Introduction
Diabetic retinopathy (DR), a leading cause of
blindness worldwide, is an important area of study in clinical and
basic research. The pathogenesis of DR is complex and to date, the
precise mechanisms involved remain unclear. Previous studies have
largely focused on the mechanisms underlying microvascular changes.
However, an increasing number of studies have reported that the
pathogenesis of DR correlates with neurodegeneration of the retina
(1). Neuronal and glial tissues,
which are sensitive to hyperglycemia, may be involved in the
pathogenesis of DR. Microvascular damage is also likely to be
associated with malfunctions of glial cell metabolism (2). Therefore, understanding the
pathological changes which correlate with DR and the mechanisms
that protect neurons, are likely to be important aims of future
studies. Studies in animal models have demonstrated that at early
stages of DR, specific retinal ganglion cells (RGCs) undergo
apoptosis and retinal neurodegeneration is likely to be associated
with a lack of brain-derived neurotrophic factor (BDNF) (3).
BDNF is a member of the neurotrophin family of
growth factors and is important in the development, differentiation
and maintenance of neurons. Previous studies have revealed that
BDNF is critical for photoreceptor cells and the repair of damage
to the retina and the optic nerve. BDNF promotes survival in
injured RGCs induced by axotomy or retinal ischemia, and also
promotes regeneration of the nerve fiber (4,5). In
addition, BDNF promotes the survival of retinal interneurons and is
important for establishing phenotypes and synaptic connections in
the developing retina (6). BDNF
has been reported to inhibit neuroretinal cell death under
conditions of ischemia and hypoxia, and to inhibit apoptosis in rat
RGCs at early stages of DR (7).
However, the mechanisms by which BDNF regulates RGCs remain unclear
(8–10).
Tropomyosin-related kinase B (TrkB) is a receptor
protein involved in the development and maturation of the central
and peripheral nervous systems. BDNF has a high affinity for TrkB
and p75 enhances the interaction between BDNF and TrkB (11). Upon ligand-binding, TrkB undergoes
homodimerization, autophosphorylation and activation. It then
recruits and activates several downstream effectors to regulate
gene expression and protect neurons. Members of the TrkB downstream
signaling cascade, including ERK/MAPK and PI3K/PKB, have been
reported to be responsive to BDNF (12,13).
Several studies have hypothesized that BDNF largely activates the
ERK/MAPK pathway (14–17). By contrast, other studies have
reported that activation of the ERK/MAPK pathway leads to cell
death and PI3K/PKB is the main pathway involved in the protection
of neurons induced by BDNF (18).
In addition, it has been reported that both pathways are critical
for neuroprotection induced by BDNF. To the best of our knowledge,
no studies to date have addressed which pathway dominates the
response to TrkB activation (13,19,20).
A number of studies have demonstrated in vivo
that the death of retinal neurons at early stages of DR correlates
with decreased levels of BDNF, and that TrkB is important for the
protection of brain neurons and RGCs induced by BDNF. However, it
is not known whether BDNF protects retinal neurons exposed to
hyperglycemia in vitro or whether the ERK/MAPK pathway is
activated in response to BDNF-induced neuroprotection.
The aim of the present study was to determine
whether conditions of hyperglycemia increase the levels of
apoptosis in retinal neurons in vitro and whether BDNF is
effective in protecting retinal neurons from hyperglycemia. In
addition, we determined whether BDNF promotes TrkB expression and
activates the TrkB/ERK/MAPK pathway to decrease levels of
apoptosis. This study is likely to provide novel insights into the
pathogenesis of DR and be useful for the development of novel
treatment strategies for this disease.
Materials and methods
Cell culture
Retinal neurons dissected from postnatal Wistar rats
were grown in Neurobasal medium containing 5 mM glucose, 4 mM
L-glutamine, 1.5 g/l sodium bicarbonate, 1.0 mM sodium pyruvate and
B-27 serum-free supplements (all purchased from Gibco-BRL,
Carlsbad, CA, USA). Retinal neurons were cultured in a
CO2 incubator with 5% CO2 at 37°C. Cells were
used at 80% confluence for the experiments. The study was approved
by the ethics committee of the Ministry of Science and Technology
of China, Beijing, China.
Immunohistochemistry
Immunohistochemistry assays were performed to
investigate the effect of BNDF on TrkB protein expression. Briefly,
retinal neuron cells were treated with BDNF (Sigma-Aldrich, St.
Louis, MO, USA) at various concentrations 2 or 3 days following
dissection, followed by the addition of 35 mmol/l glucose or buffer
1 day later. The cells were divided into 5 groups: i) 5.5 mmol/l
glucose in basal medium (Ctr); ii) 35 mmol/l glucose (HG); iii) 35
mmol/l glucose + 75 ng/ml BDNF (HG + L-BDNF); iv) 35 mmol/l glucose
+ 100 ng/ml BDNF (HG + M-BDNF); and v) 35 mmol/l glucose + 125
ng/ml BDNF (HG + H-BDNF). Following BDNF treatment (4 days), the
cells were fixed in 4% (v/v) neutral paraformaldehyde for 30 min
and blocked with 3% peroxide-methanol at room temperature for
endogenous peroxidase ablation. To block the non-specific binding
sites, samples were incubated with 1% bovine serum albumin and 0.5%
Triton X-100 in phosphate-buffered saline (PBS) for 20 min,
followed by 10% normal goat serum in PBS for 20 min. The expression
of TrkB was then detected by immunostaining with a rabbit
anti-mouse TrkB polyclonal antibody (1:50; Cell Signaling
Technology, Inc., Danvers, MA, USA). Following sequential
incubation with biotinylated secondary antibody and
avidin-biotin-peroxidase reagents, the cells were stained with 0.5
mg/ml horseradish peroxidase (HRP) substrate solution [0.05% DAB
and 0.03% H2O2 prepared in Tris-Cl buffer (pH
7.4)]. Next, the cells were counterstained with hematoxylin for 25
sec, followed by dehydration, clearing and mounting with neutral
gums. The negative control group was subjected to the same
protocol; however, the primary antibody was replaced by PBS.
Histological assessments were performed as described previously
(21).
MTT analysis
Retinal neuron cells were seeded in 96-well tissue
culture plates (1–3×104 cells/well) and then treated
with various combinations of glucose and BDNF as described. Next, 4
days following the addition of BDNF, thiazolyl blue tetrazolium
bromide (MTT; 5 mg/ml; Sigma-Aldrich) was added to each well and
incubated for 4 h. Formazan crystals formed from MTT by living
cells were dissolved in DMSO and the formazan purple solution was
detected using a spectrophotometer at 570 nm. Inhibitory ratios
were calculated using the following formula: [(control −
sample)/control] × 100.
FACS analysis
FACS analysis was performed to detect the levels of
apoptosis of retinal neurons using the Annexin V-FITC apoptosis
detection kit (Bender MedSystems GmbH, Vienna, Austria) according
to the manufacturer’s instructions. Briefly, retinal neurons with
various treatments were detached with 0.25% trypsin, followed by
10% fetal bovine serum to terminate the reaction. The cells were
then collected and resuspended in 250 μl binding buffer, followed
by staining with Annexin V-FITC and propidium iodide solution for
15 min at room temperature in the dark. The samples were analyzed
immediately using the FACSCalibur flow cytometer.
Western blot analysis
Retinal neurons were treated under the following
conditions: i) 5.5 mmol/l glucose in basal medium (Ctr); ii) 5.5
mmol/l glucose + 100 ng/ml BDNF (Ctr + BDNF); iii) 35 mmol/l
glucose (HG); and iv) 35 mmol/l glucose + 100 ng/ml BDNF (HG +
BDNF). The cells were then washed with D-PBS and lysed with cell
lysis buffer [20 mmol/l Tris-HCl (pH 7.4), 150 mmol/l NaCl, 1%
Triton X-100, 0.1% SDS, 1% sodium deoxycholate, 1 mmol/l EDTA, 1
mmol/l phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin and 1
mmol/l Na3VO4]. Total proteins were exacted
by centrifugation at 13,000 × g for 15 min and denatured in a 100°C
water-bath for 5 min. The concentration of total protein was
measured using the Bradford method.
For western blot analysis, the proteins (25 μg/lane)
were separated by electrophoresis using 10% SDS-PAGE gels and then
transferred onto polyvinylidene fluoride membranes. The membranes
were blocked with blocking buffer (Tris-buffered saline, 5% nonfat
dry milk and 0.1% Tween-20) prior to incubation with polyclonal
antibodies against ERK, pERK, TrkB, pTrkB and monoclonal antibody
against β-actin (all purchased from Cell Signaling Technology,
Inc.). Following incubation with HRP-conjugated secondary antibody
(Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA)
for 1 h, ECL substrate (Pierce Biotechnology, Inc., Rockford, IL,
USA) was used for detection. Images were captured and band density
was analyzed using Bio-Rad Quantity One software (Bio-Rad,
Hercules, CA, USA). Protein expression was normalized using β-actin
as the control.
RNA isolation and RT-PCR
Total RNA was extracted using TRIzol reagent
(Invitrogen Life Technologies, Carlsbad, CA, USA) according to the
manufacturer’s instructions. cDNA was synthesized from 1 μg RNA via
AMV Reverse Transcriptase (Promega Corporation, Madison, WI, USA).
Semi-quantitative RT-PCR was performed to evaluate the mRNA levels
of TrkB and ERK using the following primers: rat TrkB, sense
5′-TCTACAACGGAGCCATACTGAA-3′ and antisense,
5′-CAGGCAGAATCCTACCACAGAG-3′; rat ERK, sense
5′-CCGCTCTATCCCAGACTTCACG-3′ and antisense
5′-TGGTTTCCCACGGCTTCTACAC-3′; and rat β-actin, sense
5′-CCACTGCCGCATCCTCTT-3′ and antisense
5′-CTCATCGTACTCCTGCTTGCT-3′.
RT-PCR products were analyzed on 1% agarose gels.
Band density was analyzed using Band Leader 3.0 software. β-actin
levels were used to normalize mRNA expression levels.
Statistical analysis
All values are presented as the mean ± SEM. All data
were analyzed using one-way ANOVA followed by post-hoc LSD multiple
comparisons or the independent-samples t-test using SPSS version
13.0 (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Protective effect of BDNF on retinal
neurons under hyperglycemia
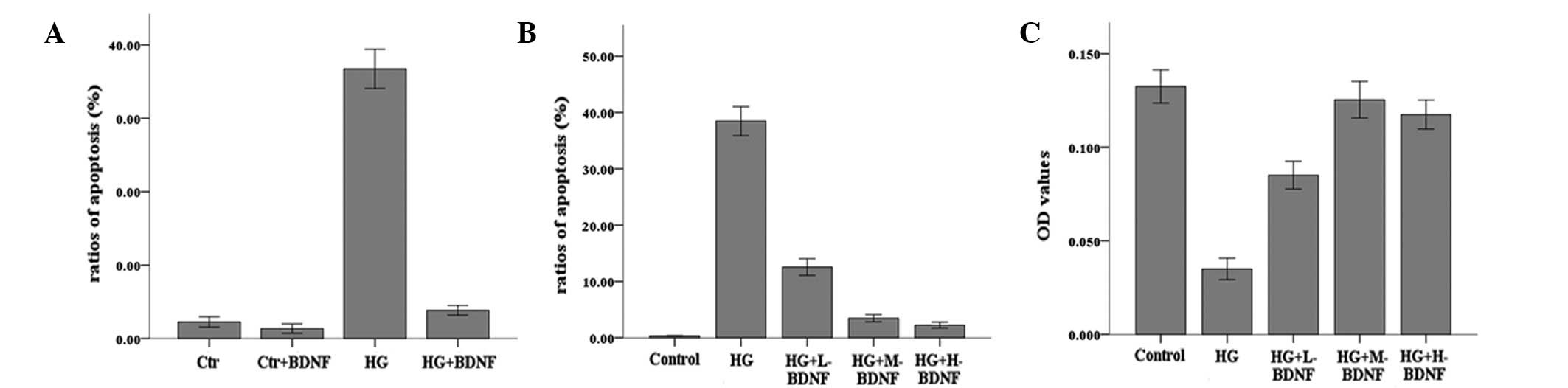
To assess the effects of hyperglycemia and the
neurotrophic factor, BDNF, on the survival of neurons, levels of
apoptosis were evaluated by FACS analysis. As demonstrated in
Fig. 1A, the cells exposed to
hyperglycemia exhibited higher levels of apoptosis, while BDNF was
observed to markedly inhibit apoptosis mediated by hyperglycemia.
In addition, the protective effects of BDNF on cells were
demonstrated to be concentration dependent and the optimal
concentration of BDNF was 100 ng/ml. There was a positive
correlation between the concentration of BDNF and the survival of
neurons when the concentration was <100 ng/ml. MTT and FACS
analysis results revealed that cell survival increased and levels
of apoptosis decreased significantly as the concentration of BDNF
increased (Fig. 1B and C).
However, when cells were treated with >100 ng/ml BDNF, cell
survival and apoptosis were largely unaltered, indicating that the
protective effect of BDNF had reached a plateau (P>0.05). These
results indicate that hyperglycemia induces retinal neuron cell
death and BDNF protects cells by decreasing levels of apoptosis in
a concentration-dependent manner.
BDNF promotes the expression of TrkB in
retinal neurons under hyperglycemia
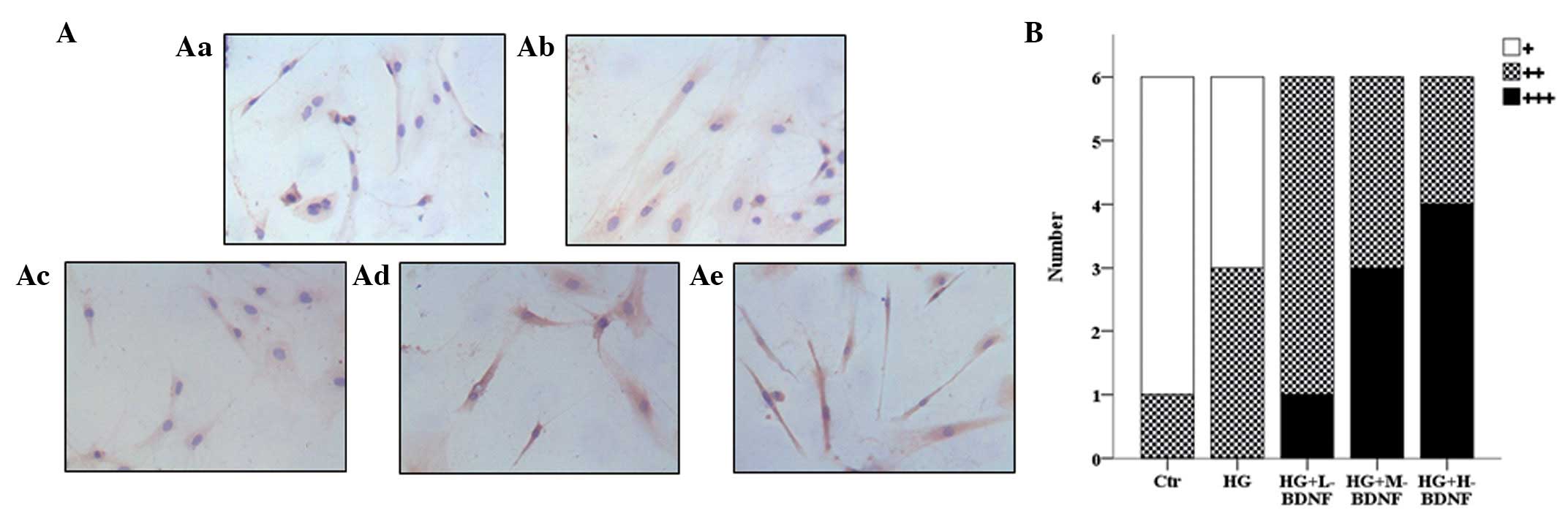
To determine the mechanism by which BDNF protects
retinal neurons from hyperglycemia, the endogenous levels of TrkB
in cells were examined using immunohistochemistry. Compared with
the control group, hyperglycemia treatment alone slightly elevated
the expression of TrkB. However, TrkB expression increased markedly
when cells were subjected to hyperglycemia combined with BDNF
treatment (Fig. 2A and B). The
effect of BDNF on TrkB expression was concentration dependent.
Staining intensity was observed to increase with the concentration
of BDNF (Fig. 2A and B). These
results indicate that BDNF promotes the expression of TrkB in a
concentration-dependent manner.
BDNF increases the mRNA levels of TrkB
but not ERK in retinal neurons under hyperglycemia
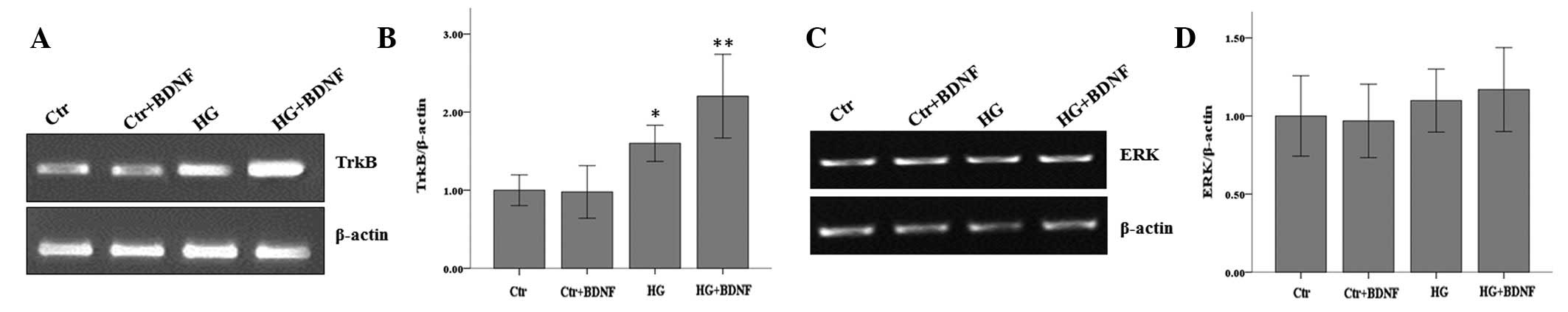
To further confirm that BDNF promotes the expression
of TrkB, RT-PCR was performed to examine mRNA levels. Following
cell culture under normal or hyperglycemic conditions, in the
presence or absence of BDNF (100 ng/ml), total RNA was extracted
and TrkB mRNA levels were examined. Consistent with our previous
results, hyperglycemia alone was observed to increase the
expression of TrkB. The results also demonstrated that BDNF
treatment alone did not alter TrkB levels; however, its expression
increased markedly when cells were subjected to hyperglycemia in
the presence of BDNF (Fig. 3A and
B).
ERK is known to be located downstream of TrkB,
therefore, the mRNA levels of ERK under various treatment
conditions were analyzed. Neither hyperglycemia and/or BDNF
treatment affected ERK mRNA levels (Fig. 3C and D). These results indicate
that in retinal neurons under hyperglycemia, BDNF treatment
elevated TrkB mRNA levels without affecting the mRNA levels of its
downstream gene, ERK.
BDNF promotes phosphorylation of TrkB and
ERK
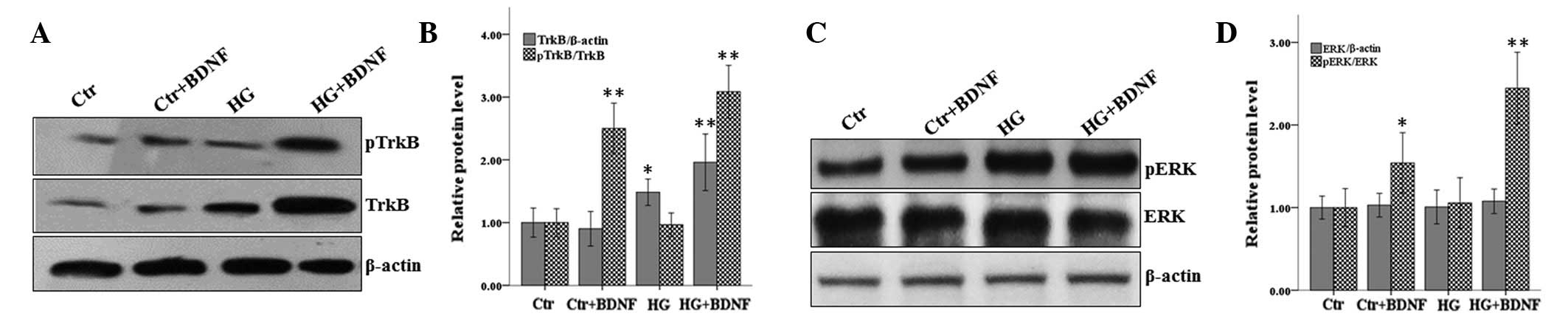
Phosphorylation of TrkB is a key step for its
activation. As BDNF was observed to increase the expression of
TrkB, the effect of BDNF on the phosphorylation of TrkB was
determined. Cells were treated as described and western blot
analysis was performed. As shown in Fig. 4, the results were consistent with
the hypothesis that BDNF and hyperglycemia affected the expression
levels of TrkB. In addition, BDNF increased the phosphorylation of
TrkB markedly while hyperglycemia treatment had no effect. The rate
of phosphorylation of TrkB also increased markedly when cells were
subjected to hyperglycemia and treated with BDNF (Fig. 4A and B). The phosphorylation of ERK
was also analyzed in these cells. Consistent with the results of
RT-PCR, BDNF and/or hyperglycemia did not affect the protein levels
of ERK and the levels of phosphorylation of ERK were unchanged when
the cells were subjected to hyperglycemia and treated with BDNF
(Fig. 4C and D). However,
treatment with BDNF alone was demonstrated to markedly increase the
phosphorylation of ERK. These results indicate that BDNF but not
hyperglycemia treatment promoted the increased phosphorylation of
TrkB and ERK.
Discussion
Previous studies have reported that BDNF and NT-4
are key factors for the survival of injured adult rat RGCs. The
neuroprotective effect of BDNF and NT-4 was observed to be mediated
by binding to the high affinity receptor, TrkB, expressed in RGCs
(5). In addition, BDNF protects
RGCs following acute high intraocular pressure by affecting the
expression and phosphorylation of the TrkB receptor. However, to
date, a limited number of studies have analyzed the protective
effect of BDNF on retinal neurons exposed to hyperglycemia in
vitro. In the present study, a model of primary hyperglycemia
in retinal neurons was used to analyze this protective effect. BDNF
was identified to increase the expression and phosphorylation of
TrkB and protect neurons in the retina from hyperglycemia. In
addition, phosphorylation of ERK was demonstrated to increase
following BDNF treatment, indicating that ERK is a downstream
effector of BDNF.
Consistent with previous studies, BDNF was observed
to protect retinal neurons from hyperglycemia and this
neuroprotective effect was concentration dependent. However, when
the concentration was >100 ng/ml, the neuroprotective effect was
not improved. An explanation for this observation may be that the
number of TrkB receptors is the limiting factor and BDNF is
saturated. Alternatively, the super-optimal concentration of BDNF
may lead to inflammation and neuronal injury.
As the receptor of BDNF, TrkB is critical for its
neuronprotective function. BDNF was shown to upregulate the
expression of TrkB at the mRNA and protein levels in retinal
neurons under conditions of hyperglycemia. Hyperglycemia also led
to higher expression levels of TrkB, which was consistent with the
findings of previous studies that mild brain injury differentially
alters neurotrophin and neurotrophin receptor levels (22,23).
Although the levels of the TrkB receptor in neurons exposed to
hyperglycemia was shown to increase significantly, apoptosis levels
in these cells were still higher compared with the control. These
results indicate that the higher expression of TrkB induced by
hyperglycemia may be a compensatory mechanism for self-protection.
As the phosphorylation of TrkB is critical for its activation,
phosphorylation of TrkB was analyzed in cells under various
treatment conditions. The results revealed that BDNF stimulated the
phosphorylation of TrkB markedly while hyperglycemia had almost no
effect (Fig. 4A and B).
Apoptosis is a form of programmed cell death that is
important in multicellular organisms. Apoptosis is regulated by
numerous proteins mainly associated with the canonical
mitochondrial or endoplasmic reticulum signaling pathways. Previous
studies have reported that normal RGCs constitutively express the
antiapoptotic molecules, Akt, Cox-2 and Mcl-1, while diabetes
induced the expression of proapoptotic molecules, including
mitochondrial proteins, cytochrome c and AIF (24). These observations indicate a marked
correlation between apoptosis and neuronal survival; however, the
mechanism of this process remains unclear (25–27).
As apoptosis in diabetic neurons is associated with the lack of
neurotrophic factors, in the present study, we hypothesized that
adding extra neurotrophin may protect diabetic neurons. The results
confirmed that BDNF protects retinal neurons from hyperglycemia by
decreasing the rate of apoptosis. As a regulator of apoptosis, the
Ras-MAPK/ERK pathway was previously reported to be downstream of
TrkB. In the present study, ERK expression was detected in neurons
exposed to hyperglycemia medium or treated with BDNF. The results
revealed that hyperglycemia and/or BDNF did not affect the
expression of ERK at the mRNA or protein levels. However, BDNF was
demonstrated to markedly increase phosphorylation of ERK, while
hyperglycemia treatment had no effect. These observations are
consistent with the findings of a previous study (28). These results indicate that the
ERK/MAPK signaling pathway is downstream of TrkB and BDNF. BDNF
activates the TrkB-ERK/MAPK pathway and inhibits apoptosis, thus
promoting the survival of neurons from hyperglycemia.
The results of the present study indicate that TrkB
and ERK/MAPK are involved in neuroprotection against hyperglycemia
induced by BDNF; however, further studies are required to determine
the mechanism of this process. The results are likely to provide an
important insight for further studies into the mechanism of BDNF
protection of retinal neurons.
Acknowledgements
This study was supported by grants from the Health
Department of Hunan Province (no. B2009-050) and the Science and
Technology Program of Hunan Province (no. 2012FJ4077).
References
|
1
|
Barber AJ: A new view of diabetic
retinopathy: a neurodegenerative disease of the eye. Prog
Neuropsychopharmacol Biol Psychiatry. 27:283–290. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lieth E, Barber AJ, Xu B, et al: Glial
reactivity and impaired glutamate metabolism in short-term
experimental diabetic retinopathy. Penn State Retina Research
Group. Diabetes. 47:815–820. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fernyhough P, Huang TJ and Verkhratsky A:
Mechanism of mitochondrial dysfunction in diabetic sensory
neuropathy. J Peripher Nerv Syst. 8:227–235. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mey J and Thanos S: Intravitreal
injections of neurotrophic factors support the survival of
axotomized retinal ganglion cells in adult rats in vivo.
Brain Res. 602:304–317. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Peinado-Ramón P, Salvador M,
Villegas-Pérez MP and Vidal-Sanz M: Effects of axotomy and
intraocular administration of NT-4, NT-3 and brain-derived
neurotrophic factor on the survival of adult rat retinal ganglion
cells. A quantitative in vivo study Invest Ophthalmol Vis
Sci. 37:489–500. 1996.PubMed/NCBI
|
|
6
|
Pinzón-Duarte G, Arango-González B,
Guenther E and Kohler K: Effects of brain-derived neurotrophic
factor on cell survival, differentiation and patterning of neuronal
connections and Müller glia cells in the developing retina. Eur J
Neurosci. 19:1475–1484. 2004.PubMed/NCBI
|
|
7
|
Seigel GM, Chiu L and Paxhia A: Inhibition
of neuroretinal cell death by insulin-like growth factor-1 and its
analogs. Mol Vis. 6:157–163. 2000.PubMed/NCBI
|
|
8
|
Müller A, Hauk TG and Fischer D:
Astrocyte-derived CNTF switches mature RGCs to a regenerative state
following inflammatory stimulation. Brain. 130:3308–3320.
2007.PubMed/NCBI
|
|
9
|
Murphy JA, Franklin TB, Rafuse VF and
Clarke DB: The neural cell adhesion molecule is necessary for
normal adult retinal ganglion cell number and survival. Mol Cell
Neurosci. 36:280–292. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu X, Clark AF and Wordinger RJ:
Expression of ciliary neurotrophic factor (CNTF) and its tripartite
receptor complex by cells of the human optic nerve head. Mol Vis.
13:758–763. 2007.PubMed/NCBI
|
|
11
|
Yoshii A and Constantine-Paton M:
Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity
and disease. Dev Neurobiol. 70:304–322. 2010.PubMed/NCBI
|
|
12
|
Hetman M, Kanning K, Cavanaugh JE and Xia
Z: Neuroprotection by brain-derived neurotrophic factor is mediated
by extracellular signal-regulated kinase and phosphatidylinositol
3-kinase. J Biol Chem. 274:22569–22580. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Almeida RD, Manadas BJ, Melo CV, et al:
Neuroprotection by BDNF against glutamate-induced apoptotic cell
death is mediated by ERK and PI3-kinase pathways. Cell Death
Differ. 12:1329–1343. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bonni A, Brunet A, West AE, Datta SR,
Takasu MA and Greenberg ME: Cell survival promoted by the Ras-MAPK
signaling pathway by transcription-dependent and -independent
mechanisms. Science. 286:1358–1362. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Han BH and Holtzman DM: BDNF protects the
neonatal brain from hypoxic-ischemic injury in vivo via the
ERK pathway. J Neurosci. 20:5775–5781. 2000.PubMed/NCBI
|
|
16
|
Satoh T, Nakatsuka D, Watanabe Y, Nagata
I, Kikuchi H and Namura S: Neuroprotection by MAPK/ERK kinase
inhibition with U0126 against oxidative stress in a mouse neuronal
cell line and rat primary cultured cortical neurons. Neurosci Lett.
288:163–166. 2000. View Article : Google Scholar
|
|
17
|
Jin K, Mao XO, Zhu Y and Greenberg DA: MEK
and ERK protect hypoxic cortical neurons via phosphorylation of
Bad. J Neurochem. 80:119–125. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Klöcker N, Kermer P, Weishaupt JH, Labes
M, Ankerhold R and Bähr M: Brain-derived neurotrophic
factor-mediated neuroprotection of adult rat retinal ganglion cells
in vivo does not exclusively depend on
phosphatidyl-inositol-3′-kinase/protein kinase B signaling. J
Neurosci. 20:6962–6967. 2000.PubMed/NCBI
|
|
19
|
Meng M, Zhiling W, Hui Z, Shengfu L, Dan Y
and Jiping H: Cellular levels of TrkB and MAPK in the
neuroprotective role of BDNF for embryonic rat cortical neurons
against hypoxia in vitro. Int J Dev Neurosci. 23:515–521.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Song XY, Li F, Zhang FH, Zhong JH and Zhou
XF: Peripherally-derived BDNF promotes regeneration of ascending
sensory neurons after spinal cord injury. PLoS One. 3:e17072008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fromowitz FB, Viola MV, Chao S, et al: ras
p21 expression in the progression of breast cancer. Hum Pathol.
18:1268–1275. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hicks RR, Martin VB, Zhang L and Seroogy
KB: Mild experimental brain injury differentially alters the
expression of neurotrophin and neurotrophin receptor mRNAs in the
hippocampus. Exp Neurol. 160:469–478. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cui Q, Tang LS, Hu B, So KF and Yip HK:
Expression of trkA, trkB and trkC in injured and regenerating
retinal ganglion cells of adult rats. Invest Ophthalmol Vis Sci.
43:1954–1964. 2002.PubMed/NCBI
|
|
24
|
Abu El-Asrar AM, Dralands L, Missotten L
and Geboes K: Expression of antiapoptotic and proapoptotic
molecules in diabetic retinas. Eye (Lond). 21:238–245.
2007.PubMed/NCBI
|
|
25
|
Santiago AR, Cristóvão AJ, Santos PF,
Carvalho CM and Ambrósio AF: High glucose induces
caspase-independent cell death in retinal neural cells. Neurobiol
Dis. 25:464–472. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li YH, Zhuo YH, Lü L, et al:
Caspase-dependent retinal ganglion cell apoptosis in the rat model
of acute diabetes. Chin Med J (Engl). 121:2566–2571.
2008.PubMed/NCBI
|
|
27
|
Oshitari T, Yamamoto S, Hata N and Roy S:
Mitochondria- and caspase-dependent cell death pathway involved in
neuronal degeneration in diabetic retinopathy. Br J Ophthalmol.
92:552–556. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Omori K, Naruishi K, Nishimura F,
Yamada-Naruishi H and Takashiba S: High glucose enhances
interleukin-6-induced vascular endothelial growth factor 165
expression via activation of gp130-mediated p44/42
MAPK-CCAAT/enhancer binding protein signaling in gingival
fibroblasts. J Biol Chem. 279:6643–6649. 2004. View Article : Google Scholar
|