Introduction
Tamoxifen (TAM) is the most commonly prescribed drug
for the prevention and treatment of breast cancer (1,2). TAM
is a non-steroidal anti-estrogen which acts, at least in part, by
compete blockage of the estrogen receptor (ER) (3,4).
However, the existence of an alternative mechanism of action for
this drug is supported by the following clinical observations: i)
30% of patients with ER-negative cancer cells respond to TAM and
ii) 30% of patients with ER-positive cancer cells are not sensitive
to this anti-estrogen (5). The ER
activates transcription from classical hormone response elements,
to which the ER binds directly and from various alternative
response elements, to which the ER does not bind. ER action upon
the ovalbumin proximal promoter and the collagenase and IGF-1 genes
traces to activator of protein 1 (AP-1) sites that bind members of
the Jun/Fos family of transcription factors (6,7). ER
action upon the quinine reductase gene traces to an electrophile
response element and these have been reported to bind ATF
transcription factors, which are potential dimerization partners
with Jun (8). ER action upon the
cyclin D gene is linked to a CRE-like element that also binds
Jun/ATF (9–11). ER also enhances the activity of
promoters that are regulated by other factors.
Protein kinase C (PKC) was originally identified as
a phospholipid- and calcium-dependent protein kinase (12). PKC affects diverse cell functions
through phosphorylation of target proteins. These cell functions
involve a wide variety of fundamental physiological processes,
including signal transduction, modulation of gene expression,
proliferation and apoptosis (13–15).
Individual PKC isozymes exhibit various tissue distributions,
subcellular localizations and biochemical properties, an indication
that each member of the family plays specialized roles (14), which ultimately translates into
unique correlations with disease.
The pAP-1(PMA)-TA-luc plasmid contains the AP-1
elements, which are designed to monitor the induction of the PKC
signal transduction pathway. PKC is involved in various biochemical
processes relevant to signal transduction pathways and linked to
lipid signaling pathways (15). By
contrast, isoenzymes of PKC affect a number of biochemical
processes, including cell growth, differentiation and
transformation and plays a key role in signal transduction from
multiple extracellular receptors (16,17).
PKC activity is regulated by multiple activators and cofactors
which associate via conserved domains within the enzyme structure
(18).
In the present study, TAM was observed to inhibit
the growth of the human ER-positive breast cancer cell line, MCF-7
and ER-negative breast cancer cell line, MDA-MB-435, in a
concentration-dependent manner. A previous study reported that the
observed effects of TAM and its active metabolite on the
proliferation of MDA-MB-435 cells were due to an ER-independent
mechanism (5). To verify the role
of c-Jun in TAM suppression of proliferation of the two breast
cancer cell lines, the expression of c-Jun mRNA and protein levels
were examined by reverse transcription polymerase chain reaction
(RT-PCR) and western blot analysis. TAM treatment of MCF-7 cells
activated the transcriptional activities of AP-1(PMA), which
responds specifically to phorbol ester. Results indicate that TAM
functions as an AP-1 activator to mediate cell functions. The
inhibition rates of MCF-7 cells were found to correlate with c-Jun
expression and stimulation of TAM, whereby TAM-stimulated MCF-7
cells were positively regulated by c-Jun overexpression and
negatively regulated by c-Jun underexpression. To the best of our
knowledge, this is the first study on the correlation between c-Jun
and antiproliferation of TAM-stimulated MCF-7 cells.
Materials and methods
Cell culture
Human breast cancer cell lines, MCF-7 and
MDA-MB-435, were obtained from Shanghai Cell Bank (Shanghai,
China). Cells were cultured in Dulbecco's modified Eagle's medium
(Gibco-BRL, Carlsbad, CA, USA) containing 10% fetal bovine serum in
a humidified atmosphere with 5% CO2 at 37°C.
Cell proliferation assays
MCF-7 and MDA-MB-435 cells were plated at
1×104 cells/well in 96-well plates with six replicate
wells, for 24 h in a humidified incubator (5% CO2 at
37°C). Cells were treated with various concentrations of TAM (The
First Affiliated Hospital of Soochow University, Suzhou, China) for
48 h. Following this, 10 μl CCK-8 (Dojindo Laboratories, Kumamoto,
Japan) solution was added to each well of the plate to measure cell
proliferation. The plate was incubated for 4 h in the incubator.
Optical density (OD) was measured at a wavelength of 450 nm using a
microplate reader. Data were presented as the mean ± SD, which was
derived from triplicate samples of at least three independent
experiments. To examine the effect of c-Jun on inhibition rates of
TAM-stimulated MCF-7 cells, cells were transfected with
pIRES2-EGFP-c-Jun or pGPU6/GFP/Neo-c-Jun-shRNA using Lipofectamine
2000 (Invitrogen, Carlsbad, CA, USA). Cells were stimulated with
TAM 48 h post-transfection.
Semiquantitative RT-PCR
Total RNA from TAM-treated or untreated MCF-7 and
MDA-MB-435 cells was extracted by TRIzol (Gibco-BRL) according to
the manufacturer's instructions. cDNA was generated from total RNA,
using M-MLV RT (MBI; Fermentas, Waltham, MA, USA). Amplification
was performed over 28 cycles consisting of 94°C for 35 sec, 53°C
for 45 sec and 72°C for 1 min. PCR products were separated by
electrophoresis on a 1.5% agarose gel and stained with ethidium
bromide to visualize the bands. To compare differences among
samples, the relative intensity of each band was normalized against
the intensity of the GAPDH band amplified from the same sample. The
primer sequences for the genes and expected product sizes were as
follows: 5′-TGAACGGGAAGCTCACTGG-3′ (sense) and
5′-TCCACCACCCTGTTGCTGTA-3 (antisense) for GAPDH (307 bp);
5′-GCCTCAGACAGTGCCCGAGAT-3′ (sense) and
5′-GTTTAAGCTGTGCCACCTGTTCC-3′ (antisense) for c-Jun (245 bp).
Western blot analysis
MCF-7 and MDA-MB-435 cells were harvested 48 h after
TAM treatment and lysed with SDS sample buffer (80 mM Tris-HCl, 2%
SDS, 300 mM NaCl and 1.6 mM EDTA). Cell extracts were separated
using 10% SDS-PAGE gel electrophoresis, transferred onto
nitrocellulose membrane and blocked with 5% skimmed milk. Following
blocking, membranes were incubated with antibodies against GAPDH or
c-Jun and then incubated with HRP-conjugated anti-mouse or -rabbit
IgG antibodies. Protein bands were visualized with ECL
solution.
Transient expression reporter gene
assay
MCF-7 and MDA-MB-435 cells grown in 24-well plates
were transfected with 1 μg reporter plasmids, pAP-1(PMA)-Luc
(Clontech, Palo Alto, CA, USA) or pAP-1-Luc, to investigate the
role of TAM in this signaling pathway. Relative luciferase activity
was normalized by co-transfection with 50 ng pRL-SV40. The cells
were then stimulated with TAM for 36 h. Cell lysates were prepared
using the Dual-Luciferase reporter assay system (Promega, Madison,
WI, USA) and luciferase activity was measured with GloMax 20/20.
Data are presented as mean ± SD of 3 independent experiments.
Results
Effect of TAM on the growth of MCF-7 and
MDA-MB-435 cells in vitro
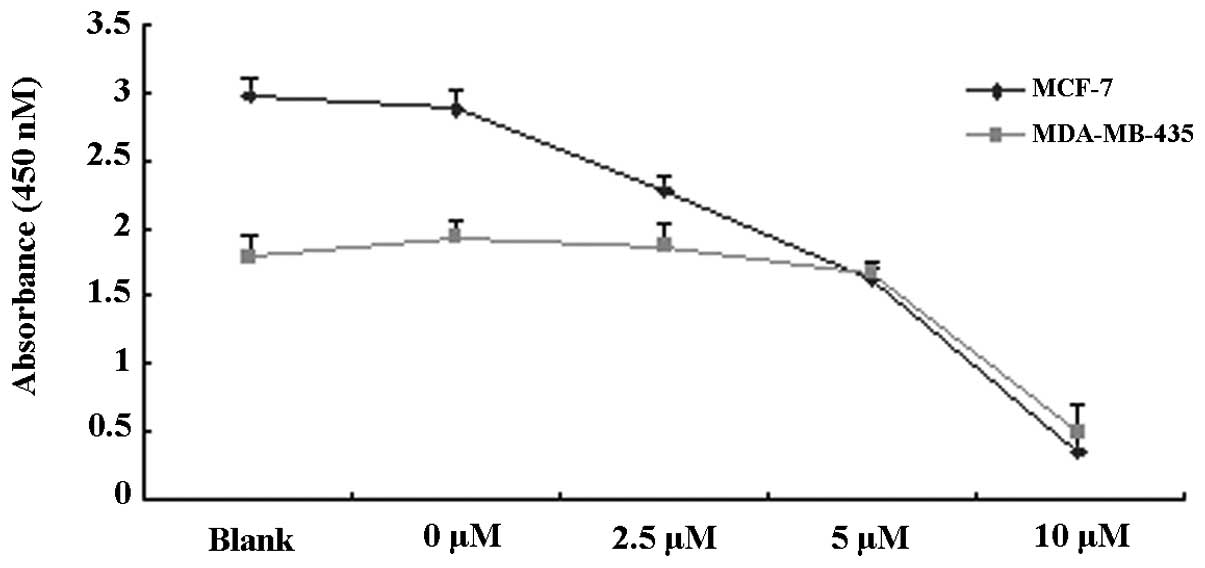
To observe the proliferation inhibitory effects of
TAM, MCF-7 and MDA-MB-435 cells were treated with various
concentrations of TAM (0, 2.5, 5 and 10 μM) for 48 h and the rate
of proliferation inhibition was detected by CCK-8 assay. As is
evident in Fig. 1A and B, TAM was
found to significantly inhibit the proliferation of MCF-7 and
MDA-MB-435 cells. The effect of inhibition was enhanced with
increased concentrations of TAM.
Expression of c-Jun in MCF-7 and
MDA-MB-435 cells stimulated by TAM
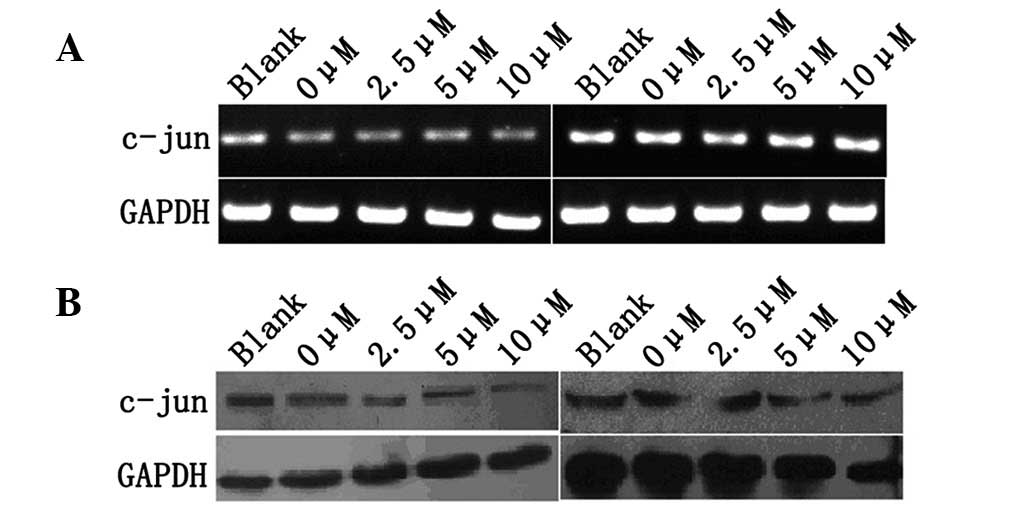
The effect of TAM on expression of c-Jun of MCF-7
and MDA-MB-435 cells was investigated using RT-PCR and western blot
analysis. MCF-7 and MDA-MB-435 cells were treated with various
concentrations of TAM (0, 2.5, 5 and 10 μM) for 48 h and c-Jun mRNA
and protein levels were examined by RT-PCR and western blot
analysis. No significant changes in MCF-7 and MDA-MB-435 cells were
observed (Fig. 2A and B).
TAM activates AP-1(PMA)-mediated
transcriptional activation in MCF-7 cells
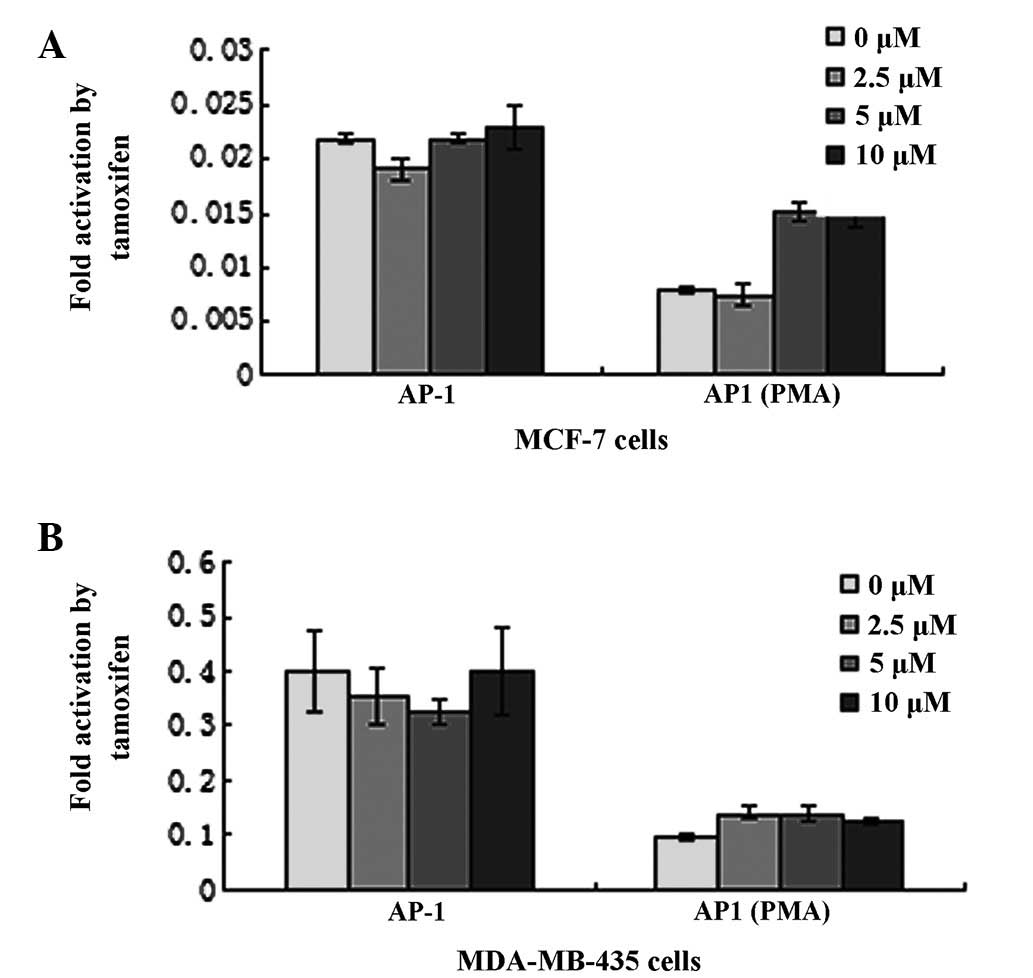
TAM is thought to function primarily by competitive
blockage of the ER. In this study, the Dual-Luciferase reporter
assay system was utilized to investigate the role of TAM on
signaling pathways, including AP-1(PMA) and AP-1. The plasmid
pAP-1(PMA)-Luc and pAP-1-Luc were transfected into MCF-7 and
MDA-MB-435 cells, which encode luciferase controlled by AP-1(PMA)
and AP-1 elements, respectively. Relative luciferase activity was
normalized by co-transfection with pRL-SV40. Cells were stimulated
with various concentrations of TAM 36 h post-transfection. In MCF-7
cells, TAM enhanced AP-1(PMA)-luciferase activity by ~200%
(Fig. 3A). In addition, TAM was
identified to induce dose-dependent activation of the AP-1(PMA)
reporter gene. However, TAM had a limited effect on AP-1. In
MDA-MB-435 cells, TAM had almost no effect on AP-1(PMA) and AP-1
(Fig. 3B).
Inhibition of proliferation of MCF-7
cells by TAM through c-Jun
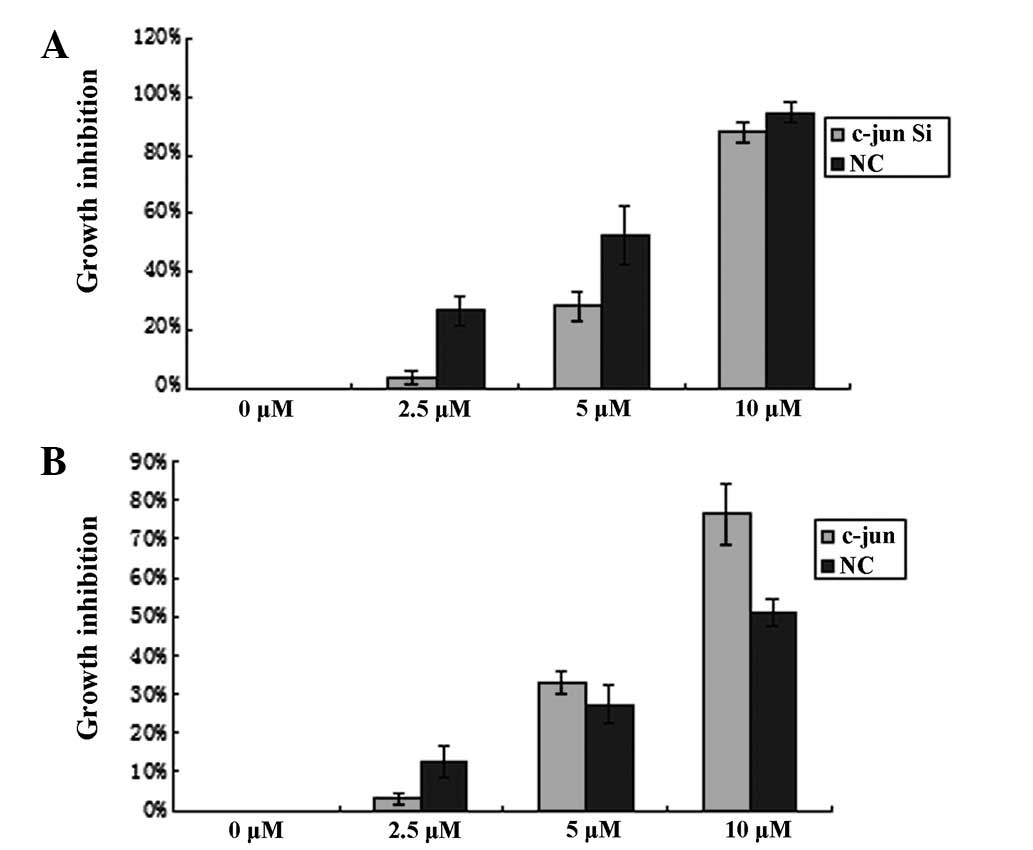
To investigate a role for c-Jun in inhibition of
TAM-stimulated MCF-7 cell growth, c-Jun vectors with upregulated or
downregulated expression of c-Jun in cells were utilized. MCF-7
cells were transfected with pIRES2-EGFP-c-Jun or
pGPU6/GFP/Neo-c-Jun-shRNA and stimulated with TAM 48 h
post-transfection. CCK-8 was used to measure cell proliferation.
Inhibition rates of TAM-stimulated MCF-7 cells were found to be
positively and negatively regulated by overexpression and
underexpression of c-Jun, respectively (Fig. 4A and B). These results indicate
that c-Jun mediates antiproliferation signals in TAM-stimulated
MCF-7 cells and TAM-effected c-Jun may also induce a protein with
antiproliferative effects.
Discussion
Antiestrogenic drugs, including TAM, provide an
effective therapeutic agent for females with hormone-dependent
cancer in neoadjuvant, adjuvant and advanced disease settings
(19). However, the existence of
an alternative mechanism of action for this drug is supported by a
number of clinical observations. For example, 30% of patients with
ER-negative cancer cells respond to TAM and 30% of patients with
ER-positive cancer cells are not sensitive to this anti-estrogen
(5). Therefore, the uses of TAM
are limited. Long-term exposure to TAM is associated with
resistance and, in specific cases, with tumor growth stimulation
(20,21). The signaling pathways by which
breast tumors acquire the ability to grow in the presence of
antiestrogens, including TAM, remain poorly understood. One of the
mechanisms by which cell lines develop resistance to antiestrogens
is through the utilization of alternative signaling pathways that
support cell growth in the presence of antiestrogens. Several
signalling molecules, including PKC-δ, are known to play a major
role in E2-mediated signaling (22,23).
The PKC family of serine/threonine kinases has been
intensively studied in cancer since their identification as major
receptors for the tumor-promoting phorbol esters. The contribution
of each individual PKC isozyme to malignant transformation is only
partially understood, but it is clear that each PKC plays a
different role in cancer progression. PKC deregulation is a common
phenomenon observed in breast cancer and PKC expression and
localization are usually dynamically regulated during mammary gland
differentiation and involution. Overexpression of several PKCs has
been reported in malignant breast tissue and breast cancer cell
lines (24). Since PKC
deregulation is observed in breast cancer (25), this kinase family is a promising
target for blocking or reverting breast cancer malignancy. PKC is
known to have at least 10 phospholipid-dependent serine-threonine
kinase isoforms, one of which is PKCδ. The role of PKCδ in breast
cancer remains ambiguous and limited information is available with
regard to expression levels of PKCδ in primary tumors. Although
altered PKCδ expression does not appear to be a prerequisite for
breast cancer progression, specific previous studies, including our
own, have identified a protumorigenic role for PKCδ overexpression
in murine mammary cells via the induction of survival and
anchorage-independent growth (26). PKCδ has been reported to promote
proliferation (27) and metastasis
development (28), whereas its
depletion is sufficient to drive murine mammary cancer cells into
apoptosis (29). By contrast,
several studies have revealed that PKCδ mediates antiproliferative
responses. For example, the antimitogenic effect of inositol
hexaphosphate in MCF-7 human breast cancer cells, which involves
inhibition of ERK and Akt as well as pRb hypophosphorylation, is
mediated by PKCδ (30). Crosstalk
between PKCδ and ER has also been hypothesized. Of note,
ER-positive breast cancer cell lines express high levels of PKCδ
and are associated with an improved endocrine response, whereas
ER-negative breast cancer cell lines express low PKCδ levels
(31). Moreover, PKCδ is likely to
play a crucial role in antiestrogen resistance in breast cancer
cells and has been associated with acquired resistance to TAM in
patients with breast cancer (32).
Based on these studies, we hypothesize that the PKC pathway is one
of the most important pathways associated with inhibition of MCF-7
cell proliferation by TAM. To investigate this hypothesis, the
signal transduction reporter vector was analyzed in MCF-7 cells
stimulated by TAM.
pAP-1(PMA)-TA-luc is a signal transduction reporter
vector. The vector contains multiple copies of the AP-1 enhancer,
located upstream of luciferase, that responds specifically to
phorbol ester treatment. Activating the PKC pathway by adding
phorbol esters, including PMA, results in transcription factors
binding AP-1 elements on the vector. It is designed for monitoring
the induction of the PKC signal transduction pathway. AP-1 is a
heterodimeric transcription factor that is composed of various
members of the Jun and Fos families and binds to DNA at specific
AP-1 binding sites. AP-1 transcriptional activity is increased by
phosphorylation at serine residues in the c-Jun component of AP-1.
In this study, no significant difference in c-Jun transcript and
protein levels was identified in MCF-7 and MDA-MB-435 cells
stimulated by TAM for 48 h. In addition, transcriptional reporter
gene assays indicated that TAM is an AP-1(PMA)-associated activator
(Fig. 3A). These data indicate
that TAM may function in a similar manner to PMA to modulate c-Jun
activity through the PKC pathway in MCF-7 cells. To determine the
role of c-Jun in antiproliferation of MCF-7 cells stimulated by
TAM, the correlation between inhibition rates of MCF-7 cells and
c-Jun expression and stimulation of TAM was analyzed. Inhibition
rates of TAM-stimulated MCF-7 cells were revealed to be positively
regulated by c-Jun overexpression and negatively regulated by c-Jun
underexpression.
Overall, these results indicate that TAM inhibits
proliferation of MCF-7 and MDA-MB-435 cells and has no
statistically significant effect on c-Jun transcript and protein
levels. However, TAM-stimulated antiproliferation of MCF-7 cells is
positively regulated by c-Jun through activation of the PKC
pathway.
References
|
1
|
Clarke M: Meta-analyses of adjuvant
therapies for women with early breast cancer: the Early Breast
Cancer Trialists' Collaborative Group overview. Ann Oncol. 17(Suppl
10): x59–x62. 2006. View Article : Google Scholar
|
|
2
|
Fisher B, Costantino JP, Wickerham DL,
Redmond CK, Kavanah M, Cronin WM, Vogel V, Robidoux A, Dimitrov N,
Atkins J, Daly M, Wieand S, Tan-Chiu E, Ford L and Wolmark N:
Tamoxifen for prevention of breast cancer: report of the National
Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer
Inst. 90:1371–1388. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Katzenellenbogen BS, Fang H, Ince BA,
Pakdel F, Reese JC, Wooge CH and Wrenn CK: William L. McGuire
Memorial Symposium. Estrogen receptors: ligand discrimination and
antiestrogen action. Breast Cancer Res Treat. 27:17–26. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Katzenellenbogen BS, Montano MM, Le Goff
P, Schodin DJ, Kraus WL, Bhardwaj B and Fujimoto N: Antiestrogens:
mechanisms and actions in target cells. J Steroid Biochem Mol Biol.
53:387–393. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Charlier C, Chariot A, Antoine N, Merville
MP, Gielen J and Castronovo V: Tamoxifen and its active metabolite
inhibit growth of estrogen receptor-negative MDA-MB-435 cells.
Biochem Pharmacol. 49:351–358. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Amin S, Kumar A, Nilchi L, Wright K and
Kozlowski M: Breast cancer cells proliferation is regulated by
tyrosine phosphatase SHP1 through c-jun N-terminal kinase and
cooperative induction of RFX-1 and AP-4 transcription factors. Mol
Cancer Res. 9:1112–1125. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mamay CL, Mingo-Sion AM, Wolf DM, Molina
MD and Van Den Berg CL: An inhibitory function for JNK in the
regulation of IGF-I signaling in breast cancer. Oncogene.
22:602–614. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Montano MM and Katzenellenbogen BS: The
quinone reductase gene: a unique estrogen receptor-regulated gene
that is activated by antiestrogens. Proc Natl Acad Sci USA.
94:2581–2586. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Umekita Y, Ohi Y, Sagara Y and Yoshida H:
Overexpression of cyclinD1 predicts for poor prognosis in estrogen
receptor-negative breast cancer patients. Int J Cancer. 98:415–418.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Altucci L, Addeo R, Cicatiello L, Dauvois
S, Parker MG, Truss M, Beato M, Sica V, Bresciani F and Weisz A:
17beta-Estradiol induces cyclin D1 gene transcription,
p36D1-p34cdk4 complex activation and p105Rb phosphorylation during
mitogenic stimulation of G(1)-arrested human breast cancer cells.
Oncogene. 12:2315–2324. 1996.
|
|
11
|
Fu XD, Cui YH, Lin GP and Wang TH:
Non-genomic effects of 17beta-estradiol in activation of the
ERK1/ERK2 pathway induces cell proliferation through upregulation
of cyclin D1 expression in bovine artery endothelial cells. Gynecol
Endocrinol. 23:131–137. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Urtreger AJ, Kazanietz MG and Bal de Kier
Joffe ED: Contribution of individual PKC isoforms to breast cancer
progression. IUBMB Life. 64:18–26. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Orchel A, Dzierzewicz Z, Parfiniewicz B,
Weglarz L and Wilczok T: Butyrate-induced differentiation of colon
cancer cells is PKC and JNK dependent. Dig Dis Sci. 50:490–498.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu DS, Krebs CE and Liu SJ: Proliferation
of human breast cancer cells and anti-cancer action of doxorubicin
and vinblastine are independent of PKC-alpha. J Cell Biochem.
101:517–528. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li Z, Wang N, Fang J, Huang J, Tian F, Li
C and Xie F: Role of PKC-ERK signaling in tamoxifen-induced
apoptosis and tamoxifen resistance in human breast cancer cells.
Oncol Rep. 27:1879–1886. 2012.PubMed/NCBI
|
|
16
|
Stabel S and Parker PJ: Protein kinase C.
Pharmacol Ther. 51:71–95. 1991. View Article : Google Scholar
|
|
17
|
Newton AC: Protein kinase C: structure,
function and regulation. J Biol Chem. 270:28495–28498. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Newton AC: Regulation of protein kinase C.
Curr Opin Cell Biol. 9:161–167. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fisher B, Dignam J, Bryant J, DeCillis A,
Wickerham DL, Wolmark N, Costantino J, Redmond C, Fisher ER, Bowman
DM, Deschênes L, Dimitrov NV, Margolese RG, Robidoux A, Shibata H,
et al: Five versus more than five years of tamoxifen therapy for
breast cancer patients with negative lymph nodes and estrogen
receptor-positive tumors. J Natl Cancer Inst. 88:1529–1542.
1996.
|
|
20
|
Berstein LM, Zheng H, Yue W, Wang JP,
Lykkesfeldt AE, Naftolin F, Harada H, Shanabrough M and Santen RJ:
New approaches to the understanding of tamoxifen action and
resistance. Endocr Relat Cancer. 10:267–277. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Santen RJ: Long-term tamoxifen therapy:
can an antagonist become an agonist? J Clin Endocrinol Metab.
81:2027–2029. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shanmugam M, Krett NL, Maizels ET, Cutler
RE Jr, Peters CA, Smith LM, O'Brien ML, Park-Sarge OK, Rosen ST and
Hunzicker-Dunn M: Regulation of protein kinase C delta by estrogen
in the MCF-7 human breast cancer cell line. Mol Cell Endocrinol.
148:109–118. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Keshamouni VG, Mattingly RR and Reddy KB:
Mechanism of 17-beta-estradiol-induced Erk1/2 activation in breast
cancer cells. A role for HER2 AND PKC-delta. J Biol Chem.
277:22558–22565. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tanaka Y, Gavrielides MV, Mitsuuchi Y,
Fujii T and Kazanietz MG: Protein kinase C promotes apoptosis in
LNCaP prostate cancer cells through activation of p38 MAPK and
inhibition of the Akt survival pathway. J Biol Chem.
278:33753–33762. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jarzabek K, Laudanski P, Dzieciol J,
Dabrowska M and Wolczynski S: Protein kinase C involvement in
proliferation and survival of breast cancer cells. Folia Histochem
Cytobiol. 40:193–194. 2002.PubMed/NCBI
|
|
26
|
Liu JF, Crepin M, Liu JM, Barritault D and
Ledoux D: FGF-2 and TPA induce matrix metalloproteinase-9 secretion
in MCF-7 cells through PKC activation of the Ras/ERK pathway.
Biochem Biophys Res Commun. 293:1174–1182. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Grossoni VC, Falbo KB, Kazanietz MG, de
Kier Joffe ED and Urtreger AJ: Protein kinase C delta enhances
proliferation and survival of murine mammary cells. Mol Carcinog.
46:381–390. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kiley SC, Clark KJ, Duddy SK, Welch DR and
Jaken S: Increased protein kinase C delta in mammary tumor cells:
relationship to transformation and metastatic progression.
Oncogene. 18:6748–6757. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lonne GK, Masoumi KC, Lennartsson J and
Larsson C: Protein kinase Cdelta supports survival of MDA-MB-231
breast cancer cells by suppressing the ERK1/2 pathway. J Biol Chem.
284:33456–33465. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vucenik I, Ramakrishna G, Tantivejkul K,
Anderson LM and Ramljak D: Inositol hexaphosphate (IP6) blocks
proliferation of human breast cancer cells through a
PKCdelta-dependent increase in p27Kip1 and decrease in
retinoblastoma protein (pRb) phosphorylation. Breast Cancer Res
Treat. 91:35–45. 2005. View Article : Google Scholar
|
|
31
|
Assender JW, Gee JM, Lewis I, Ellis IO,
Robertson JF and Nicholson RI: Protein kinase C isoform expression
as a predictor of disease outcome on endocrine therapy in breast
cancer. J Clin Pathol. 60:1216–1221. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nabha SM, Glaros S, Hong M, Lykkesfeldt
AE, Schiff R, Osborne K and Reddy KB: Upregulation of PKC-delta
contributes to antiestrogen resistance in mammary tumor cells.
Oncogene. 24:3166–3176. 2005. View Article : Google Scholar : PubMed/NCBI
|