Introduction
Reactive oxygen species (ROS) are a group of oxygen
moieties that are formed by the incomplete one-electron reduction
of oxygen. The major ROS include hydrogen peroxide
(H2O2), superoxide anion radical
(O2•−) and hydroxyl radical (•OH).
Among ROS, H2O2 diffuses freely over cell
membranes prior to reacting with specific molecular targets due to
its solubility in both lipid and aqueous environments and its
fairly low reactivity. O2•− is metabolized to
H2O2 by superoxide dismutases (1). H2O2 is further
detoxified to O2 and H2O by catalase or
glutathione (GSH) (2).
ROS might affect the activity of mitogen-activated
protein kinases (MAPKs), which are involved in important signaling
pathways in cell proliferation, differentiation and cell death in
response to a variety of stimuli (3,4). The
decision to proliferate, arrest or die depends on the relative
strengths of cell survival and apoptotic signals triggered by ROS.
The three main signaling modules of MAPKs are the extracellular
signal-regulated kinase 1/2 (ERK1/2), c-Jun N-terminal
kinase/stress-activated protein kinase (JNK/SAPK) and p38, which
has emerged as an important signaling pathway from the membrane to
nucleus (3). Each MAPK pathway has
different upstream activators and the activated MAPKs promote
differential transcriptional stimulation of multiple genes via the
phosphorylation of unambiguous transcription factors (5). MAPKs also sense the cellular redox
status and are common targets for ROS. JNK and p38 are mainly
activated by ROS or a mild oxidative shift, initiating procedures
related to apoptosis (6,7). However, the two kinases
differentially affect the levels of apoptosis (8). ROS also provoke or inhibit ERK
pathway (9,10). In most cases, ERK activation has
been shown to have a pro-survival rather than a pro-apoptotic
effect (11). In addition, MAPK
pathways are also activated by the direct inhibition of MAPK
phosphatases by ROS. Since opposite effects of MAPKs by various ROS
can occur in cells, the association between ROS and MAPKs needs to
be further elucidated, particularly signaling pathways related to
cell survival and death.
Cervical neoplasia is the major cause of
cancer-related death in women worldwide. The carcinogenesis of
cervical cancer is associated with excessive inflammation mediated
by ROS. Tissue concentrations of H2O2 during
inflammation can reach millimolar levels, while small amounts of
H2O2 produced by nicotinamide adenine
dinucleotide phosphate (NADPH) oxidase have been suggested to
affect the microenvironments of the plasma membrane (12,13).
H2O2 affects essential functions, including
cell growth, proliferation and differentiation, by altering
signaling cascades and gene expression. H2O2
might also exert severe effects such as cell apoptosis and
necrosis. The effects of H2O2 on the
activities of MAPKs differ depending on the cell type and the
experimental conditions, resulting in various cell responses.
Exogenous H2O2 is often utilized as the
representative ROS for regulating oxidative stress in cells.
H2O2-induced cell death in cervical cancer
cells may be toxicologically attractive in relation to the
intracellular ROS and MAPKs.
Thus, in the present study, the effects of exogenous
H2O2 on the cell growth and death of human
cervical adenocarcinoma HeLa cells were investigated. The effects
of various MAPK inhibitors, including N-acetyl cysteine
(NAC) and propyl gallate (PG) (well-known antioxidants), and
L-buthionine sulfoximine (BSO; an inhibitor of GSH synthesis), were
also evaluated in H2O2-treated HeLa cells
with respect to cell growth and death, as well as ROS and GSH
levels.
Materials and methods
Cell culture
Human cervical adenocarcinoma HeLa cells were
obtained from the American Type Culture Collection (ATCC; Manassas,
VA, USA) and maintained in a humidified incubator containing 5%
CO2 at 37ºC. The cells were cultured in RPMI-1640 medium
(Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal
bovine serum (FBS; Sigma-Aldrich) and 1% penicillin-streptomycin
(Gibco-BRL, Grand Island, NY, USA). The cells were then routinely
grown in 100-mm plastic tissue culture dishes (Nunc A/S, Roskilde,
Denmark) and harvested with a solution of trypsin-EDTA while in a
logarithmic phase of growth.
Reagents
H2O2 was purchased from
Sigma-Aldrich. JNK (SP600125), MEK (PD98059) and p38 inhibitors
(SB203580) were purchased from Calbiochem (San Diego, CA, USA). The
inhibitors were dissolved in Dulbecco's modified Eagle's medium
(DMSO) at 10 mM as a stock solution. NAC, PG and BSO were obtained
from Sigma-Aldrich. NAC was dissolved in 20 mM HEPES buffer (pH
7.0), PG was dissolved in ethanol at 200 mM as a stock solution and
BSO was dissolved in water. Based on previous studies (8,14),
the cells were pretreated with 10 μM of each MAPK inhibitor, 2 mM
NAC, 100 μM PG or 10 μM BSO for 1 h prior to treatment with
H2O2. Ethanol (0.2%) and DMSO (0.2%) were
used as a control vehicle and they did not affect cell growth or
death.
Cell growth assays
Cell growth changes were determined by measuring
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide dye
(MTT; Sigma-Aldrich) absorbance in living cells as previously
described (15). Briefly,
4×104 cells/well were seeded in 96-well microtiter
plates (Nunc A/S). Following exposure to 100 μM
H2O2 for 24 h in the presence or absence of
10 μM of each MAPK inhibitor (2 mM NAC, 100 μM PG or 10 μM BSO) MTT
solution [20 μl: 2 mg/ml in phosphate-buffered saline (PBS)] was
added to each well of the 96-well plates. The plates were incubated
for 4 h at 37ºC. Medium was withdrawn from the plates by pipetting
and 200 μl DMSO was added to each well to solubilize the formazan
crystals. Optical density was measured at 570 nm using a microplate
reader (Synergy™ 2; BioTek Instruments Inc., Winooski, VT,
USA).
Cell cycle and sub-G1 analysis
Cell cycle and sub-G1 analysis were determined by
propidium iodide (PI, Ex/Em=488/617 nm; Sigma-Aldrich) staining as
previously described (16).
Briefly, 1×106 cells in 60-mm culture dishes (Nunc A/S)
were incubated with 100 μM H2O2 for 24 h in
the presence or absence of 10 μM of each MAPK inhibitor, 2 mM NAC,
100 μM PG or 10 μM BSO. Total cells, including floating cells, were
then washed with PBS and fixed in 70% (v/v) ethanol. The cells were
washed again with PBS, and then incubated with PI (10 μg/ml) with
simultaneous RNase treatment at 37ºC for 30 min. Cellular DNA
content was measured using a FACStar flow cytometer
(Becton-Dickinson, Franklin Lakes, NJ, USA) and analyzed using
Lysis II and CellFit software (Becton-Dickinson).
Annexin V-fluorescein isothiocyanate
(FITC) staining for cell death detection
Apoptotic cell death was determined by staining the
cells with Annexin V-FITC (Ex/Em=488/519 nm; Invitrogen Life
Technologies, Camarillo, CA, USA) as previously described (17). Briefly, 1×106 cells in
60-mm culture dishes were incubated with 100 μM
H2O2 for 24 h in the presence or absence of
10 μM of each MAPK inhibitor, 2 mM NAC, 100 μM PG or 10 μM BSO. The
cells were washed twice with cold PBS and then resuspended in 500
μl binding buffer (10 mM HEPES/NaOH pH 7.4, 140 mM NaCl, 2.5 mM
CaCl2) at a concentration of 1×106 cells/ml.
Annexin V-FITC (5 μl) was then added, and the cells were analyzed
with a FACStar flow cytometer.
Measurement of the mitochondrial membrane
potential (MMP; ΔΨm)
MMP (ΔΨm) levels were measured using a
rhodamine 123 fluorescent dye (Sigma-Aldrich; Ex/Em=485/535 nm) as
described previously (17,18). Briefly, 1×106 cells in
60-mm culture dishes were incubated with 100 μM
H2O2 for 24 h in the presence or absence of
10 μM of each MAPK inhibitor, 2 mM NAC, 100 μM PG or 10 μM BSO. The
cells were washed twice with PBS and incubated with rhodamine 123
(0.1 μg/ml) at 37ºC for 30 min. Rhodamine 123 staining intensity
was determined using a FACStar flow cytometer. Rhodamine
123-negative cells were characterized by loss of MMP
(ΔΨm).
Detection of the intracellular ROS
levels
Intracellular ROS levels were detected using an
oxidation-sensitive fluorescent probe dye,
2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA,
Ex/Em=495/529 nm; Invitrogen Life Technologies) and dihydroethidium
(DHE, Ex/Em=518/605 nm; Invitrogen Life Technologies) as previously
described (17,19). DHE is highly selective for
O2•− among ROS. Briefly, 1×106
cells/ml in FACS tube (Becton-Dickinson) were treated with 100 μM
H2O2 with or without 10 μM of each MAPK
inhibitor in the presence of 20 μM H2DCFDA or DHE. The
levels of DCF and DHE fluorescence dyes were evaluated using a
FACStar flow cytometer at 1 h of treatment. DCF (ROS) and DHE
(O2•−) levels were expressed as mean
fluorescence intensity (MFI), which was calculated using CellQuest
software (Becton-Dickinson). Moreover, 1×106 cells in
60-mm culture dishes (Nunc A/S) were incubated with 100 μM
H2O2 for 24 h in the presence or absence of
10 μM of each MAPK inhibitor, 2 mM NAC, 100 μM PG or 10 μM BSO. The
cells were then incubated with 20 μM H2DCFDA or DHE at
37ºC for 30 min. H2DCFDA or DHE fluorescence was
assessed using a FACStar flow cytometer.
Detection of the intracellular GSH
Cellular GSH levels were analyzed using a
5-chloromethylfluorescein diacetate dye (CMFDA, Ex/Em=522/595 nm;
Invitrogen Life Technologies) as previously described (19,20).
Briefly, 1×106 cells/ml in FACS tube were treated with
100 μM H2O2 with or without 10 μM of each
MAPK inhibitor in the presence of 5 μM CMFDA. The levels of CMF
fluorescence were evaluated using a FACStar flow cytometer at 1 h
of treatment. CMF (GSH) levels were expressed as MFI, which was
calculated using CellQuest software. In addition, 1×106
cells in 60-mm culture dishes (Nunc A/S) were incubated with 100 μM
H2O2 for 24 h in the presence or absence of
10 μM of each MAPK inhibitor, 2 mM NAC, 100 μM PG or 10 μM BSO. The
cells were then incubated with 5 μM CMFDA at 37ºC for 30 min. CMF
fluorescence was assessed using a FACStar flow cytometer. Negative
CMF staining (GSH-depletion) of cells is expressed as the
percentage of (−) CMF cells.
Statistical analysis
Results were the mean of at least two independent
experiments (mean ± SD). Data were analyzed using GraphPad Prism4
software (GraphPad Prism, Inc., San Diego, CA, USA). Student's
t-test or one-way analysis of variance with post hoc analysis using
Tukey's multiple comparison test was used for parametric data.
P<0.05 was considered to indicate a statistically significant
difference.
Results
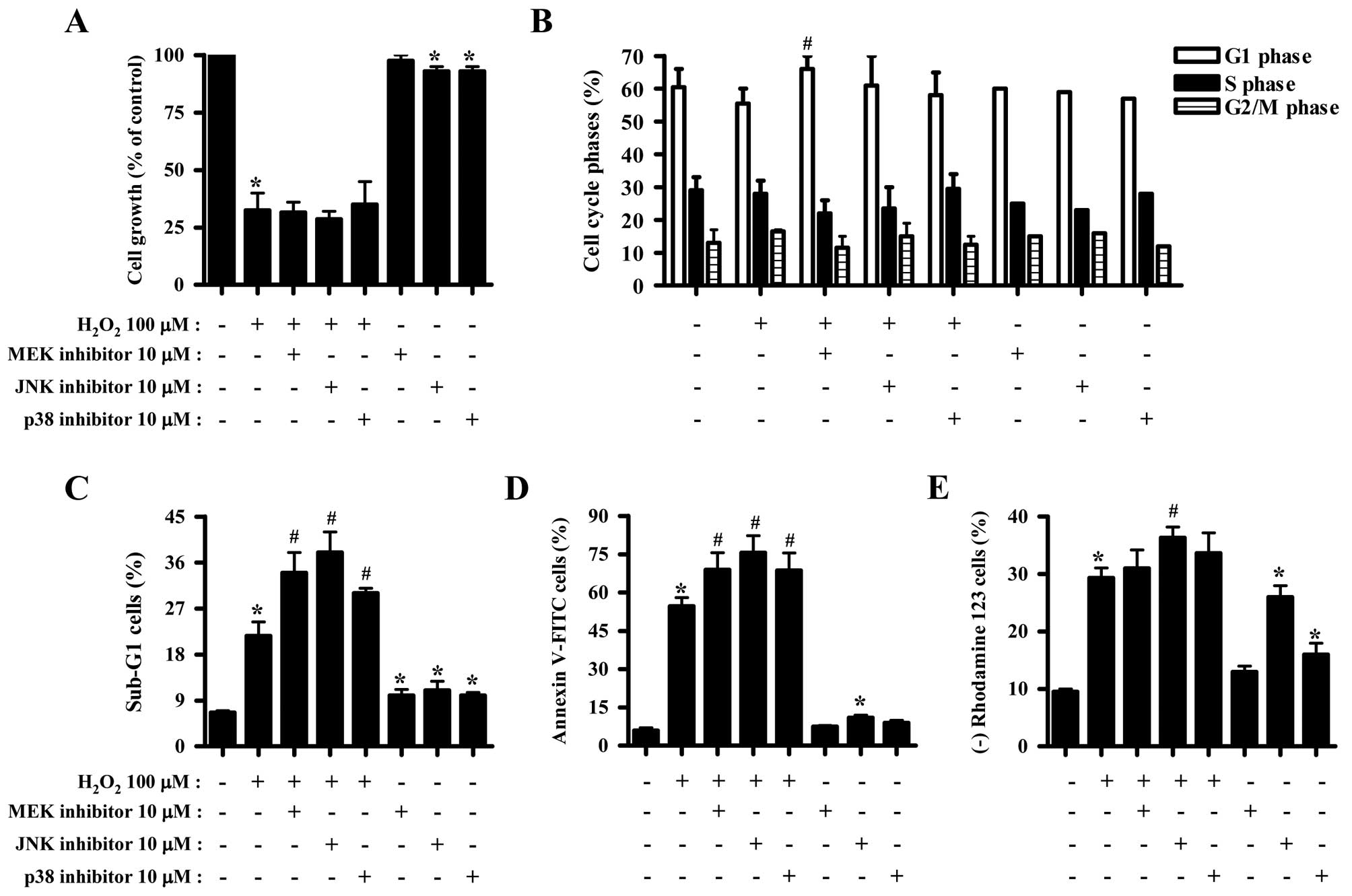
Effects of MAPK inhibitors on cell growth
and death of H2O2-treated HeLa cells
The effect of H2O2 on the
growth inhibitions of HeLa cells was examined using MTT assays. A
concentration of 100 μM H2O2 was considered
sufficient to differentiate the levels of cell growth and death in
the presence or absence of each MAPK inhibitor. Exposure to 100 μM
H2O2 for 24 h inhibited the growth of HeLa
cells by ~70% (Fig. 1A). None of
the MAPK inhibitors significantly affected the growth inhibition
induced by H2O2 (Fig. 1A). JNK and p38 inhibitors slightly
reduced the growth of HeLa control cells (Fig. 1A). Moreover,
H2O2 did not significantly affect HeLa cell
cycle distribution (Fig. 1B).
While JNK and p38 inhibitors did not affect the cell cycle
distribution of H2O2-treated HeLa cells, MEK
inhibitor was found to increase the number of HeLa cells in the G1
phase of the cell cycle (Fig. 1B).
H2O2 increased the number of HeLa cells in
the sub-G1 phase by ~15% compared with
H2O2-untreated HeLa control cells (Fig. 1C). All the MAPK inhibitors
increased the number of H2O2-treated and
control HeLa cells in the sub-G1 phase of the cell cycle (Fig. 1C). H2O2
increased the number of Annexin V-positive HeLa cells by ~50%,
indirectly indicating that HeLa cell death induced by
H2O2 occurred via apoptosis (Fig. 1D). MAPK inhibitors were found to
significantly increase the number of Annexin V-FITC-positive
H2O2-treated HeLa cells (Fig. 1D). Particularly, JNK inhibitor was
found to exert a strong effect on cell death (Fig. 1C and D). JNK inhibitor alone
increased the number of Annexin V-FITC-positive HeLa control cells
(Fig. 1D). Cell death has been
closely associated with the collapse of MMP (ΔΨm)
(21). As expected, loss of MMP
(ΔΨm) was observed in H2O2-treated
HeLa cells (Fig. 1E). However, the
percentage of cells with MMP (ΔΨm) loss was lower
compared with that of Annexin V-FITC-positive cells. All the MAPK
inhibitors slightly enhanced the loss of MMP (ΔΨm) in
H2O2-treated HeLa cells, and JNK inhibitor
exerted the most significant effect (Fig. 1E). JNK or p38 inhibitor alone
triggered MMP (ΔΨm) loss in HeLa control cells (Fig. 1E).
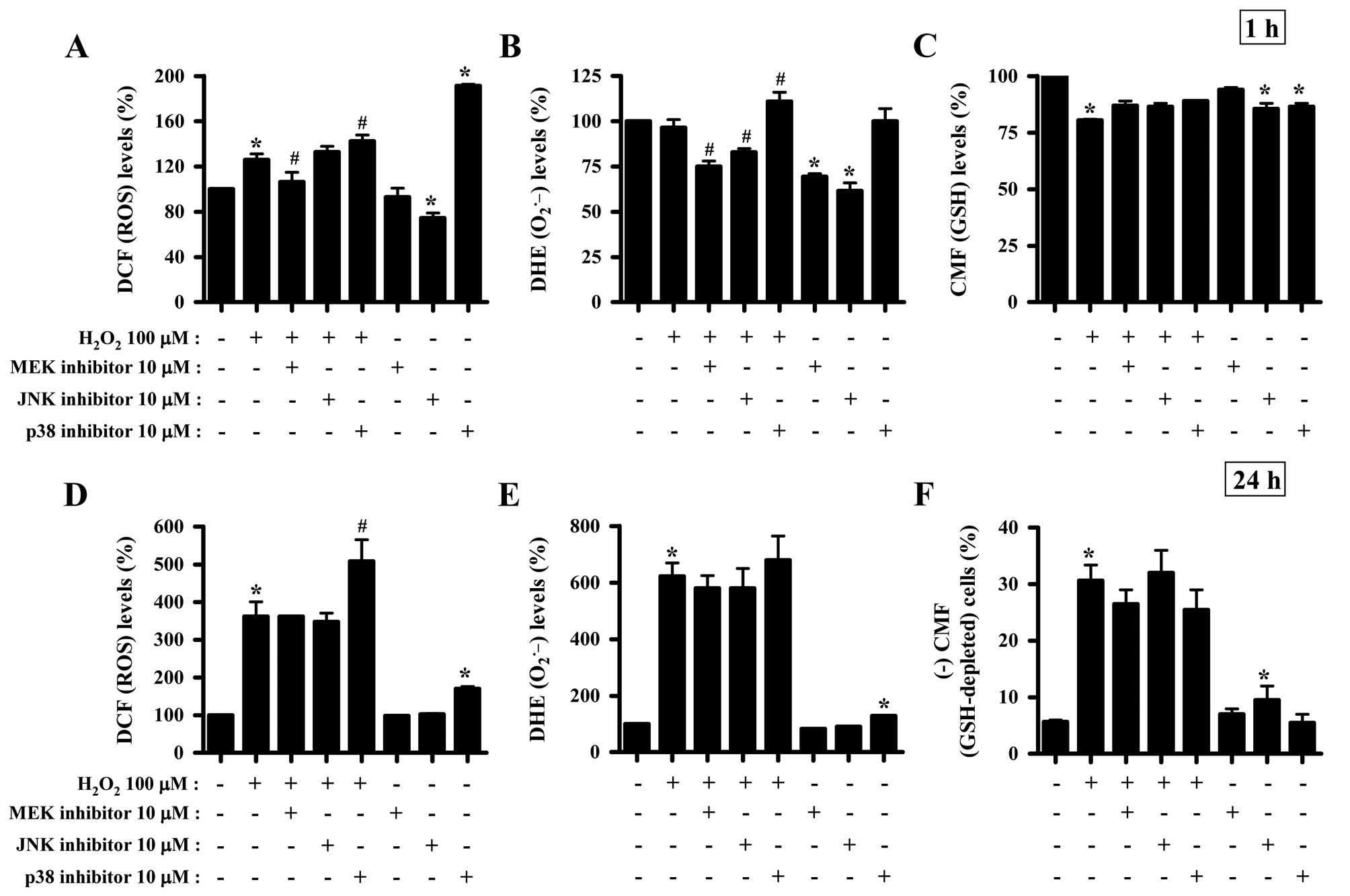
Effects of MAPK inhibitors on the
intracellular ROS and GSH levels in
H2O2-treated HeLa cells
To determine whether the levels of intracellular ROS
and GSH in H2O2-treated HeLa cells were
changed by treatment with each MAPK inhibitor, ROS and GSH levels
in HeLa cells were assessed at 1 and 24 h of
H2O2 treatment (Fig. 2). Intracellular ROS (DCF) levels
were increased in H2O2-treated cells at 1
(Fig. 1A) and 24 h (Fig. 2D). The MEK inhibitor appeared to
attenuate the increased ROS (DCF) levels induced by
H2O2 treatment for 1 h (Fig. 2A). The p38 inhibitor enhanced the
increased ROS (DCF) levels induced by H2O2
treatment for 1 (Fig. 2A) and 24 h
(Fig. 2D). The JNK inhibitor was
found to significantly decrease ROS levels in HeLa control cells at
1 h, while the p38 inhibitor increased ROS levels in these cells at
1 (Fig. 2A) and 24 h (Fig. 2D). Moreover, red fluorescence
derived from DHE reflecting the intracellular
O2•− levels was not altered in
H2O2-treated HeLa cells at 1 h (Fig. 2B), while it was significantly
increased at 24 h of H2O2 treatment (Fig. 2E). The MEK and JNK inhibitors
decreased DHE (O2•−) levels in
H2O2-treated and -untreated HeLa cells at 1
h, while the p38 inhibitor increased the DHE
(O2•−) levels in
H2O2-treated HeLa cells (Fig. 2B). At 24 h of
H2O2 treatment, none of the MAPK inhibitors
significantly changed DHE (O2•−) levels in
H2O2-treated HeLa cells, and p38 inhibitor
alone increased DHE (O2•−) levels in HeLa
control cells (Fig. 2E).
H2O2 decreased GSH levels in HeLa cells at 1
h of treatment (Fig. 2C) as
measured using a CMF fluorescence dye. All the MAPK inhibitors were
shown to attenuate the decreased GSH levels induced by
H2O2 treatment for 1 h and to decrease the
basal levels of GSH in HeLa control cells (Fig. 2C). H2O2
increased the percentages of GSH-depleted HeLa cells at 24 h of
treatment by ~25% compared with the
H2O2-untreated HeLa control cells (Fig. 2F). MEK and p38 inhibitors were
found to attenuate the depletion of GSH in
H2O2-treated HeLa cells, and JNK inhibitor
alone induced the depletion of GSH in HeLa control cells (Fig. 2F).
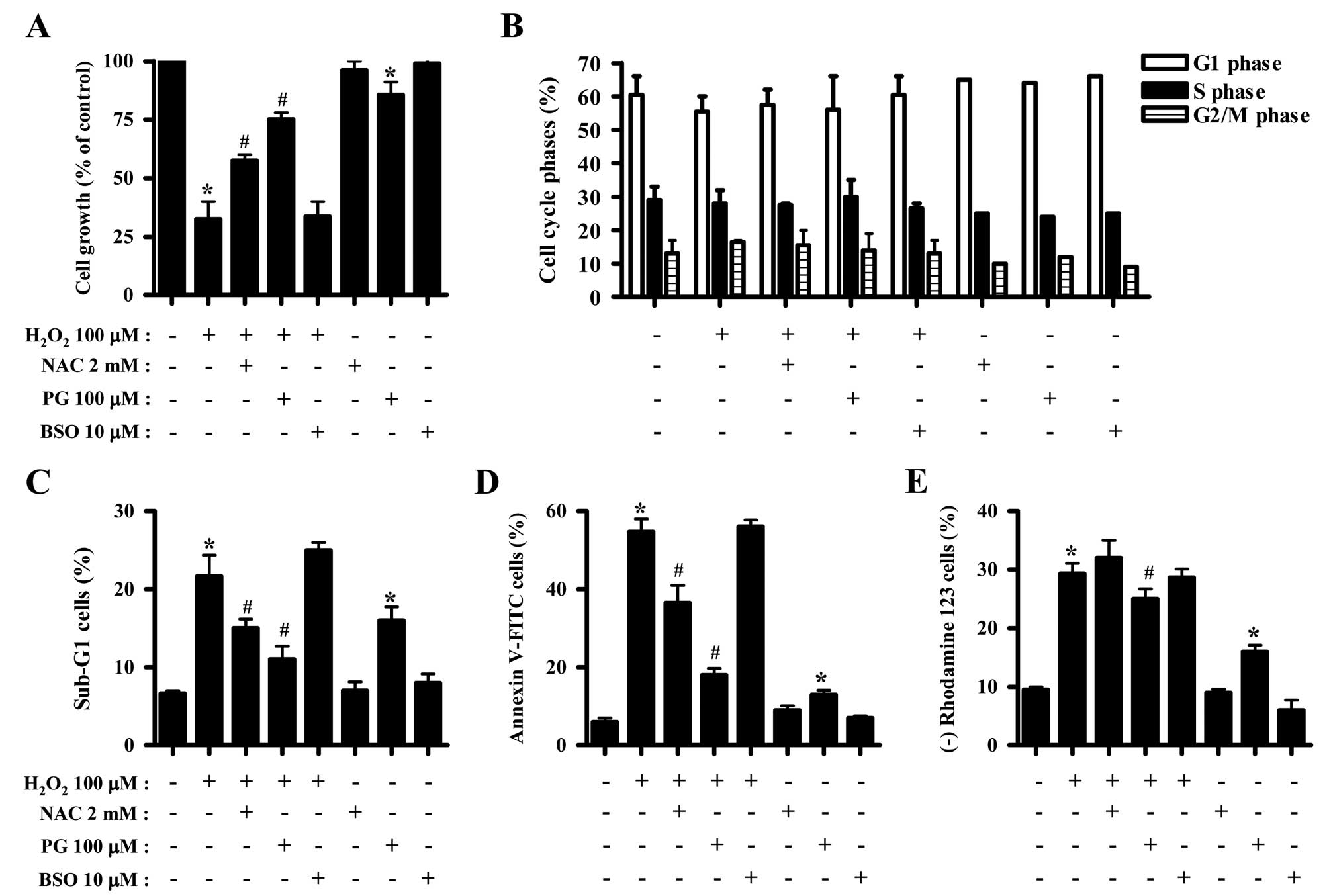
Effects of NAC, PG and BSO on cell growth
and death of H2O2-treated HeLa cells
The effects of NAC, PG or BSO on cell growth, cell
death and MMP (ΔΨm) in
H2O2-treated HeLa cells were assessed at 24 h
of treatment. NAC and PG significantly attenuated the growth
inhibition induced by H2O2, where PG exerted
a stronger effect (Fig. 3A).
However, BSO did not affect cell growth of the
H2O2-treated HeLa cells (Fig. 3A). Only PG reduced the cell growth
in HeLa control cells (Fig. 3A).
Concerning cell cycle analysis, NAC, PG or BSO did not alter the
cell cycle distribution of H2O2-treated HeLa
cells (Fig. 3B). NAC and PG
decreased the percentage of H2O2-treated HeLa
cells in the sub-G1 phase, while BSO slightly increased the
percentage in sub-G1 cells (Fig.
3C). Notably, PG significantly increased the percentage of
sub-G1 HeLa control cells (Fig.
3C). Moreover, NAC and PG significantly reduced the percentage
of Annexin V-FITC-positive H2O2-treated HeLa
cells, and PG markedly prevented HeLa cell death induced by
H2O2 (Fig.
3D). In addition, PG alone increased the percentage of Annexin
V-FITC-positive HeLa control cells (Fig. 3D). With respect to MMP
(ΔΨm), PG decreased the loss of MMP (ΔΨm)
induced by H2O2 to some extent, while NAC and
BSO did not significantly affect the loss of MMP (ΔΨm)
(Fig. 3E). PG also increased the
percentage of HeLa control cells with MMP (ΔΨm) loss
(Fig. 3E).
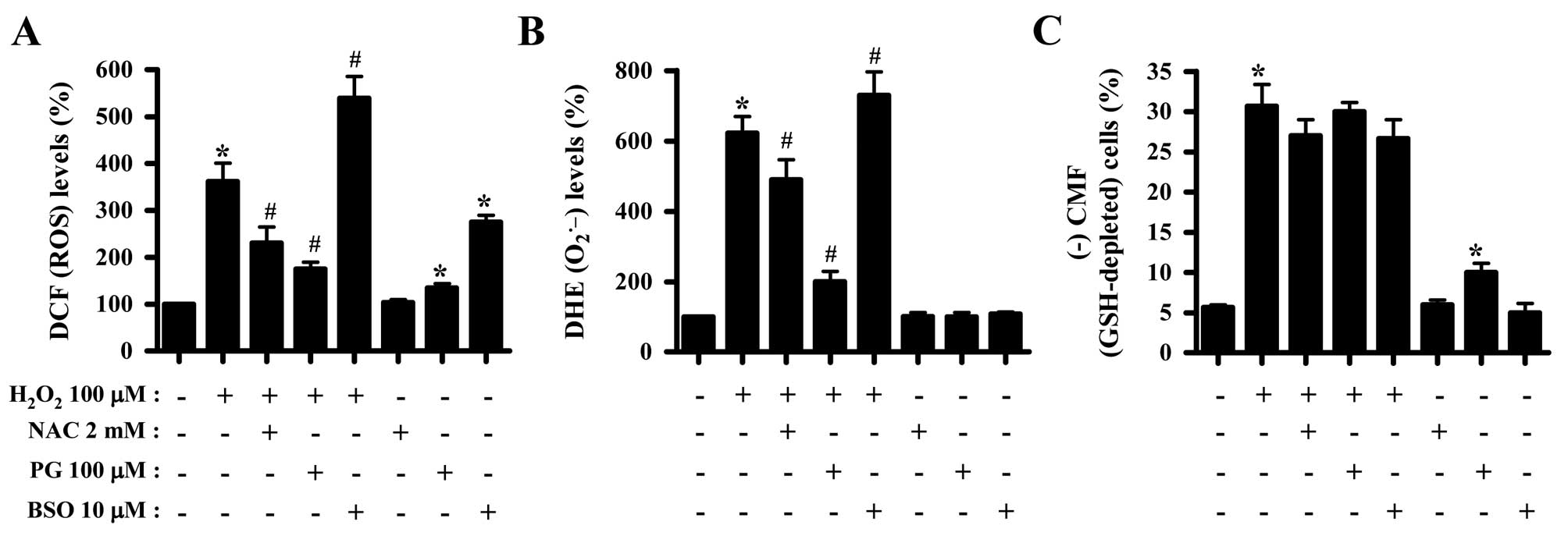
Effects of NAC, PG and BSO on
intracellular ROS and GSH levels in
H2O2-treated HeLa cells
Whether the levels of intracellular ROS and GSH in
H2O2-treated HeLa cells were changed by
treatment with NAC, PG or BSO was subsequently investugated. Both
NAC and PG suppressed the increased ROS levels in
H2O2-treated HeLa cells, while BSO enhanced
the increased ROS levels induced by H2O2
(Fig. 4A). Additionally, PG and
BSO significantly increased ROS (DCF) levels in HeLa control cells
(Fig. 4A). Similarly, NAC and PG
decreased the augmented O2•− levels in
H2O2-treated HeLa cells, in contrast to BSO
which increased O2•− levels (Fig. 4B). Concerning assessment of the GSH
levels, NAC appeared to reduce the percentage of GSH-depleted
H2O2-treated HeLa cells, while PG did not
affect the depletion of GSH (Fig.
4C). PG alone significantly induced the depletion of GSH in
HeLa control cells (Fig. 4C).
Notably, treatment with 10 μM BSO failed to enhance the depletion
of GSH in H2O2-treated HeLa cells, which
instead slightly attenuated GSH depletion in these cells (Fig. 4C).
Discussion
Since H2O2 inhibited HeLa
cell growth and induced HeLa cell death, the present study aimed to
evaluate the toxicological effect of H2O2 on
the cell growth and death of HeLa cells following treatment with
MAPK inhibitors, NAC, PG or BSO. H2O2
increased the number of Annexin V-FITC-positive HeLa cells. The
activity of caspase-3 was also found to be increased in
H2O2-treated HeLa cells (data not shown),
indicating that the H2O2-induced HeLa cell
death occurred via apoptosis. In addition,
H2O2 triggered the loss of MMP
(ΔΨm) in HeLa cells, suggesting that cell death by
H2O2 was correlated with the collapse of MMP
(ΔΨm). H2O2 did not induce any
specific phase arrests of the HeLa cell cycle, indicating that the
H2O2-induced oxidative stress did not affect
particular proteins related to cell cycle arrest and
progression.
ERK activation was observed in
H2O2-treated HeLa cells (22). In the present study, the MEK
inhibitor, which presumably inactivates ERK, did not affect the
H2O2-induced inhibition of HeLa cell growth,
while it increased cell death. In addition, the MEK inhibitor alone
increased the number of HeLa control cells in the sub-G1 phase of
the cell cycle. Thus, ERK is likely to be associated with cell
survival rather than cell death and proliferation. JNK and p38
MAPKs are known to be related to cell death (6,7).
H2O2 has been shown to increase the activity
of JNK and p38 in HeLa cells (22,23).
Yamagishi et al(23)
demonstrated that HeLa cell apoptosis induced by 500 μM
H2O2 was suppressed by treatment with p38
inhibitor but not JNK inhibitor, suggesting that apoptosis occurs
through a p38 MAPK-dependent signaling pathway. However, the
results of the present study indicate that treatment with JNK and
p38 inhibitors significantly increased the cell death of HeLa cells
treated with 100 μM H2O2. These results
suggest that JNK and p38 signaling pathways in HeLa cells treated
with 100 μM H2O2 are involved in a
pro-survival function. The difference potentially resulted from the
different concentrations and incubation times used in each
experiment since the effects of MAPKs are altered by the different
types of oxidative stress. JNK and p38 inhibitors significantly
induced cell growth inhibition, cell death and MMP (ΔΨm)
loss in HeLa control cells, indicating that the basal activities of
these MAPKs are involved in the cell growth and survival of HeLa
cells. Regarding the loss of MMP (ΔΨm), none of the MAPK
inhibitors markedly enhanced the loss of MMP (ΔΨm) in
H2O2-treated HeLa cells compared with HeLa
cell death. Thus, the loss of MMP (ΔΨm) was not likely
to correlate with apoptosis in HeLa cells treated with
H2O2 and each MAPK inhibitor. Moreover, JNK
and p38 inhibitors did not change the cell cycle distributions in
H2O2-treated HeLa cells, while the MEK
inhibitor increased the number of cells in the G1 phase of the cell
cycle. The underlying mechanisms of the cell cycle regulation by
MAPKs should be further investigated under oxidative stress.
ROS levels, including O2•−,
were significantly increased in HeLa cells treated with
H2O2 for 24 h. It is suggested that exogenous
H2O2 strongly generates
O2•− by damaging the mitochondria, and both
H2O2 and O2•− can be
efficiently converted into the toxic •OH via the Fenton
reaction to kill HeLa cells. However, H2O2
did not increase O2•− (DHE) levels in HeLa
cells at 1 h of treatment, suggesting that it does not affect the
mitochondrial respiratory transport chain and the activity of
various oxidases to generate O2•−. MEK
inhibitor showing a proapoptotic effect on
H2O2-treated HeLa cells did not alter ROS
levels, including O2•−, at 24 h of treatment,
while it decreased ROS levels in H2O2-treated
and -untreated HeLa cells at 1 h of treatment. MEK inhibitor
appeared to act as an antioxidant at 1 h of treatment, while it did
not suppress HeLa cell death at 24 h of treatment. The JNK
inhibitor also did not alter the ROS levels in
H2O2-treated and -untreated HeLa cells at 24
h. Instead, this inhibitor reduced O2•−
levels in H2O2-treated HeLa cells at 1 h of
treatment and it also decreased basal ROS levels in HeLa control
cells. Thus, ERK and JNK signaling pathways in
H2O2-treated and -untreated HeLa cells did
not significantly influence redox state to affect HeLa cell death.
The p38 inhibitor enhanced ROS levels in
H2O2-treated and -untreated HeLa cells at 1
and 24 h of treatment, suggesting that p38 signaling is involved in
cell survival and the antioxidant system in HeLa cells. Since
changes in ROS levels regulated by each MAPK inhibitor and outcomes
of these signaling by different types of oxidant stress are complex
in cells, the diverse functions of each MAPK inhibitor under the
different oxidative states were then investigated with regard to
cell survival or death. NAC, a well-known antioxidant, attenuated
the growth inhibition and cell death of
H2O2-treated HeLa cells. As expected, NAC
markedly decreased ROS levels, including
O2•−, in H2O2-treated
HeLa cells. BSO increased ROS levels in
H2O2-treated and -untreated HeLa cells.
However, growth inhibition and cell death were not affected. The
BSO-increased ROS levels may not be sufficient to increase cell
death in these cells.
PG is known to be a synthetic antioxidant (24,25),
while it has been suggested to possess prooxidant properties
(26-28). Antioxidant and cytoprotective
properties of PG may change to prooxidant, cytotoxic and genotoxic
in the presence of Cu(II) (29).
According to a previous study, ROS levels, including
O2•−, were demonstrated to be increased or
decreased in PG-treated HeLa cells depending on the incubation
times and doses (19). The results
of the present study indicate that PG alone slightly inhibited the
growth of HeLa cells and induced cell death accompanied by the loss
of MMP (ΔΨm). In addition, PG slightly increased ROS
levels in HeLa cells at 24 h and it also increased
O2•− (DHE) levels at 1 h of treatment (data
not shown). Thus, it is conceivable that PG generates
O2•− in HeLa cells by impairing the
mitochondrial function, consequently leading to HeLa cell death via
oxidative stress. Notably, PG significantly attenuated growth
inhibition and cell death in H2O2-treated
HeLa cells. Moreover, PG markedly reduced the increased ROS levels,
including O2•−, by H2O2
treatment. Therefore, PG was found to protect HeLa cells against
exogenous H2O2 by reducing
H2O2-induced oxidative stress. Thus, PG acts
as an antioxidant (24,25) or prooxidant (26-28)
depending on various conditions, such as incubation times and
doses, co-incubation drugs and cell types. In addition, PG did not
strongly attenuate the loss of MMP (ΔΨm) following
H2O2 treatment. Moreover, NAC failed to
prevent the loss of MMP (ΔΨm) by
H2O2 treatment. Since the levels of MMP
(ΔΨm) loss in H2O2-treated HeLa
cells was relatively low compared with that of Annexin
V-FITC-positive cells, the loss of MMP (ΔΨm) by
H2O2 was expected to be essential but not
sufficient to induce apoptosis in HeLa cells. Regarding cell cycle
changes, none of the NAC, PG or BSO significantly altered the cell
cycle distributions in H2O2-treated HeLa
cells, suggesting that changes in ROS levels did not specifically
regulate the cell cycle to induce particular phase arrests in HeLa
cells.
Apoptotic effects are inversely proportional to GSH
content (20,30,31).
Similarly, H2O2 increased the percentages of
GSH-depleted HeLa cells at 24 h of treatment. The JNK inhibitor and
PG also significantly induced the depletion of GSH in HeLa control
cells. These results support the hypothesis that the intracellular
GSH content has a decisive impact on cell death (18,20,32).
However, none of the MAPK inhibitors enhanced the depletion of GSH
in H2O2-treated HeLa cells, while NAC failed
to prevent the depletion of GSH. Furthermore, PG partially
recovered the depletion of GSH in
H2O2-treated HeLa cells. Therefore, the loss
of GSH content appeared to be necessary, but not sufficient to
induce apoptosis in H2O2-treated HeLa cells.
H2O2 decreased GSH levels at 1 h of
treatment. The decreased GSH levels were likely to decrease in
order to reduce ROS (DCF) levels. In addition, MAPK inhibitors
partially recovered GSH levels in
H2O2-treated HeLa cells and reduced basal GSH
levels in HeLa control cells. Thus, these results suggest that MAPK
inhibitors differentially regulate the intracellular GSH levels in
HeLa cells depending on the presence or absence of
H2O2. Notably, treatment with 10 μM BSO
showing an increased effect on ROS levels did not intensify the
depletion of GSH in H2O2-treated HeLa cells.
Previous studies have demonstrated that 1 or 10 μM BSO
significantly enhanced the depletion of GSH in arsenic
trioxide-treated HeLa (30) and
A549 cells (33). Additional
studies have shown that >100 μM BSO decreased GSH levels in
breast cancer (34) and leukemia
cells (35). Therefore, these
results suggest that BSO differentially affects GSH levels
depending on the concentration used, the cell type and
co-incubation drugs.
In conclusion, H2O2 induced
growth inhibition and death in HeLa cells, which was accompanied by
intracellular increase in ROS levels and GSH depletion. MAPK
inhibitors generally enhanced H2O2-induced
HeLa cell death. Particularly, the p38 inhibitor increased ROS
levels in H2O2-treated HeLa cells. NAC and PG
attenuated H2O2-induced HeLa cell growth
inhibition and death together with the suppression of ROS levels.
The present study provides insight into the toxicological effects
of exogenous H2O2 on HeLa cells with respect
to MAPK signaling and antioxidants.
Acknowledgements
This study was supported by a grant from the
Ministry of Science and Technology (MoST)/Korea Science and
Engineering Foundation (KOSEF) through the Diabetes Research Center
at Chonbuk National University (2012-0009323) and research funds of
Chonbuk National University in 2013.
Abbreviations:
|
ROS
|
reactive oxygen species
|
|
GSH
|
glutathione
|
|
MAPK
|
mitogen-activated protein kinase
|
|
MEK
|
MAP kinase or ERK kinase
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
JNK
|
c-Jun N-terminal kinase
|
|
MMP (ΔΨm)
|
mitochondrial membrane potential
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
FITC
|
fluorescein isothiocyanate
|
|
PI
|
propidium iodide
|
|
H2DCFDA
|
2′,7′-dichlorodihydrofluorescein
diacetate
|
|
DHE
|
dihydroethidium
|
|
CMFDA
|
5-chloromethylfluorescein
diacetate
|
|
NAC
|
N-acetyl cysteine
|
|
PG
|
propyl gallate
|
|
BSO
|
L-buthionine sulfoximine
|
References
|
1
|
Zelko IN, Mariani TJ and Folz RJ:
Superoxide dismutase multigene family: a comparison of the CuZn-SOD
(SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures,
evolution, and expression. Free Radic Biol Med. 33:337–349. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wilcox CS: Reactive oxygen species: roles
in blood pressure and kidney function. Curr Hypertens Rep.
4:160–166. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Genestra M: Oxyl radicals, redox-sensitive
signalling cascades and antioxidants. Cell Signal. 19:1807–1819.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Blenis J: Signal transduction via the MAP
kinases: proceed at your own RSK. Proc Natl Acad Sci USA.
90:5889–5892. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kusuhara M, Takahashi E, Peterson TE, Abe
J, Ishida M, Han J, Ulevitch R and Berk BC: p38 Kinase is a
negative regulator of angiotensin II signal transduction in
vascular smooth muscle cells: effects on
Na+/H+ exchange and ERK1/2. Circ Res.
83:824–831. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hsin YH, Chen CF, Huang S, Shih TS, Lai PS
and Chueh PJ: The apoptotic effect of nanosilver is mediated by a
ROS- and JNK-dependent mechanism involving the mitochondrial
pathway in NIH3T3 cells. Toxicol Lett. 179:130–139. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mao X, Yu CR, Li WH and Li WX: Induction
of apoptosis by shikonin through a ROS/JNK-mediated process in
Bcr/Abl-positive chronic myelogenous leukemia (CML) cells. Cell
Res. 18:879–888. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Han YH, Moon HJ, You BR, Kim SZ, Kim SH
and Park WH: JNK and p38 inhibitors increase and decrease
apoptosis, respectively, in pyrogallol-treated calf pulmonary
arterial endothelial cells. Int J Mol Med. 24:717–722.
2009.PubMed/NCBI
|
|
9
|
Lee YJ, Kang IJ, Bünger R and Kang YH:
Enhanced survival effect of pyruvate correlates MAPK and NF-kappaB
activation in hydrogen peroxide-treated human endothelial cells. J
Appl Physiol. 96:792–801. 2004.PubMed/NCBI
|
|
10
|
Guyton KZ, Liu Y, Gorospe M, Xu Q and
Holbrook NJ: Activation of mitogen-activated protein kinase by
H2O2. Role in cell survival following oxidant
injury. J Biol Chem. 271:4138–4142. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Henson ES and Gibson SB: Surviving cell
death through epidermal growth factor (EGF) signal transduction
pathways: implications for cancer therapy. Cell Signal.
18:2089–2097. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rhee SG, Kang SW, Jeong W, Chang TS, Yang
KS and Woo HA: Intracellular messenger function of hydrogen
peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol.
17:183–189. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vilhardt F and van Deurs B: The phagocyte
NADPH oxidase depends on cholesterol-enriched membrane microdomains
for assembly. EMBO J. 23:739–748. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Han YH and Park WH: The effects of
N-acetyl cysteine, buthionine sulfoximine,
diethyldithiocarbamate or 3-amino-1,2,4-triazole on antimycin
A-treated Calu-6 lung cells in relation to cell growth, reactive
oxygen species and glutathione. Oncol Rep. 22:385–391. 2009.
|
|
15
|
Han YH, Moon HJ, You BR, Kim SZ, Kim SH
and Park WH: Effects of carbonyl cyanide p-(trifluoromethoxy)
phenylhydrazone on the growth inhibition in human pulmonary
adenocarcinoma Calu-6 cells. Toxicology. 265:101–107. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Han YH, Kim SZ, Kim SH and Park WH:
Pyrogallol inhibits the growth of human lung cancer Calu-6 cells
via arresting the cell cycle arrest. Toxicol In Vitro.
22:1605–1609. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Han YH, Moon HJ, You BR and Park WH: The
effect of MG132, a proteasome inhibitor on HeLa cells in relation
to cell growth, reactive oxygen species and GSH. Oncol Rep.
22:215–221. 2009.PubMed/NCBI
|
|
18
|
Han YH, Kim SH, Kim SZ and Park WH:
Carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP)
as an O2•− generator induces apoptosis via
the depletion of intracellular GSH contents in Calu-6 cells. Lung
Cancer. 63:201–209. 2009.
|
|
19
|
Han YH and Park WH: Propyl gallate
inhibits the growth of HeLa cells via regulating intracellular GSH
level. Food Chem Toxicol. 47:2531–2538. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Han YH, Kim SZ, Kim SH and Park WH:
Intracellular GSH level is a factor in As4.1 juxtaglomerular cell
death by arsenic trioxide. J Cell Biochem. 104:995–1009. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: release of cytochrome c from mitochondria blocked.
Science. 275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Singh M, Sharma H and Singh N: Hydrogen
peroxide induces apoptosis in HeLa cells through mitochondrial
pathway. Mitochondrion. 7:367–373. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yamagishi N, Saito Y and Hatayama T:
Mammalian 105 kDa heat shock family proteins suppress hydrogen
peroxide-induced apoptosis through a p38 MAPK-dependent
mitochondrial pathway in HeLa cells. FEBS J. 275:4558–4570. 2008.
View Article : Google Scholar
|
|
24
|
Reddan JR, Giblin FJ, Sevilla M,
Padgaonkar V, Dziedzic DC, Leverenz VR, Misra IC, Chang JS and Pena
JT: Propyl gallate is a superoxide dismutase mimic and protects
cultured lens epithelial cells from H2O2
insult. Exp Eye Res. 76:49–59. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu TW, Fung KP, Zeng LH, Wu J and Nakamura
H: Propyl gallate as a hepatoprotector in vitro and in vivo.
Biochem Pharmacol. 48:419–422. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kobayashi H, Oikawa S, Hirakawa K and
Kawanishi S: Metal-mediated oxidative damage to cellular and
isolated DNA by gallic acid, a metabolite of antioxidant propyl
gallate. Mutat Res. 558:111–120. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kawanishi S, Oikawa S and Murata M:
Evaluation for safety of antioxidant chemopreventive agents.
Antioxid Redox Signal. 7:1728–1739. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Han YH, Moon HJ, You BR, Yang YM, Kim SZ,
Kim SH and Park WH: Propyl gallate inhibits the growth of
endothelial cells, especially calf pulmonary arterial endothelial
cells via caspase-independent apoptosis. Int J Mol Med. 25:937–944.
2010.PubMed/NCBI
|
|
29
|
Jacobi H, Eicke B and Witte I: DNA strand
break induction and enhanced cytotoxicity of propyl gallate in the
presence of copper(II). Free Radic Biol Med. 24:972–978. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Han YH, Kim SZ, Kim SH and Park WH:
Enhancement of arsenic trioxide-induced apoptosis in HeLa cells by
diethyldithiocarbamate or buthionine sulfoximine. Int J Oncol.
33:205–213. 2008.PubMed/NCBI
|
|
31
|
Han YH, Kim SZ, Kim SH and Park WH:
Suppression of arsenic trioxide-induced apoptosis in HeLa cells by
N-acetylcysteine. Mol Cells. 26:18–25. 2008.PubMed/NCBI
|
|
32
|
Estrela JM, Ortega A and Obrador E:
Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci.
43:143–181. 2006. View Article : Google Scholar
|
|
33
|
Han YH, Kim SZ, Kim SH and Park WH:
Induction of apoptosis in arsenic trioxide-treated lung cancer A549
cells by buthionine sulfoximine. Mol Cells. 26:158–164.
2008.PubMed/NCBI
|
|
34
|
Lewis-Wambi JS, Kim HR, Wambi C, Patel R,
Pyle JR, Klein-Szanto AJ and Jordan VC: Buthionine sulfoximine
sensitizes antihormone-resistant human breast cancer cells to
estrogen-induced apoptosis. Breast Cancer Res. 10:R1042008.
View Article : Google Scholar
|
|
35
|
Ramos AM and Aller P: Quercetin decreases
intracellular GSH content and potentiates the apoptotic action of
the antileukemic drug arsenic trioxide in human leukemia cell
lines. Biochem Pharmacol. 75:1912–1923. 2008. View Article : Google Scholar
|