Introduction
Cone-rod dystrophy (CORD) is a heterogeneous
inherited retinal disease characterized by reduced visual acuity,
photophobia and color vision defects. Fundus observation usually
identifies temporal pallor of the optic disc, attenuation of
retinal arterioles and macular atrophy. Recordings on an
electroretinogram (ERG) usually reveal the predominant functional
impairment of cones over rods during the early stages (1). The prevalence of CORD is
approximately 1 in 40,000 individuals (2).
The disease may be transmitted as an autosomal
dominant, autosomal recessive or X-linked trait. At least 24 genes
have been identified to be responsible for CORD (RetNet: https://sph.uth.tmc.edu/Retnet/). The genes for
autosomal dominant CORD are AIPL1(3), CRX(4), GUCA1A(5), GUCY2D(6), PITPNM3(7), PROM1(8), PRPH2(9), RIMS1(10), SEMA4A(11) and UNC119(12). The genes for autosomal recessive
CORD are ABCA4(13),
ADAM9(14),
CACNA2D4(15),
CDHR1(16),
CERKL(17),
CNGB3(18),
CNNM4(19),
KCNV2(20),
PDE6C(21),
RAX2(22),
RPGRIP1(23) and
RDH5(24). The genes for
X-linked CORD are CACNA1F(25) and RPGR(26). Although studies on individual genes
have been reported, the systemic analysis of these genes in a
cohort of patients is rare, with the exception of a few studies on
the genes for autosomal dominant CORD (27) or the genes for autosomal recessive
CORD (17,28). Extensive analysis may provide
insight into the mutation frequency and spectrum of the majority of
CORD-related genes (29). In this
study, we comprehensively screened 58 exons in 20 genes for
mutations in 130 unrelated Chinese patients with CORD, mostly on
the coding regions with reported mutations.
Materials and methods
Data from 130 unrelated patients with CORD were
collected at the Pediatric and Genetic Eye Clinic, Eye Hospital of
Zhongshan Ophthalmic Center, Guangzhou, China. Of the 130 patients,
111 were isolated cases, 8 had an autosomal dominant trait and 11
had an autosomal recessive trait. This study was performed in
accordance with the guidelines set out in the Declaration of
Helsinki and was approved by the Institutional Review Board of the
Zhongshan Ophthalmic Center. Informed consent was obtained from all
participants or their guardians prior to the collection of clinical
data and genomic samples. Genomic DNA was extracted from the
leukocytes of venous blood using previously reported methods
(30).
Of the 24 genes responsible for CORD, 4 genes,
CRX, GUCA1A, CACNA1F and RDH5, were not
analyzed in this study, as they already have been analyzed in
independent studies [(31) and
unpublished data]. When this study was initiated in April 2011, all
coding exons with a previously reported mutation in the 20 genes
(Table I) were selected as targets
for further analysis, with the exception of ABCA4, in which
a large number of variations have previously been identified both
in patients and controls (32).
Furthermore, in 3 genes, GUCY2D, PRPH2 and
KCNV2, all exons were analyzed, as mutations in
GUCY2D and PRPH2 are frequently observed in patients
with CORD (27,33), while mutations in both exons of
KCNV2 have been reported (20). In this study, a total of 58 exons
in 20 genes were analyzed (Table
I). For the 58 coding exons, DNA fragments encompassing
individual exons were amplified by PCR using corresponding primer
pairs (available upon request). The sequences of amplicons were
determined by Sanger sequencing using an ABI BigDye Terminator
Cycle Sequencing kit v3.1 on an ABI 3130 Genetic analyzer (Applied
Biosystems, Foster City, CA, USA). The results from the patients
were aligned with the reference sequences from the NCBI database
using SeqManII (DNAstar, Madison, WI, USA) to determine the
variations. Each variant was bidirectionally sequenced and any
novel variant was further evaluated using 192 normal controls (384
chromosomes). The mutation descriptions are in accordance with the
recommendations from the Human Genomic Variation Society
(http://www.hgvs.org/mutnomen/).
 | Table IThe genes and targeted exons analyzed
in this study. |
Table I
The genes and targeted exons analyzed
in this study.
| Genes | OMIM | cDNA | Trait | Total coding
exonsa | Exons for
sequencingb |
|---|
| GUCY2D | 600179 | NM_000180.3 | AD | 18 | 1–18c |
| PRPH2 | 179605 | NM_000322.4 | AD | 3 | 1–3 |
| RIMS1 | 606629 | NM_014989.4 | AD | 34 | 6, 34 |
| AIPL1 | 604392 | NM_014336.3 | AD | 6 | 5, 6 |
| PITPNM3 | 608921 | NM_031220.3 | AD | 20 | 9, 14 |
| UNC119 | 604011 | NM_005148.3 | AD | 5 | 1, 2 |
| SEMA4A | 607292 | NM_022367.3 | AD | 14 | 9 |
| PROM1 | 604365 | NM_006017.2 | AD | 26 | 11, 13 |
| ADAM9 | 602713 | NM_003816.2 | AR | 22 | 6, 9, 12 |
| CNGB3 | 605080 | NM_019098.4 | AR | 18 | 6, 8, 11 |
| KCNV2 | 607604 | NM_133497.3 | AR | 2 | 1, 2 |
| PDE6C | 600827 | NM_006204.3 | AR | 22 | 1 |
| CDHR1 | 609502 | NM_033100.2 | AR | 17 | 6 |
|
CACNA2D4 | 608171 | NM_172364.4 | AR | 38 | 25, 30 |
| RPGRIP1 | 605446 | NM_020366.3 | AR | 24 | 13, 16 |
| RAX2 | 610362 | NM_032753.3 | AR | 2 | 2 |
| ABCA4 | 601691 | NM_000350.2 | AR | 50 | 6 |
| CERKL | 608381 | NM_201548.4 | AR | 10 | 1, 2, 6, 8 |
| CNNM4 | 607805 | NM_020184.3 | AR | 7 | 1, 4, 7 |
| RPGR | 312610 | NM_000328.2 | X-LINKED | 19 | 4, 6, 7 |
| Total | | | | 357 | 58 |
Four online computational algorithms (34–37),
PANTHER (http://www.pantherdb.org/), PMut
(http://mmb2.pcb.ub.es:8080/PMut/), SIFT
(http://sift.jcvi.org/) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/),
respectively, were used to predict the functional impact of the
detected missense mutations.
Results
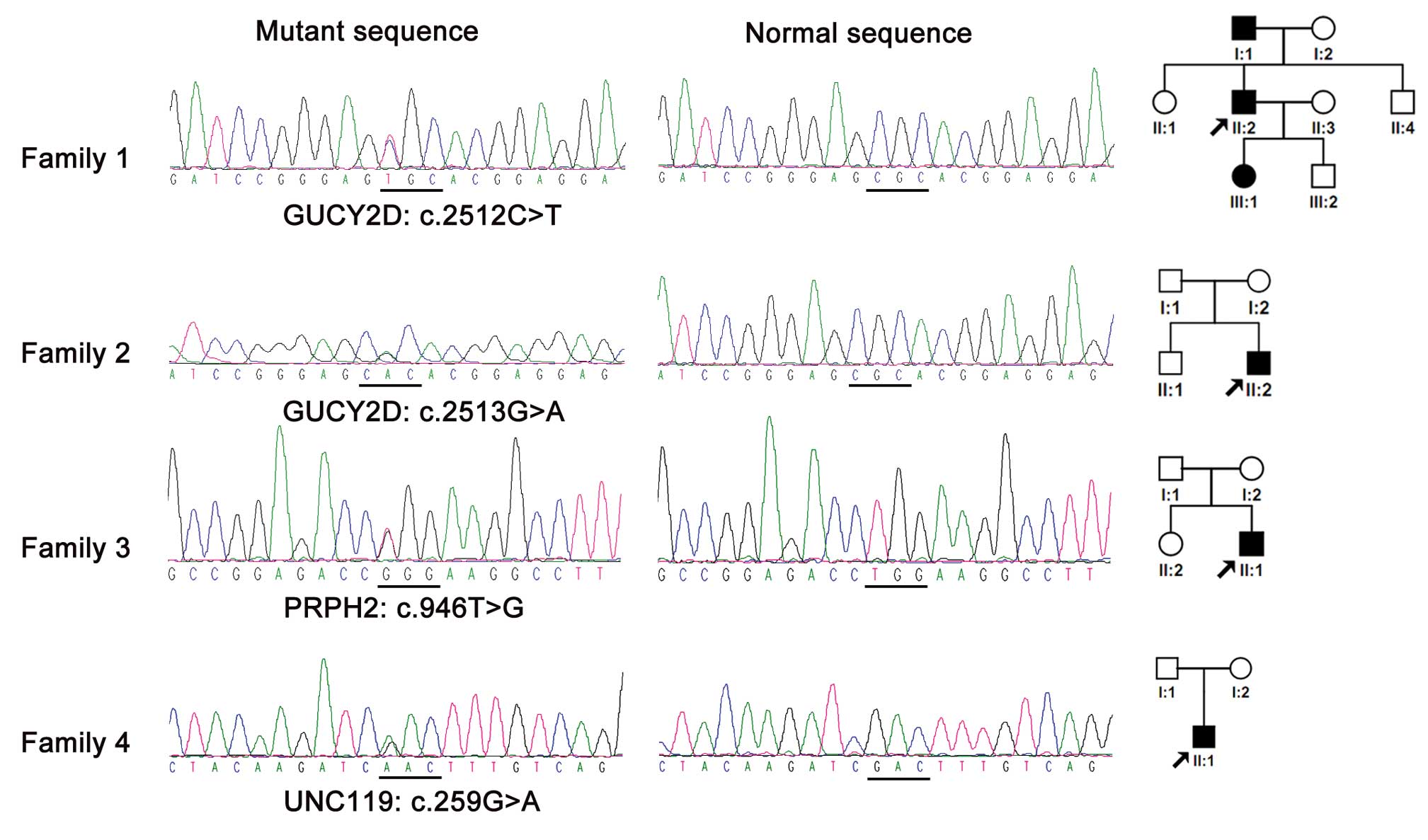
Upon the sequencing analysis of 58 exons in 20
genes, 4 mutations, 1 novel and 3 known (38–40),
in 3 genes were discovered in 4/130 unrelated probands
(4/130=3.08%) (Table II). All 4
mutations were heterozygous and detected in genes known to cause
autosomal dominant CORD: c.259G>A (p.Asp87Asn) in UNC119,
c.2512C>T (p.Arg838Cys) and c.2513G>A (p.Arg838His) in
GUCY2D and c.946T>G (p.Trp316Gly) in PRPH2
(Fig. 1). In addition to the 4
mutations, a number of possible non-pathogenic variants were also
detected in KCNV2, CERKL, PITPNM3,
RPGRIP1, AIPL1, RPGR, ABCA4,
RIMS1, CNGB3, PDE6C, CDHR1,
RAX2, CNNM4, GUCY2D and PRPH2 (Table III).
| Table IIMutations detected in 130 unrelated
cone-rod dystrophy (CORD) patients and 192 healthy controls. |
Table II
Mutations detected in 130 unrelated
cone-rod dystrophy (CORD) patients and 192 healthy controls.
| | Changes | Description | Computational
prediction | | | |
|---|
| |
|
|
| | | |
|---|
| Family | Gene | DNA | Protein | State | Cons | Blosum62a | PolyPhen-2 | SIFT | Pmut | PANTHERb | Cases | Controls | Refs |
|---|
| 1 | GUCY2D | c.2512C>T | p.Arg838Cys | Hetero | Yes | 8 | PD | D | PA | −8.7 | 1/130 | ND | (38) |
| 2 | GUCY2D | c.2513G>A | p.Arg838His | Hetero | Yes | 5 | PD | D | PA | −5.5 | 1/130 | ND | (39) |
| 3 | PRPH2 | c.946T>G | p.Trp316Gly | Hetero | Yes | 13 | Benign | D | PA | NA | 1/130 | ND | (40) |
| 4 | UNC119 | c.259G>A | p.Asp87Asn | Hetero | Yes | 5 | PD | Tolerated | Neutral | −3.2 | 1/130 | 0/192 | This study |
| Table IIIPolymorphisms detected in 130
unrelated patients. |
Table III
Polymorphisms detected in 130
unrelated patients.
| Gene | Exon | Variations | Status | Bioinformation
analysis | Frequency in
cases/controlsa | Refsc |
|---|
|
|
|---|
| Nucleotide | Amino acid | Conservation | PolyPhen-2 | Splice site |
|---|
| KCNV2 | 1 | c.612G>A | p.(=) | Hetero/Homo | Yes | NA | No change | 11/4 | This study |
| 1 | c.645G>C | p.Lys215Asp | Hetero/Homo | Yes | PD | No change | 8/6 | This study |
| 1 | c.920T>G | p.Met307Arg | Hetero | No | Benign | No change | 5/8 | This study |
| 1 | c.759A>G | p.(=) | Hetero | Yes | NA | No change | 2/14 | rs10967709 |
| 1 | c.795C>G | p.(=) | Hetero/Homo | No | NA | No change | 91/170 | rs12237048 |
| 2 | c.1513G>T | p.Ala505Ser | Hetero | Yes | Benign | No change | 1/1 | This study |
| 2 | c.1638+6T>C | - | Hetero | NA | NA | Change | 8/NA | rs41306094 |
| 2 | c.1386C>T | p.(=) | Hetero | Yes | NA | No change | 8/NA | rs41312842 |
| 2 | c.1597C>G | p.Val533Leu | Hetero | No | Benign | No change | 8/NA | rs12352254 |
| CERKL | 2 | c.242A>C | p.Asp81Ala | Hetero/Homo | No | Benign | No change | 43/NA | rs61750041 |
| 2 | c.239-12T>A | - | Hetero/Homo | NA | NA | No change | 27/NA | rs6433923 |
| 2 | c.313C>T | p.Arg105Trp | Hetero | Yes | PD | No change | 2/NA | rs149078111 |
| PITPNM3 | 9 | c.901-10G>C | - | Hetero/Homo | NA | NA | Change | 34/44 | rs77580616 |
| 9 | c.1016C>G | p.Pro339Arg | Hetero | Yes | Benign | No change | 1/1 | This study |
| RPGRIP1 | 16 | c.2592T>C | p.(=) | Hetero | Yes | NA | No change | 1/NA | This study |
| AIPL1 | 5 | c.726G>A | p.(=) | Hetero | Yes | NA | No change | 9/NA | This study |
| 5 | c.784+18G>A | - | Hetero | NA | NA | No change | 6/NA | rs7222126 |
| RPGR | 7 | c.732G>A | p.(=) | Hetero | No | NA | No change | 1/NA | This study |
| 7 | c.762T>C | p.(=) | Hetero | No | NA | No change | 1/NA | This study |
| ABCA4 | 6 | c.635G>A | p.Arg212His | Hetero | Yes | PD | Change | 10/7 | This study |
| 6 | c.673G>A | p.Val225Met | Hetero | Yes | PD | Change | 2/1 | This study |
| 6 | c.634C>T | p.Arg212Cys | Hetero | Yes | PD | No change | 1/1 | This study |
| RIMS1 | 6 | c.942G>A | p.(=) | Hetero | No | NA | No change | 2/NA | This study |
| 6 | c.1209G>A | p.(=) | Hetero | No | NA | No change | 28/NA | This study |
| 6 | c.1311G>A | p.(=) | Hetero | No | NA | No change | 1/NA | This study |
| CNGB3 | 8 | c.919A>G | p.Val307Ile | Hetero | No | Benign | Change | 14/NA | rs13265557 |
| 8 | c.912C>T | p.(=) | Hetero | No | NA | No change | 1/NA | rs117806701 |
| PDE6C | 1 | c.252G>A | p.(=) | Hetero | Yes | NA | No change | 26/NA | rs1131978 |
| 1 | c.471T>G | p.Asp157Glu | Hetero/Homo | Yes | PD | No change | 5/NA | rs76999928 |
| CDHR1 | 6 | c.477A>G | p.(=) | Hetero/Homo | Yes | NA | No change | 22/NA | rs4933975 |
| RAX2 | 2 | c.282C>T | p.(=) | Hetero | No | NA | No change | 3/NA | This study |
| 2 | c.217-8C>T | - | Hetero | NA | NA | No change | 3/NA | rs79588413 |
| CNNM4 | 1 | c.47G>Ab | p.Arg16His | Hetero | Yes | Unknown | No change | 0/0 | This study |
| GUCY2D | 1 | c.154G>T | p.Ala52Ser | Hetero/Homo | No | Benign | No change | 80/NA | rs61749665 |
| 1 | c.61T>C | p.Trp21Arg | Herero | No | PD | No change | 2/NA | rs9905402 |
| 1 | c.164C>T | p.Thr55Met | Hetero | Yes | PD | No change | 2/NA | rs201414567 |
| 1 | c.340G>A | p.Val114Met | Hetero | No | PD | No change | 1/0 | This study |
| 1 | c.343T>C | p.Ser115Pro | Hetero/Homo | No | PD | No change | 2/3 | This study |
| 1 | c.459delC |
p.Trp154GlyfsX12 | Hetero | NA | NA | NA | 1/0 | This study |
| 2 | c.741C>T | p.(=) | Hetero | Yes | NA | No change | 22/NA | rs3829789 |
| 9 | c.2101C>T | p.Pro701Ser | Hetero/Homo | No | Benign | No change | 38/NA | rs34598902 |
| 11 | c.2282G>A | p.Arg761Gln | Hetero | No | Benign | No change | 1/0 | This study |
| PRPH2 | 1 | c.318T>C | p.(=) | Hetero/Homo | No | NA | No change | 106/NA | This study |
| 3 | c.910C>G | p.Gln304Glu | Hetero/Homo | No | Benign | No change | 116/NA | This study |
| 3 | c.1013A>G | p.Asp338Gly | Hetero/Homo | No | Benign | No change | 116/NA | rs434102 |
| 3 |
c.1041+13C>T | - | Hetero | NA | NA | No change | 40/NA | This study |
The clinical data of the 4 patients with a mutation
in GUCY2D, PRPH2 or UNC119 are summarized in
Table IV. Affected members had
poor vision, photophobia or nystagmus as initial symptoms. The
onset age varied from the first few months after birth to 16 years
of age. Fundus examination revealed attenuated vessels, macular
atrophy and temporal pallor of the optic disc. ERG recordings
revealed severely reduced or extinguished cone responses
accompanied by normal to mildly reduced rod responses in 3 patients
with these mutations.
| Table IVClinical information of the cone-rod
dystrophy (CORD) patients with mutations. |
Table IV
Clinical information of the cone-rod
dystrophy (CORD) patients with mutations.
| | | | Age | | | BCVA | Fundus changes | ERG responses |
|---|
| | | |
| | |
|
|
|
|---|
| Family | Gene | Mutations | Gender | Exam | Onset | Inheritance | First symptom | OD | OS | OU | Rod | Cone |
|---|
| 1 | GUCY2D | c.2512C>T | M | 36 | 16 | Dominant | PV | 0.10 | 0.10 | APM, TDP | Normal | Extinguished |
| 2 | GUCY2D | c.2513G>A | M | 5 | EC | Isolated | PV, PP, NYS | 0.06 | 0.06 | AV | Mildly reduced | Extinguished |
| 3 | PRPH2 | c.946T>G | M | 0.3 | FMB | Isolated | NYS | LP | LP | NA | Mildly reduced | Severely
Reduced |
| 4 | UNC119 | c.259G>A | M | 3.5 | 3.25 | Isolated | PP | NA | NA | APM, AV | NA | NA |
Discussion
In this study, 4 mutations in 58 exons from 20 genes
were detected in 4/130 patients with CORD, which suggests that the
frequency of mutations in these regions is rare in Chinese
patients. All coding exons of GUCY2D and PRPH2 were
analyzed in this study. The mutation frequency for GUCY2D
was 1.54% (2/130), 0.77% for PRPH2 and 0.77% for
UNC119.
The mutation spectrum and frequency for certain
CORD-related genes have previously been reported (17,21,28,31,41).
The systematic screening of 10 genes (AIPL1, CRX,
GUCA1A, GUCY2D, PITPNM3, PROM1,
PRPH2, RIMS1, SEMA4A and UNC119)
responsible for autosomal dominant CORD identified mutations in
25/52 (48.1%) families. The mutation frequency of individual genes
in this cohort is as follows: GUCY2D (23.0%), PRPH2
(11.0%), GUCA1A (8.0%), CRX (4.0%) and PROM1
(2.0%) (27). For individual gene
analysis in different populations, the frequency of CORD-associated
GUCY2D mutations has been detected in 11.0% of Japanese
patients (42) and in 40.0% of
European and American patients (33). Mutations in several other genes
have been detected in a small proportion of patients with CORD,
such as CNGB3 mutations in 5.0% of patients from the
Netherlands (43), AIPL1
mutations in 3.6% of patients from the USA (3) and SEMA4A mutations in 8.0% of
patients from Pakistan (11).
However, the mutation spectrum and frequency for the majority of
CORD-related genes have not been well evaluated. For a few genes,
mutations have only been reported in 1 or 2 CORD families, such as
the c.2459G>A mutation of RIMS1 in a British family
(44), the c.1878G>C mutation
of PITPNM3 in 2 Swedish families (7) and the c.524dup1 mutation of
CDHR1 in a family from the Faroe Islands (16). It is unclear as to whether this is
due to the rare variants in these genes or a lack of subsequent
studies. Comprehensive evaluation of these genes in various ethnic
populations based on a large number of cases would provide a better
overview of the mutation spectrum and frequency, which would be
beneficial for use in personalized gene diagnosis and genetic
counseling.
Using a similar strategy to this study, our previous
study on Leber’s congenital amaurosis (LCA) detected mutations in
approximately half of the 87 families tested, based on Sanger
sequencing of exons with reported mutations in 15 LCA-related genes
(29); this correlated with other
reports based on the individual analysis of one or several genes.
However, in the present study, only 4 mutations were identified in
4/130 families with CORD, which is lower than previously reported.
It is possible that the mutation spectrum and frequency of these
genes may differ in Chinese patients than in those with different
ethnic backgrounds, with frequent mutations in exons not covered in
this study. It is also possible that the genetic causes of CORD in
Chinese patients have not yet been identified. To answer these
questions, additional comprehensive evaluation of these patients
with other methods, such as exome sequencing, is required.
Acknowledgements
The authors are grateful to the patients for their
participation. This study was supported by the National Natural
Science Foundation of China (81170881), National 973 plan
(2010CB529904) and the Fundamental Research Funds of State Key
Laboratory of Ophthalmology.
References
|
1
|
Hamel CP: Cone rod dystrophies. Orphanet J
Rare Dis. 2:72007. View Article : Google Scholar
|
|
2
|
Hamel CP, Griffoin JM, Bazalgette C, et
al: Molecular genetics of pigmentary retinopathies: identification
of mutations in CHM, RDS, RHO, RPE65, USH2A and XLRS1 genes. J Fr
Ophtalmol. 23:985–995. 2000.(In French).
|
|
3
|
Sohocki MM, Perrault I, Leroy BP, et al:
Prevalence of AIPL1 mutations in inherited retinal degenerative
disease. Mol Genet Metab. 70:142–150. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Freund CL, Gregory-Evans CY, Furukawa T,
et al: Cone-rod dystrophy due to mutations in a novel
photoreceptor-specific homeobox gene (CRX) essential for
maintenance of the photoreceptor. Cell. 91:543–553. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Payne AM, Downes SM, Bessant DA, et al: A
mutation in guanylate cyclase activator 1A (GUCA1A) in an autosomal
dominant cone dystrophy pedigree mapping to a new locus on
chromosome 6p21.1. Hum Mol Genet. 7:273–277. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kelsell RE, Evans K, Gregory CY, Moore AT,
Bird AC and Hunt DM: Localisation of a gene for dominant cone-rod
dystrophy (CORD6) to chromosome 17p. Hum Mol Genet. 6:597–600.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Köhn L, Kadzhaev K, Burstedt MS, et al:
Mutation in the PYK2-binding domain of PITPNM3 causes autosomal
dominant cone dystrophy (CORD5) in two Swedish families. Eur J Hum
Genet. 15:664–671. 2007.
|
|
8
|
Yang Z, Chen Y, Lillo C, et al: Mutant
prominin 1 found in patients with macular degeneration disrupts
photoreceptor disk morphogenesis in mice. J Clin Invest.
118:2908–2916. 2008.PubMed/NCBI
|
|
9
|
Fishman GA, Stone EM, Alexander KR,
Gilbert LD, Derlacki DJ and Butler NS: Serine-27-phenylalanine
mutation within the peripherin/RDS gene in a family with cone
dystrophy. Ophthalmology. 104:299–306. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Johnson S, Halford S, Morris AG, et al:
Genomic organisation and alternative splicing of human RIM1, a gene
implicated in autosomal dominant cone-rod dystrophy (CORD7).
Genomics. 81:304–314. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Abid A, Ismail M, Mehdi SQ and Khaliq S:
Identification of novel mutations in the SEMA4A gene associated
with retinal degenerative diseases. J Med Genet. 43:378–381. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kobayashi A, Higashide T, Hamasaki D, et
al: HRG4 (UNC119) mutation found in cone-rod dystrophy causes
retinal degeneration in a transgenic model. Invest Ophthalmol Vis
Sci. 41:3268–3277. 2000.PubMed/NCBI
|
|
13
|
Cremers FP, van de Pol DJ, van Driel M, et
al: Autosomal recessive retinitis pigmentosa and cone-rod dystrophy
caused by splice site mutations in the Stargardt’s disease gene
ABCR. Hum Mol Genet. 7:355–362. 1998.PubMed/NCBI
|
|
14
|
Parry DA, Toomes C, Bida L, et al: Loss of
the metalloprotease ADAM9 leads to cone-rod dystrophy in humans and
retinal degeneration in mice. Am J Hum Genet. 84:683–691. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wycisk KA, Budde B, Feil S, et al:
Structural and functional abnormalities of retinal ribbon synapses
due to Cacna2d4 mutation. Invest Ophthalmol Vis Sci. 47:3523–3530.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ostergaard E, Batbayli M, Duno M,
Vilhelmsen K and Rosenberg T: Mutations in PCDH21 cause autosomal
recessive cone-rod dystrophy. J Med Genet. 47:665–669. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Littink KW, Koenekoop RK, van den Born LI,
et al: Homozygosity mapping in patients with cone-rod dystrophy:
novel mutations and clinical characterizations. Invest Ophthalmol
Vis Sci. 51:5943–5951. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Michaelides M, Aligianis IA, Ainsworth JR,
et al: Progressive cone dystrophy associated with mutation in
CNGB3. Invest Ophthalmol Vis Sci. 45:1975–1982. 2004. View Article : Google Scholar
|
|
19
|
Polok B, Escher P, Ambresin A, et al:
Mutations in CNNM4 cause recessive cone-rod dystrophy with
amelogenesis imperfecta. Am J Hum Genet. 84:259–265. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu H, Cowing JA, Michaelides M, et al:
Mutations in the gene KCNV2 encoding a voltage-gated potassium
channel subunit cause ‘cone dystrophy with supernormal rod
electroretinogram’ in humans. Am J Hum Genet. 79:574–579.
2006.PubMed/NCBI
|
|
21
|
Thiadens AA, den Hollander AI, Roosing S,
et al: Homozygosity mapping reveals PDE6C mutations in patients
with early-onset cone photoreceptor disorders. Am J Hum Genet.
85:240–247. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang QL, Chen S, Esumi N, et al: QRX, a
novel homeobox gene, modulates photoreceptor gene expression. Hum
Mol Genet. 13:1025–1040. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hameed A, Abid A, Aziz A, Ismail M, Mehdi
SQ and Khaliq S: Evidence of RPGRIP1 gene mutations associated with
recessive cone-rod dystrophy. J Med Genet. 40:616–619. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wada Y, Abe T, Sato H and Tamai M: A novel
Gly35Ser mutation in the RDH5 gene in a Japanese family with fundus
albipunctatus associated with cone dystrophy. Arch Ophthalmol.
119:1059–1063. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jalkanen R, Mäntyjärvi M, Tobias R, et al:
X linked cone-rod dystrophy, CORDX3, is caused by a mutation in the
CACNA1F gene. J Med Genet. 43:699–704. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Demirci FY, Rigatti BW, Wen G, et al:
X-linked cone-rod dystrophy (locus COD1): identification of
mutations in RPGR exon ORF15. Am J Hum Genet. 70:1049–1053. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kohl S, Kitiratschky V, Papke M, Schaich
S, Sauer A and Wissinger B: Genes and mutations in autosomal
dominant cone and cone-rod dystrophy. Adv Exp Med Biol.
723:337–343. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
den Hollander AI, Black A, Bennett J and
Cremers FP: Lighting a candle in the dark: advances in genetics and
gene therapy of recessive retinal dystrophies. J Clin Invest.
120:3042–3053. 2010.PubMed/NCBI
|
|
29
|
Li L, Xiao X, Li S, et al: Detection of
variants in 15 genes in 87 unrelated Chinese patients with Leber
congenital amaurosis. PLoS One. 6:e194582011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang Q, Wang P, Li S, et al: Mitochondrial
DNA haplogroup distribution in Chaoshanese with and without myopia.
Mol Vis. 16:303–309. 2010.PubMed/NCBI
|
|
31
|
Huang L, Xiao X, Li S, et al: CRX variants
in cone-rod dystrophy and mutation overview. Biochem Biophys Res
Commun. 426:498–503. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Webster AR, Héon E, Lotery AJ, et al: An
analysis of allelic variation in the ABCA4 gene. Invest Ophthalmol
Vis Sci. 42:1179–1189. 2001.PubMed/NCBI
|
|
33
|
Kitiratschky VB, Wilke R, Renner AB, et
al: Mutation analysis identifies GUCY2D as the major gene
responsible for autosomal dominant progressive cone degeneration.
Invest Ophthalmol Vis Sci. 49:5015–5023. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mi H, Lazareva-Ulitsky B, Loo R, et al:
The PANTHER database of protein families, subfamilies, functions
and pathways. Nucleic Acids Res. 33:D284–288. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ferrer-Costa C, Gelpi JL, Zamakola L,
Parraga I, de la Cruz X and Orozco M: PMUT: a web-based tool for
the annotation of pathological mutations on proteins.
Bioinformatics. 21:3176–3178. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ng PC and Henikoff S: SIFT: Predicting
amino acid changes that affect protein function. Nucleic Acids Res.
31:3812–3814. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ramensky V, Bork P and Sunyaev S: Human
non-synonymous SNPs: server and survey. Nucleic Acids Res.
30:3894–3900. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kelsell RE, Gregory-Evans K, Payne AM, et
al: Mutations in the retinal guanylate cyclase (RETGC-1) gene in
dominant cone-rod dystrophy. Hum Mol Genet. 7:1179–1184. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Payne AM, Morris AG, Downes SM, et al:
Clustering and frequency of mutations in the retinal guanylate
cyclase (GUCY2D) gene in patients with dominant cone-rod
dystrophies. J Med Genet. 38:611–614. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jin ZB, Mandai M, Yokota T, et al:
Identifying pathogenic genetic background of simplex or multiplex
retinitis pigmentosa patients: a large scale mutation screening
study. J Med Genet. 45:465–472. 2008. View Article : Google Scholar
|
|
41
|
Thiadens AA, Phan TM, Zekveld-Vroon RC, et
al: Clinical course, genetic etiology, and visual outcome in cone
and cone-rod dystrophy. Ophthalmology. 119:819–826. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ito S, Nakamura M, Nuno Y, Ohnishi Y,
Nishida T and Miyake Y: Novel complex GUCY2D mutation in Japanese
family with cone-rod dystrophy. Invest Ophthalmol Vis Sci.
45:1480–1485. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Thiadens AA, Roosing S, Collin RW, et al:
Comprehensive analysis of the achromatopsia genes CNGA3 and CNGB3
in progressive cone dystrophy. Ophthalmology. 117:825–830. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kelsell RE, Gregory-Evans K, Gregory-Evans
CY, et al: Localization of a gene (CORD7) for a dominant cone-rod
dystrophy to chromosome 6q. Am J Hum Genet. 63:274–279. 1998.
View Article : Google Scholar : PubMed/NCBI
|