhibits TLR and
FcR responses in bone-marrow-derived macrophages in
vitro(18), and Turnbull et
al showed that TREM-2 is expressed on newly differentiated and
alternatively activated macrophages, functioning to restrain
macrophage activation (8). In an
animal model of LPS-induced acute lung injury (ALI), TREM-2 was
highly decreased in lung tissues (19), which was accordant with our
findings that TREM-2 levels in alveolar macrophages were
significantly downregulated in response to LPS treatment. However,
the exact mechanism by which TREM-2 affects alveolar macrophages
during LPS stimulation is not well defined.
TLR-4 has been established as a primary receptor for
LPS, which initiates the production of pro-inflammatory mediators
such as IL-1β and TNF-α, leading to cellular injury and clinical
manifestations of sepsis (1,3).
Previous studies suggest that TREM expression is associated with
TLR-4 (20). Evidence has shown
that activation of TREM-1 amplifies the TLR-4 signaling pathway by
activating downstream mediators such as MyD88, CD14, IL-1β, MCP-1,
IκBα and IL-10 (7). In TLR-4
mutant C3H/HeJ mice, LPS treatment has little effect on both TREM-1
and TREM-2 in hepatic macrophages compared to TLR-4 normal cells in
which TREM-1 expression was significantly increased, whereas TREM-2
was decreased (21). This evidence
indicates that LPS-induced TREM alteration is dependent on TLR-4.
As for TREM-2, production of TNF-α is inhibited in a TREM-2+DAP12
chimera in bone marrow-derived macrophages stimulated by TLR and
FcR (including CpG DNA, zymosan and anti-FcγRII/III mAb) (18). It is possible that the
anti-inflammatory role of TREM-2 in LPS-induced immune responses
occurs due to downregulation of TLR-4 and this is proved by the
present study. Our study demonstrated that silencing of TREM-2
using recombinant lentivirus vector significantly upregulated
levels of TLR-4 with LPS treatment. However, the mechanism remains
to be further investigated.
After binding of LPS, TLR-4 initiates cascades of
signaling, resulting in excessive production of inflammatory
mediators including TNF-α and IL-10 (1,3).
Turnbull et al have shown that peritoneal macrophages from
TREM-2−/− mice produced increased TNF-α in response to
LPS (8). Another study
demonstrated that DAP-12-deficient macrophages treated with
TREM-2+DAP12 chimeras inhibited the production of TNF-α with TLR
and FcR stimulation (19).
However, in regard to levels of IL-10 in response to LPS-induced
inflammation, Turnbull et al observed no difference between
wild-type and TREM-2−/− macrophages (8).
In the present study, we demonstrated high levels of
TNF-α and IL-10 but decreased expression of TREM-2 in alveolar
macrophages after LPS stimulation. By silencing TREM-2, production
of TNF-α and IL-10 were increased >1-fold. These results
indicate that TREM-2 is reduced in the early stage of LPS-induced
inflammation in order to increase the production of inflammatory
mediators and activate the immune response. Silencing of TREM-2
leads to excessive production of inflammatory mediators, which
aggravates the inflammatory response induced by LPS. This implies
that TREM-2 is one of the major signaling pathways that negatively
regulates the production of inflammatory mediators, which is
consistent with the findings in other studies (4,8,18).
In conclusion, this study has shown that silencing
of TREM-2 activated the expression of TLR-4 and its downstream
cytokines TNF-α and IL-10 in response to LPS in alveolar
macrophages. Moreover, the anti-inflammatory effect of TREM-2 may
be dependent on the TLR-4 pathway, and this requires further
study.
References
|
1
|
Dauphinee SM and Karsan A:
Lipopolysaccharide signaling in endothelial cells. Lab Invest.
86:9–22. 2006. View Article : Google Scholar
|
|
2
|
Chen LC, Gordon RE, Laskin JD and Laskin
DL: Role of TLR-4 in liver macrophage and endothelial cell
responsiveness during acute endotoxemia. Exp Mol Pathol.
83:311–326. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Adib-Conquy M, Moine P, Asehnoune K, et
al: Toll-like receptor-mediated tumor necrosis factor and
interleukin-10 production differ during systemic inflammation. Am J
Respir Crit Care Med. 168:158–164. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ford JW and McVicar DW: TREM and TREM-like
receptors in inflammation and disease. Curr Opin Immunol. 21:38–46.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bouchon A, Facchetti F, Weigand MA and
Colonna M: TREM-1 amplifies inflammation and is a crucial mediator
of septic shock. Nature. 410:1103–1107. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zanzinger K, Schellack C, Nausch N and
Cerwenka A: Regulation of triggering receptor expressed on myeloid
cells 1 expression on mouse inflammatory monocytes. J Immunol.
128:185–195. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ornatowska M, Azim AC, Wang X, et al:
Functional genomics of silencing TREM-1 on TLR4 signaling in
macrophages. Am J Physiol Lung Cell Mol Physiol. 293:1377–1384.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Turnbull IR, Gilfillan S, Cella M, et al:
Cutting edge: TREM-2 attenuates macrophage activation. J Immunol.
177:3520–3524. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Takahashi K, Rochford CD and Neumann H:
Clearance of apoptotic neurons without inflammation by microglial
triggering receptor expressed on myeloid cells-2. J Exp Med.
201:647–657. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lanier LL: DAP10- and DAP12-associated
receptors in innate immunity. Immunol Rev. 227:150–160. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Manjunath N, Wu H, Subramanya S and
Shankar P: Lentiviral delivery of short hairpin RNAs. Adv Drug
Deliv Rev. 61:732–745. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Singer O and Verma IM: Applications of
lentiviral vectors for shRNA delivery and transgenesis. Curr Gene
Ther. 8:483–488. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Egan PJ, Andrews AE, Barcham GJ, Brandon
MR and Nash AD: The extraction, purification and activity detection
of murine alveolar macrophages. The public health of China.
19:1087–1088. 2003.
|
|
14
|
Singer O, Marr RA, Rockenstein E, et al:
Targeting BACE1 with siRNAs ameliorates Alzheimer disease
neuropathology in a transgenic model. Nat Neurosci. 8:1343–1349.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schenk M, Bouchon A, Seibold F and Mueller
C: TREM-1-expressing intestinal macrophages crucially amplify
chronic inflammation in experimental colitis and inflammatory bowel
diseases. J Clin Invest. 117:3097–3106. 2007. View Article : Google Scholar
|
|
16
|
Daws MR, Sullam PM, Niemi EC, Chen TT,
Tchao NK and Seaman WE: Pattern recognition by TREM-2: binding of
anionic ligands. J Immunol. 171:594–599. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
N’Diaye EN, Branda CS, Branda SS, et al:
TREM-2 (triggering receptor expressed on myeloid cells 2) is a
phagocytic receptor for bacteria. J Cell Biol. 184:215–223.
2008.
|
|
18
|
Hamerman JA, Jarjoura JR, Humphrey MB,
Nakamura MC, Seaman WE and Lanier LL: Cutting edge: inhibition of
TLR and FcR responses in macrophages by triggering receptor
expressed on myeloid cells (TREM)-2 and DAP12. J Immunol.
177:2051–2055. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun GY, Guan CX, Zhou Y, et al: Vasoactive
intestinal peptide re-balances TREM-1/TREM-2 ratio in acute lung
injury. Regul Pept. 167:56–64. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Klesney-Tait J and Colonna M: Uncovering
the TREM-1-TLR connection. Am J Physiol Lung Cell Mol Physiol.
293:1374–1376. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen LC, Laskin JD, Gordon MK and Laskin
DL: Regulation of TREM expression in hepatic macrophages and
endothelial cells during acute endotoxemia. Exp Mol Pathol.
84:144–145. 2008.PubMed/NCBI
|