Introduction
Von Hippel-Lindau (VHL) disease (OMIM 193300) is an
autosomal dominant familial cancer syndrome that increases
susceptibility to several benign and malignant tumors, including
central nervous system (CNS) hemangioblastomas, renal cysts and
renal cell carcinoma (RCC), as well as pheochromocytoma (PCC).
Additional tumors, such as pancreatic and endolymphatic sac tumors
and epididymal and broad ligament cystadenomas, have also been
previously described (1–6). VHL syndrome accounts for ~1/3 of
patients with CNS hemangioblastoma, >50% of those with retinal
angioma, 1% of those with RCC, 50% of those with apparently
isolated familial PCC and 11% of those with an apparently sporadic
PCC (7–9). VHL is present at a frequency of 1 in
36,000 live births in Eastern England (2,5).
Similar to other hereditary syndromes, VHL has its own clinical
classification based on genotype-phenotype associations. Based on
clinical symptoms, patients with low-risk PCC (<5%) are
classified as VHL type 1 and those with high-risk PCC (>60%) as
VHL type 2. VHL type 2, according to 7–20% of VHL kindreds, is
further subdivided into three hypotypes, 2A (patients with a low
risk for RCC), 2B [patients with an increased risk (~70%) for RCC]
and 2C (affected members with isolated PCC) (1,2,10).
VHL, the gene responsible for VHL disease,
was identified in 1993 (11). It
is a pleiotropic tumor suppressor gene located on chromosome 3
(3p25.3) that is widely expressed in fetal and adult tissues
(11,12). Several VHL germline
mutations have been identified in individuals with VHL disease, as
well as somatic mutations in sporadic tumors (5,13–16).
VHL type 1 families frequently harbor larger deletions and nonsense
mutations in addition to missense variants in the VHL gene,
and patients with type 2 disease carry missense mutations almost
exclusively (5,10). Up to 96% of patients with PCC have
missense mutations (10).
The first VHL case with a germline mutation in China
was reported by Huang et al(17) in 2004. In the present study, two
frequent VHL germline mutations, c.A233G (p.N78S) and
c.G482A (p.R161Q), are described in two families with type 2A and
2C VHL syndrome. Patients underwent surgery and were subsequently
followed up.
Materials and methods
Patients
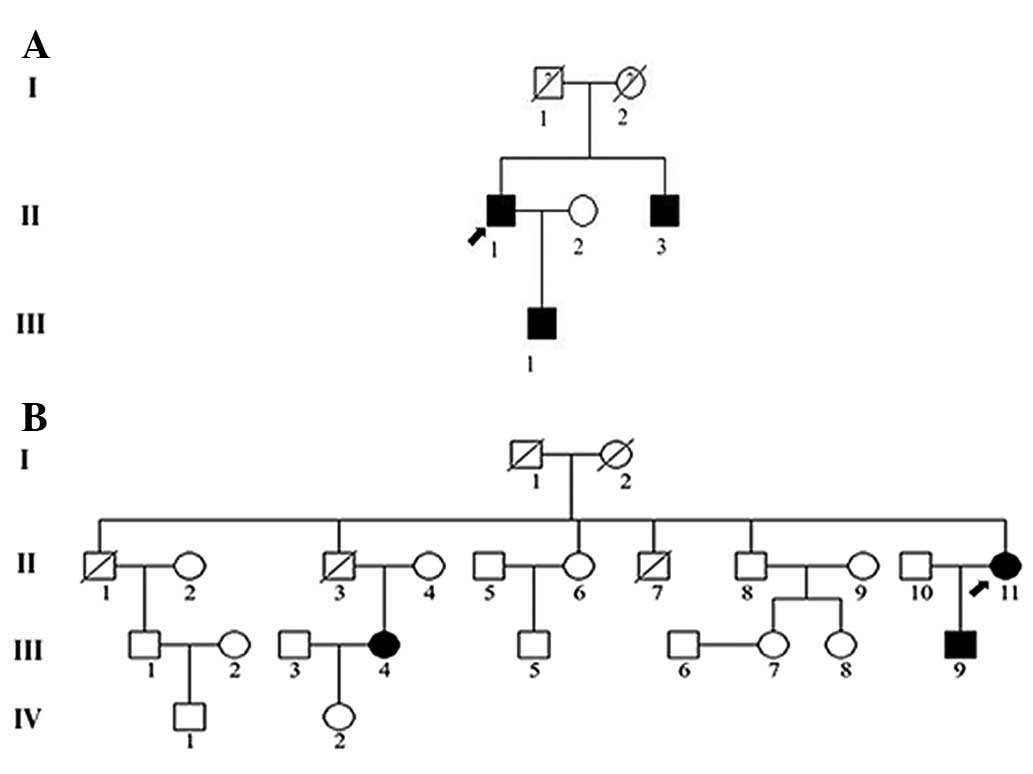
Two VHL syndrome pedigrees were investigated in this
study; family 1, a three-generation pedigree including 6
individuals (Fig. 1A), and family
2, a four-generation pedigree containing 24 individuals (Fig. 1B). The present study was conducted
in accordance with the Declaration of Helsinki and was approved by
the Ethics Committees of the 117th PLA Hospital and Zhejiang
University (Hangzhou, China). Written informed consent was provided
by all the subjects in this study.
In family 1, the proband (F1-II-1), a 31-year-old
male patient, presented with headache, paroxysmal hypertension
(150–178/100–120 mmHg) and dizzy spells in 2009 with a 4-year
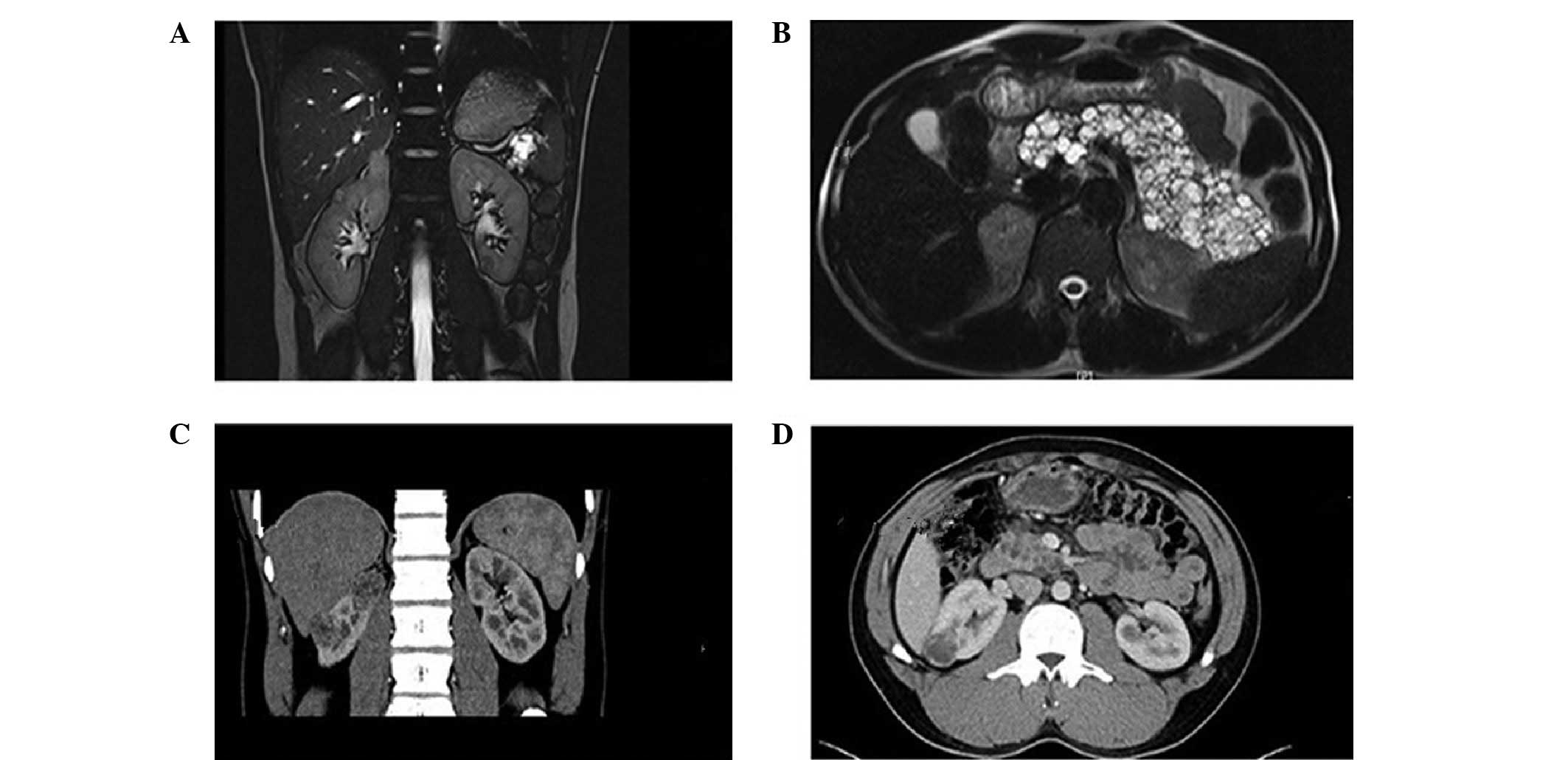
history. Ultrasound (US) and magnetic resonance imaging (MRI) scans
confirmed single masses, multiple tumors and multiple cysts in the
right adrenal glands, bilateral kidney and pancreas, respectively
(Fig. 2A and B). Following
treatment with α-blockers, the patient underwent right adrenal
tumorectomy and right nephron-sparing surgery (NSS). Right PCC and
multiple right RCCs (Fuhrman Grade 2) were confirmed by
histopathological examination. In 2012, the patient was diagnosed
with bilateral epididymal head multiple nodules. Bilateral
epididymal tubercle resection was performed and epididymal
papillary adenoma was confirmed by histopathological examination.
Additionally, the patient's younger brother (F1-II-3, 30 years old)
presented with multiple right renal masses, epididymal nodules and
multiple pancreatic cysts (Fig. 2C and
D). Radical right nephrectomy was performed and
histopathological analysis revealed right RCC with hemorrhagic cyst
(Fuhrman Grade 2; Table I).
 | Table IClinical presentations of RCC and PCC
with VHL mutations. |
Table I
Clinical presentations of RCC and PCC
with VHL mutations.
| Individual | Gender | Age at onset
(years) | VHL
mutation | Catecholamine serum
level (ng/l) (normal: DA, 100; E, 600; NE, 100) | Tumor size
(cm) | Operation | Histology |
|---|
|
|---|
| PCC | RCC |
|---|
| F1-II-1 | M | 31 | p.N78S | - | R, 4.1×3.4×3.9 | R, 1.0×1.0×1.0; L,
1.0×1.0×0.8 | R-Tumorectomy;
R-NSS; Scheduled left NSS later | R-PCC, Bi-RCC |
| F1-II-3 | M | 30 | p.N78S | - | - | R, 4.0×3.0×2.5 | Radical
R-nephrectomy | R-RCC |
| F1-III-1 | M | 5 | p.N78S | - | - | - | - | - |
| F2-II-11 | F | 18 | p.R161Q | - | R, 5.5×4.5×2.8 | - | Tumorectomy | R-PCC |
| F2-III-9 | M | 9(R)/17(L) | p.R161Q | Elevated (DA,
88.64; E, 106.23; NE, 827.15)/Elevated (DA, 600.36; E, 297.94; NE,
1093.84) | L, 7.0×5.5×3.4; R,
5.6×3.8×3.0 | - | Tumorectomy | Bi-PCC/He |
| F2-III-4 | F | 28 | p.R161Q | Elevated (DA,
268.63; E, 1270.3; NE, 1802.25) | L, 6.0×4.7×3.5; R,
3.5×2.8×2.5 | - | Bi-partial
adrenalectomy | Bi-PCC/Si |
In family 2, the proband (F2-II-11), a 45-year-old
female patient, presented with paroxysmal hypertension
(180–210/100–120 mmHg), headache, palpitations and hyperhidrosis in
1985 at the age of 18. US scans showed an isolated mass in the
right adrenal gland. Following treatment with α-blockers and
phenoxybenzamine orterazosin hydrochloride, the patient underwent
right adrenal tumorectomy, and PCC was confirmed by
histopathological examination. In 2002, the son of the proband
(F2-III-9, 9 years old) also presented with paroxysmal hypertension
(140–160/100–110 mmHg) and headache. Biochemical examination
indicated increased serum catecholamines, and an isolated mass in
the right adrenal gland was confirmed by US and CT scans. Following
treatment with α-blockers, right adrenal tumorectomy was performed.
Histopathological analysis revealed a right adrenal PCC. In 2010,
when the patient was 17 years old, laparoscopic adrenal tumorectomy
with carbon dioxide pneumoperitoneum was performed based on the
evidence of paroxysmal hypertension, headache, elevated serum
catecholamines and a mass in the left adrenal gland, but without
right adrenal gland abnormalities. PCC was confirmed by
histopathological examination. Another patient, the niece of the
proband (F2-III-4, 33 years old), presented with headache,
sustained hypertension, dizzy spells and excess serum
catecholamines. Bilateral masses in the adrenal glands were
confirmed by US and CT scans when the patient was 28 years old in
2007. High-dosage α1 blockade with doxazosin mesylate and
hydrocortisone cortisol were administered prior to operation, and
partial bilateral laparoscopic adrenalectomy with carbon dioxide
pneumoperitoneum was performed. Histopathological examination
indicated bilateral PCC. The 38-year-old father of this patient
(F2-II-3, the proband's brother) succumbed to hypertensive
intracerebral hemorrhage 21 years prior to this study (Table I). Among the members of family 2,
three were diagnosed with PCC as the only symptom and no other
systematic tumors, including CNS hemangioblastomas, renal cysts or
RCC, were identified.
All five patients (F1-II-1, F1-II-3, F2-II-11,
F2-III-4 and F2-III-9) were followed up. Biochemical and imaging
examination did not reveal any evidence of recurrence of PCC and
RCC and blood pressure returned to normal, with the exception of
one case (patient F1-II-1) where left RCC remained evident and the
patient agreed to undergo scheduled left NSS at a later stage.
Patient F2-III-4 required steroid replacement therapy. However,
patients F2-III-4 and F2-III-9 with bilateral tumorectomy or
partial adrenalectomy did not develop adrenocortical insufficiency
or Addisonian crisis during the period of observation.
Mutation analysis
Peripheral blood samples were collected for mutation
analysis of the RET proto-oncogene and VHL tumor
suppressor gene. Genomic DNA was extracted from whole-blood samples
using the QIAamp blood kit (Qiagen, Hilden, Germany) according to
the manufacturer's instructions. Polymerase chain reaction (PCR)
amplification of all exons and the flanking splice junctions of
RET and VHL was performed, followed by direct
bidirectional DNA sequencing. One hundred unrelated controls with a
similar ethnic background were also included in this study.
Results
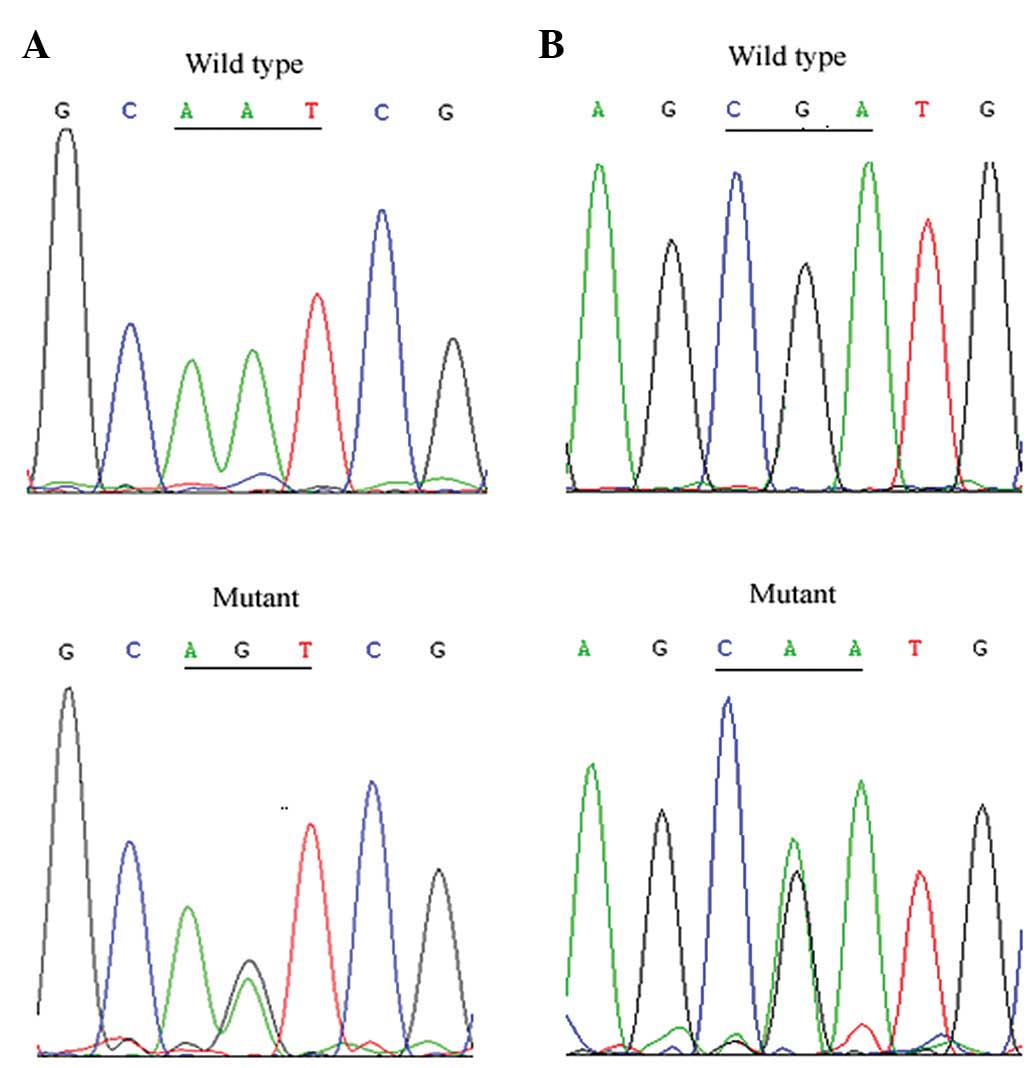
In family 1 with type 2A VHL syndrome, we identified
a heterozygous variant, c.A233G (p.N78S), within exon 1 of the
VHL gene in two affected cases and one clinically normal
family member (F1-III-1, the proband's 5-year-old son; Fig. 3A). A heterozygous nucleotide
substitution within exon 3 of the VHL gene, c.G482A
(p.R161Q), was identified in the proband (F2-II-11) and two
additional cases (F2-III-4 and F2-III-9) of family 2 with VHL type
2C (Fig. 3B). No mutants of the
RET gene were identified in these cases.
Discussion
VHL disease-related PCC is a tumor of the main
sympathetic paraganglion, the adrenal medulla, which is complicated
by hypertension. PCC is usually benign, with ~20% malignant cases
(18). Molecular genetic studies
suggest that ~50% of individuals with apparent familial
non-syndromic PCC have VHL germline mutations (18). Previous studies have shown that PCC
occurs sporadically or as part of hereditary tumor-predisposition
syndromes caused by germline mutations affecting one of the ten
following susceptibility genes: VHL, RET,
SDHA, SDHB, SDHC, SDHD, SDHAF2,
NF1, TMEM127 or MAX (Table II) (1,2,19–27).
The majority of familial cases of PCC and 10–20% of sporadic cases
carry germline mutations of these genes (4). RCC represents 90–95% of neoplasms
arising from the kidney, which is an important cause of death in
VHL disease (7). RCC is rare in
children and young adults, and occurs in both a nonfamilial and
familial form. Hereditary RCC accounts for ~4% of RCC, including
the following disorders: VHL disease; hereditary leiomyomatosis
related to FH gene mutation; tuberous sclerosis, which
results from the germline mutation of TSC1 and TSC2
genes; hereditary papillary renal cell carcinoma (HPRCC), which is
associated with MET gene; and the gene responsible for Birt
Hogg-Dubé syndrome, which has been reported to be the FLCN
gene (Table II) (28–31).
In a familial form, RCC occurs at an early age (with the exception
of HPRCC) and usually presents bilaterally with a high incidence
(7,31). According to a recent study, 20/255
patients (7.8%) with hereditary leiomyomatosis and RCC were
reported to have primary adrenal lesions, including 4 patients with
bilateral adrenal lesions and 4 with multiple nodules (28). Therefore, in future studies on PCC
and RCC patients, targeted regions (all exons of VHL,
RET, SDHA, SDHB, SDHC, SDHD,
SDHAF2, NF1, TMEM127, MAX, FH,
TSC1, TSC2, MET and FLCN) or exome
sequencing should be analyzed (32).
| Table IISummary of susceptibility genes
associated with PCC and RCC. |
Table II
Summary of susceptibility genes
associated with PCC and RCC.
| Gene
(location) | Protein
function | Clinical
phenotype | (Ref.) |
|---|
| RET
(10q11.2) | Tyrosine kinase
receptor that plays a crucial role in regulating cell
proliferation, migration, differentiation and survival through
embryogenesis. | Multiple endocrine
neoplasia type 2 (MEN 2)-autosomal dominantly inherited
predisposition to medullary thyroid carcinoma and PCC. MEN 2A is
characterized by medullary thyroid carcinoma, PCC and primary
hyperparathyroidism. MEN 2B is characterized by medullary thyroid
carcinoma, PCC and developmental anomalies (e.g., Marfanoid habitus
and mucosal neuromas). PCCs in MEN 2 are characteristically adrenal
and benign. | (22) |
| VHL
(3p25) | Short protein with
potent tumor suppressive activity. | Von Hippel-Lindau
disease, autosomal dominantly inherited predisposition to PCC,
retinal and cerebellar hemangioblastomas, clear RCC, non-secretory
pancreatic neuroendocrine tumors, endolymphatic tumors and visceral
cysts (renal, pancreatic and epididymal). Rarely head and neck
paragangliomas. Epididymal cystadenomas. | (1) |
| SDHA
(5p15) | Flavoprotein
catalytic subunit of Complex II. | Autosomal
dominantly inherited predisposition to PCC, head and neck
paragangliomas (though infrequent cause of these tumors) (autosomal
recessively inherited juvenile encephalopathy or neonatal
cardiomyopathy). | (23) |
| SDHB
(1p36) | Iron-sulfur protein
catalytic subunit of Complex II. | Autosomal
dominantly inherited predisposition to PCC, head and neck
paragangliomas and renal cell carcinoma. High frequency of
extra-adrenal PCC and malignancy. | (24) |
| SDHC
(1q21) | One of the two
transmembrane subunits of Complex II of the respiratory chain. | Autosomal
dominantly inherited predisposition to head and neck
paragangliomas, PCC. | (19) |
| SDHD
(11q23) | One of the two
transmembrane subunits of Complex II of the respiratory chain. | Autosomal
dominantly inherited predisposition to parasympathetic head and
neck paragangliomas and PCC. Tumor development generally occurs
only in individuals who have inherited the mutation from their
father (i.e., parent-of-origin effects on disease phenotype). | (20) |
| SDHAF2
(11q12.2) | Mitochondrial
assembly factor for Complex II, interacts directly with SDHA. | Autosomal
dominantly inherited predisposition to head and neck paragangliomas
and PCC. | (25) |
| NF1
(17q11.2) | Studies on animal
models show that homozygous mutant mice for the NF1 gene display a
variety of developmental abnormalities leading to midgestation
lethality. | Neurofibromatosis
type 1 (von Recklinghausen disease) autosomal dominantly inherited
predisposition to PCC, peripheral nervous system tumors (cutaneous,
nodular and plexiform neurofibromas), intestinal and
gastrointestinal stromal cell tumors, malignant gliomas and
juvenile chromic leukemias. | (26) |
| TMEM127
(2q11.2) | Transmembrane
protein involved in protein trafficking. | Autosomal
dominantly inherited predisposition to PCC and head and neck
paragangliomas. Relatively late mean age of diagnosis (compared to
VHL disease and SDHB mutations). | (27) |
| MAX
(14q23) | Component of the
MYC-MAX-MXD1 network and regulates cell proliferation,
differentiation and apoptosis. | Autosomal dominant
inheritance of bilateral PCC, confirming the association between
genetic abnormalities and multiple tumors. An association with
malignancy was also found. | (21) |
| FH
(1q42–43) | FH is a Krebs cycle
enzyme that converts fumarate to malate. | Hereditary
leiomyomatosis and RCC is an autosomal dominant disorder
characterized familial with an aggressive form of papillary kidney
cancer and the development of cutaneous and uterine
leiomyomas. | (28) |
| TSC1 (9q34 )
and TSC2 (16p13.3) | Proteins act as
tumor growth suppressors, agents that regulate cell proliferation
and differentiation. | Tuberous sclerosis
or tuberous sclerosis complex is a rare multi-system genetic
disease that causes non-malignant tumors to grow in the brain and
on other vital organs such as the kidneys, heart, eyes, lungs and
skin. | (29) |
| MET
(7q31) | MET transmembrane
protein is a tyrosine kinase receptor which induces proliferation
and differentiation of epithelial and endothelial cells, cell
branching and invasion. | Hereditary
papillary renal carcinoma is an autosomal dominant renal cancer
syndrome in which affected individuals are at risk of developing
multifocal bilateral type 1 papillary renal carcinoma. | (30) |
| FLCN
(17q11.2) | FLCN forms a
complex with folli- culin-interacting proteins (FNIP1 and FNIP2).
These components intern bind to AMP-activated protein kinase. | Birt-Hogg-Dubé
syndrome is an autosomal dominant disease, and affected individuals
develop cutaneous fibrofolliculomas, pulmonary cysts, spontaneous
pneumothoraces and renal tumors. | (31) |
The VHL gene has been conserved throughout
evolution and is found in all multicellular organisms. The two VHL
gene products, pVHL, a 30-kDa full-length protein [p30, 213 amino
acids, NM_000551.2 (variant 1 mRNA)], and a 19-kDa protein [p19,
160 amino acids, NM_198156.1 (variant 2 mRNA)], suppress tumor
formation in nude mice and regulate hypoxia-inducible factor-α
(HIF-α) (33,34). pVHL has two domains, the β domain
(N-terminal β-sheet domain), which contains a macromolecular
binding site, and an α domain (N-terminal α-helical domain), which
interacts with elongin C (35).
The region between codon 75–82 is located in the β-binding domain
of VHL protein; thus, the p.N78S mutation interferes with HIF
binding, which results in the excessive transcription of HIF target
genes, such as vascular endothelial growth factor (13). Codon 161 is located in the α domain
and interacts with elongin C. The mutation in the α domain affects
VHL tumor suppressor activity (36,37).
Through errors in chromosomal replication during cell division,
additional genes or modifying factors may be involved in the
genetic basis of VHL (37).
VHL genotype-phenotype correlations have been
previously established (5,17). The p.N78S mutation has been
reported to be associated with PCC, RCC and CNS hemangioblastomas
(8). The p.R161Q mutation was
first reported by Tong et al(38) in Chinese VHL patients who had
non-syndromic familial PCC without hypertension and is also known
to be associated with retinal and CNS hemangioblastoma and RCC
(10). For patients included in
the present study, VHL mutations were consistent with the
type 2A and 2C phenotypes. In particular, p.R161Q was associated
with PCC with hypertension syndrome, and p.N78S had a more severe
clinical phenotype (RCC and bilateral PCC) compared with p.R161Q.
However, none of the mutations were associated with CNS
hemangioblastomas.
The identification of the VHL tumor
suppressor gene facilitates the diagnosis of VHL disease,
particularly in patients who do not meet the clinical diagnostic
criteria. It also has important implications on the estimation of
risk for the development of further tumors in patients and of VHL
disease in their relatives. In family 1, a young family member
(F1-III-1, 5 years old) did not have an evident clinical phenotype
but carried the p.N78S mutation. The identification of the
VHL tumor suppressor gene is beneficial for the early
prevention and treatment of VHL syndrome. Additionally, the mean
age at diagnosis of PCC and RCC in VHL is ~30 and ~40 years,
respectively (39,40). However, according to a clinical
report, a 2.75-year-old female patient with PCC was found to carry
a p.R161Q germline mutation (40).
This is the youngest patient with PCC reported to date. For
patients in the present study, the age of onset for patient
F2-III-9 was 9 years, while the other two patients were 18 and 28
years old when they were diagnosed with PCC. Thus, molecular
genetic testing and genetic counseling for patients with PCC and
RCC should be performed at an early age. Early diagnosis of VHL
would be beneficial, since regular surveillance for tumors and
early intervention are suggested to result in better prognosis.
Numerous studies have shown that VHL patients
treated with NSS have a high incidence of local recurrence of new
primary tumors, but a low risk of distant metastasis. Approximately
25% of VHL patients with more advanced RCC (>3 cm) develop
metastatic disease (41,42). Radical bilateral nephrectomy should
be performed when RCC >3 cm, and renal transplantation is
required. Bilateral PCC is usually treated by CSA (adrenal
tumorectomy or partial adrenalectomy). Patients may avoid hormone
replacement, but there would be the risk of late recurrence. Total
adrenalectomy may be considered as an alternative when there is a
high incidence of multiple bilateral adrenal masses and large
tumors; however, life-long steroid replacement is required. All the
patients should be followed up regularly to aid the identification
of the processing adrenocortical insufficiency or Addisonian crisis
(3).
In the present study, the p.N78S mutation patient
(F1-II-1) with multiple bilateral RCC (1.0 and 1.0 cm) and right
PCC (4.1 cm) was subjected to right NSS and right CSA. The left
RCCs were observed using imaging and exhibited no marked
enlargement, while locoregional recurrence, distant metastasis
and/or CNS were not found in two cases (F1-II-1 and F1-II-3) 39 and
6 months after the surgery, respectively. Among the three p.R161Q
mutation patients, one (F2-III-9) had a left PCC 8 years after
presenting with a right PCC at 9 years of age, one (F2-III-4) had
simultaneous bilateral PCC and the other (F2-II-11) only presented
with a right PCC at 18 years of age. All of them underwent
bilateral or unilateral CSA and adrenocortical function was normal
without steroid replacement. Therefore, identification of the
underlying mutation not only confirms the diagnosis, but also
provides useful information for genetic counseling and subsequent
management decision making.
Acknowledgements
The authors thank all the patients and their
families who agreed to participate in this study. This study was
supported by the Key Scientific Research Project of Nanjing
Military Command, China (09Z038 and 10Z036) and the National 973
Basic Research Program of China (2013CB911300)
References
|
1
|
Huang Y, Zhou D, Liu J, Zhou P, Li X and
Wang Z: Germline mutations of the VHL gene in seven Chinese
families with von Hippel-Lindau disease. Int J Mol Med. 29:47–52.
2012.PubMed/NCBI
|
|
2
|
Qi XP, Ma JM, Du ZF, et al: RET germline
mutations identified by exome sequencing in a Chinese multiple
endocrine neoplasia type 2A/familial medullary thyroid carcinoma
family. PLoS One. 6:e203532011. View Article : Google Scholar
|
|
3
|
Qi XP, Chen XL, Ma JM, et al: RET
proto-oncogene genetic screening of families with multiple
endocrine neoplasia type 2 optimizes diagnostic and clinical
management in China. Thyroid. 22:1257–1265. 2012. View Article : Google Scholar
|
|
4
|
Gimenez-Roqueplo AP, Dahia PL and Robledo
M: An update on the genetics of paraganglioma, pheochromocytoma,
and associated hereditary syndromes. Horm Metab Res. 44:328–333.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vicha A, Holzerova M, Krepelova A, et al:
Molecular cytogenetic characterization in four pediatric
pheochromocytomas and paragangliomas. Pathol Oncol Res. 17:801–808.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Santarpia L, Lapa D and Benvenga S:
Germline mutation of von Hippel-Lindau (VHL) gene 695 G>A
(R161Q) in a patient with a peculiar phenotype with type 2C VHL
syndrome. Ann N Y Acad Sci. 1073:198–202. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Richards FM, Payne SJ, Zbar B, Affara NA,
Ferguson-Smith MA and Maher ER: Molecular analysis of de novo
germline mutations in the von Hippel-Lindau disease gene. Hum Mol
Genet. 4:2139–2143. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Neumann HP, Bausch B, McWhinney SR, et al:
Germline mutations in nonsyndromic phaeochromocytoma. N Engl J Med.
346:1459–1466. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Neumann HP, Bender BU, Berger DP, et al:
Prevalence, morphology and biology of renal cell carcinoma in von
Hippel-Lindau disease compared to sporadic renal cell carcinoma. J
Urol. 160:1248–1254. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gong K, Zhang N, Zhang K and Na Y: The
relationship of erythropoietin overexpression with von
Hippel-Lindau tumour suppressor gene mutations between
hypoxia-inducible factor-1α and -2α in sporadic clear cell renal
carcinoma. Int J Mol Med. 26:907–912. 2010.PubMed/NCBI
|
|
11
|
Latif F, Tory K, Gnarra J, et al:
Identification of the von Hippel-Lindau disease tumor suppressor
gene. Science. 260:1317–1320. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cantley J, Selman C, Shukla D, et al:
Deletion of the von Hippel-Lindau gene in pancreatic beta cells
impairs glucose homeostasis in mice. J Clin Invest. 119:125–135.
2009.PubMed/NCBI
|
|
13
|
Crossey PA, Richards FM, Foster K, et al:
Identification of intragenic mutations in the von Hippel-Lindau
disease tumour suppressor gene and correlation with disease
phenotype. Hum Mol Genet. 3:1303–1308. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Glavac D, Neumann HP, Wittke C, et al:
Mutations in the VHL tumor suppressor gene and associated lesions
in families with von Hippel-Lindau disease from central Europe. Hum
Genet. 99:271–280. 1996.PubMed/NCBI
|
|
15
|
Eng C, Crossey PA, Mulligan LM, et al:
Mutations in the RET protooncogene and the von Hippel-Lindau
disease tumour suppressor gene in sporadic and syndromic
phaeochromocytomas. J Med Genet. 32:934–937. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kondo K, Yao M, Yoshida M, et al:
Comprehensive mutational analysis of the VHL gene in sporadic renal
cell carcinoma: relationship to clinicopathological parameters.
Genes Chromosomes Cancer. 34:58–68. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang YR, Zhang J, Wang JD and Fan XD:
Genetic study of a large Chinese kindred with von Hippel-Lindau
disease. Chin Med J (Engl). 117:552–557. 2004.PubMed/NCBI
|
|
18
|
Opocher G and Schiavi F: Genetics of
pheochromocytoma and paragangliomas. Best Pract Res Clin Endocrinol
Metab. 24:943–956. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Peczkowska M, Cascon A, Prejbisz A, et al:
Extra-adrenal and adrenal phaeochromocytomas associated with a
germline SDHC mutation. Nat Clin Pract Endocrinol Metab. 4:111–115.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Astuti D, Douglas F, Lennard TW, et al:
Germline SDHD mutation in familial phaeochromocytoma. Lancet.
357:1181–1182. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Comino-Mendez I, Gracia-Aznarez FJ,
Schiavi F, et al: Exome sequencing identifies MAX mutations as a
cause of hereditary pheochromocytoma. Nat Genet. 43:663–667. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Eng C, Clayton D, Schuffeneckeret I, et
al: The relationship between specific RET proto-oncogene mutations
and disease phenotype in multiple endocrine neoplasia type 2.
International RET mutation consortium analysis. JAMA.
276:1575–1579. 1996. View Article : Google Scholar
|
|
23
|
Korpershoek E, Favier J, Gaal J, et al:
SDHA immunohistochemistry detects germline SDHA gene mutations in
apparently sporadic paragangliomas and pheochromocytoma. J Clin
Endocrinol Metab. 96:E1472–E1476. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Astuti D, Latif F, Dallol A, et al: Gene
mutations in the succinate dehydrogenase subunit SDHB cause
susceptibility to familial pheochromocytoma and to familial
paraganglioma. Am J Hum Genet. 69:49–54. 2001. View Article : Google Scholar
|
|
25
|
Bayley JP, Kunst HP, Cascon A, et al:
SDHAF2 mutations in familial and sporadic paraganglioma and
phaeochromocytoma. Lancet Oncol. 11:366–372. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bausch B, Koschker AC, Fassnacht M, et al:
Comprehensive mutation scanning of NF1 in apparently sporadic cases
of pheochromocytoma. J Clin Endocrinol Metab. 91:3478–3481. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Neumann HP, Sullivan M, Winter A, et al:
Germline mutations of the TMEM127 gene in patients with
paraganglioma of head and neck and extraadrenal abdominal sites. J
Clin Endocrinol Metab. 96:E1279–E1282. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shuch B, Ricketts CJ, Vocke CD, et al:
Adrenal nodular hyperplasia in hereiditary leiomyomatosis and renal
cell cancer. J Urol. 189:430–435. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dabora SL, Jozwiak S, Franz DN, et al:
Mutational analysis in a cohort of 224 tuberous sclerosis patients
indicates increased severity of TSC2, compared with TSC1, disease
in multiple organs. Am J Hum Genet. 68:64–80. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schmidt L, Junker K, Weirich G, et al: Two
North American families with hereditary papillary renal carcinoma
and identical novel mutations in the MET proto-oncogene. Cancer
Res. 58:1719–1722. 1998.PubMed/NCBI
|
|
31
|
Toro JR, Wei MH, Glenn GM, et al: BHD
mutations, clinical and molecular genetic investigations of
Birt-Hogg-Dubé syndrome: a new series of 50 families and a review
of published reports. J Med Genet. 45:321–331. 2008.PubMed/NCBI
|
|
32
|
Qi XP, Du ZF, Ma JM, et al: Genetic
diagnosis of autosomal dominant polycystic kidney disease by
targeted capture and next-generation sequencing: utility and
limitations. Gene. 516:93–100. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Iliopouluos O, Levy AP, Jiang C, Kaelin WG
Jr and Goldberg MA: Negative regulation of hypoxia-inducible genes
by the von Hippel-Lindau protein. Proc Natl Acad Sci USA.
93:10595–10599. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stebbins CE, Kaelin WG Jr and Pavletich
NP: Structure of the VHL-ElonginC-ElonginB complex: implications
for VHL tumor-suppressor function. Science. 284:455–461. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Okuda H, Hirai S, Takaki Y, et al: Direct
interaction of the beta-domain of VHL tumor suppressor protein with
the regulatory domain of atypical PKC isotypes. Biochem Biophys Res
Commun. 263:491–497. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Domene C and Illingworth CJ: Effects of
point mutations in pVHL on the binding of HIF-1α. Proteins.
80:733–746. 2012.
|
|
37
|
Arjumand W and Sultana S: Role of VHL gene
mutation in human renal cell carcinoma. Tumour Biol. 33:9–16. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tong AL, Zeng ZP, Li HZ, et al: von
Hippel-Lindau gene mutation in non-syndromic familial
pheochromocytomas. Ann N Y Acad Sci. 1073:203–207. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Akcaglar S, Yavascaoglu I, Vuruskan H and
Oktay B: Genetic evaluation of von Hippel-Lindau disease for early
diagnosis and improved prognosis. Int Urol Nephrol. 40:615–620.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sovinz P, Urban C, Uhrig S, et al:
Pheochromocytoma in a 2.75-year-old-girl with a germline von
Hippel-Lindau mutation Q164R. Am J Med Genet A. 152A:1752–1755.
2010.PubMed/NCBI
|
|
41
|
Walther MM, Choyke PL, Glenn G, Lyne JC,
Rayford W, Venzon D and Linehan WM: Renal cancer in families with
hereditary renal cancer: Prospective analysis of a tumor size
threshold for renal parenchymal sparing surgery. J Urol.
161:1475–1479. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Steinbach F, Novick AC, Zincke H, et al:
Treatment of renal-cell carcinoma in von Hippel-Lindau disease: a
multicenter study. J Urol. 153:1812–1816. 1995. View Article : Google Scholar : PubMed/NCBI
|