1. Introduction
Chronic lymphocytic leukemia (CLL) is the most
common type of leukemia among adults in the western world. The
incidence of CLL in the United States is ~3.9/100,000 and the
median age of diagnosis is 72 (1).
It is characterized by a malignant clone of B cells in the bone
marrow, blood and secondary lymphoid tissues, which are primarily
arrested in the G0/G1 cell-cycle phase. The clinical course of CLL
is heterogeneous and survival times range from a few years to
several decades. However, ~5–20% of CLL patients develop Richter’s
syndrome, an aggressive lymphoma which results in a short survival
time (2).
microRNAs (miRNAs) are a family of small,
evolutionarily conserved, single-stranded non-coding RNAs and are
~18–25 nucleotides in length. The first miRNAs were discovered by
Lee et al(3) in 1993. It
has been demonstrated that miRNAs act as biological regulators
involved in numerous cell processes, such as development,
differentiation, proliferation, survival and death. They function
by directly binding to the 3′ untranslated regions (UTRs) of
specific target messenger RNAs (mRNAs), leading to the inhibition
of expression or degradation of target mRNAs. Recently, intensive
studies on miRNA biogenesis and mechanisms by which target mRNAs
are regulated by miRNAs have been conducted. A simple model of
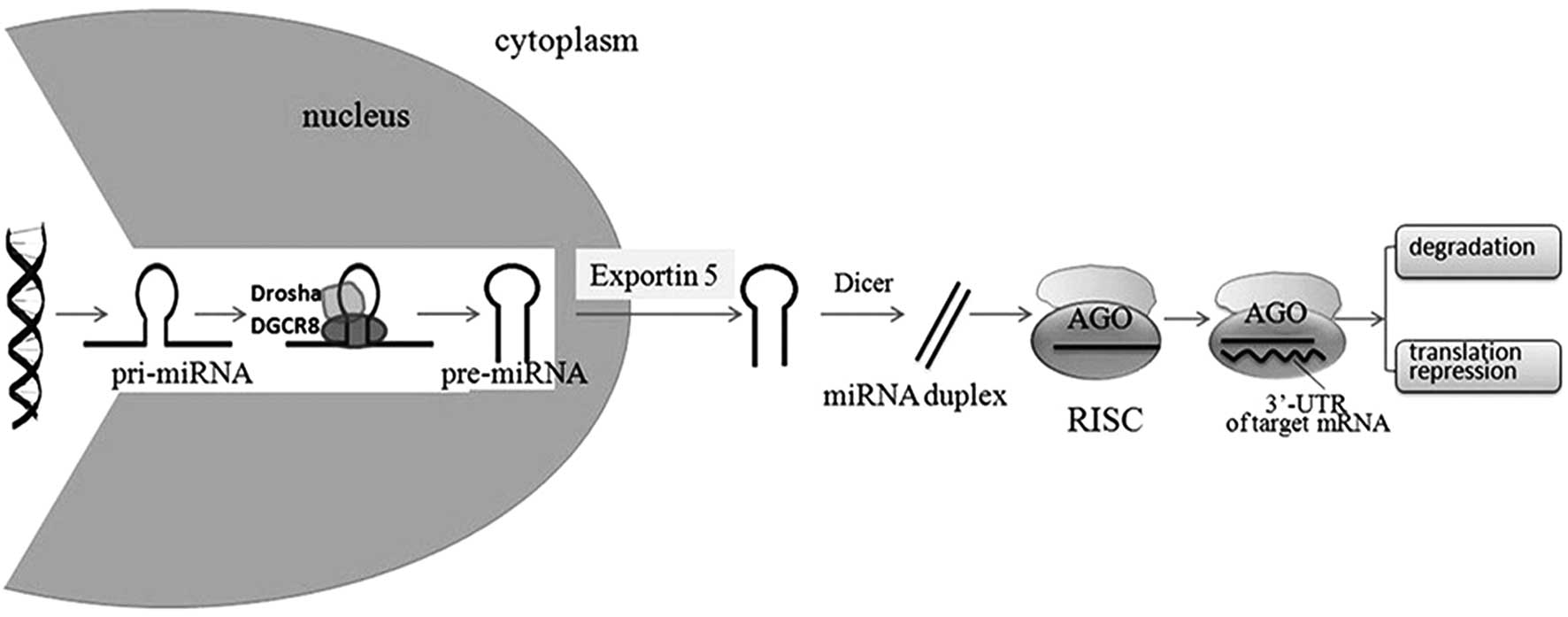
miRNA biosynthesis and function is shown in Fig. 1. miRNAs are encoded by genomic DNA
in the nucleus and are transcribed by either RNA polymerase II (RNA
Pol II) or III into primary miRNA transcripts (pri-miRNAs).
pri-miRNA are processed by Drosha (also termed ribonuclease 3) and
DiGeorge syndrome chromosomal region 8, and further cleaved into a
hairpin-shaped 70–100 nucleotide precursor (pre-miRNA). Pre-miRNA
is then actively transported into the cytoplasm by exportin 5 and
processed by Dicer (another RNase III) and transactivating response
RNA-binding protein into a 19–24 nucleotide, mature miRNA duplex by
removal of the terminal loop. Generally, only one strand of the
miRNA duplex is functional and the other strand is degraded. By
associating with Argonaute proteins, mature miRNAs are incorporated
into the RNA-induced silencing complex (RISC). miRNAs then guide
the RISC to the 3′UTR of target mRNAs, leading to the inhibition of
translation or the decrease in the stability of the target mRNAs
(4,5). Perfect or near perfect
complementarity of miRNA with mRNA leads to the degradation of
target mRNA and imperfect complementarity inhibits the translation
of target mRNA (6). It has been
demonstrated that miRNAs act as biomarkers in numerous types of
cancer, such as breast and colorectal cancer (7). Furthermore, miRNAs are also important
in almost every stage of hematopoietic cell development and signal
transduction, which suggests the potential significance of miRNAs
in CLL, the most common type of leukemia in adults (8). In this review, we focus on previous
studies which have investigated the involvement of miRNAs in the
onset, prognosis and chemoresistance of CLL. Further understanding
of miRNAs may provide novel diagnostic and therapeutic strategies
for CLL.
2. miRNAs in the pathogenesis of CLL
miRNAs associated with the genetic
aberration of CLL
Genetic aberrations are observed in >80% of CLL
patients (9). The most frequent
alteration is del13q14, which accounts for >50% of CLL cases.
Calin et al(10) determined
that a cluster of two miRNA genes, miR-15a and miR-16-1, were
absent or downregulated in CLL patients with a 13q deletion and
this downregulation correlated with allelic loss at 13q14. This was
the first study to demonstrate that two miRNAs may act as
suppressor genes of human cancer. A minimally deleted region
containing deletions in the leukemia 2 (DLEU2) gene and the
miR-15a/miR-16-1 genes has been identified in this region (10,11).
However, Lia et al(12)
determined that the size of the 13q14 deletion was related to the
penetrance and aggressiveness of lymphoproliferative diseases,
which suggested another tumor suppressor function of genetic
elements in addition to DLEU2/miR-15a/miR-16-1. Therefore, other
genes are located in 13q14 as well as miR-15a and miR-16-1. miR-15a
and miR-16-1 function as tumor suppressor genes through targeting
several molecules involved in cell proliferation and apoptosis,
such as B-cell CLL/lymphoma 2 (BCL2), cyclin D1 and D3 (CCND1 and
CCND3) and cyclin-dependent kinase 6 (CDK6) (13,14).
Another common aberration in CLL is trisomy 12
(10–20%). It has previously been determined that NOTCH1 mutations
were detected in approximately half of IGVH
unmutated/ZAP70+ trisomy 12 CLL patients (41.9%), which
indicated that NOTCH1 activation is strongly correlated with
trisomy 12 (15). However, López
et al(16) suggested that
the distribution of NOTCH1 mutations in CLL with trisomy 12 is
heterogeneous and other chromosomal abnormalities including trisomy
18 altered the prognosis of these patients. Further studies are
required to determine the correlation between NOTCH1 and trisomy 12
in CLL. The involvement of miRNAs in NOTCH1 mutations have been
identified in T cell acute lymphocytic leukemia (T-ALL), including
miR-181-a-1, miR-181-b-1 and miR-223 (17,18).
Deletion of 11q22-23 is observed in ~20% of patients
with advanced CLL (9) and
predominantly impacts the ataxia-telangiectasia mutated (ATM) gene.
Mutations of the ATM gene were detected in 12% of all CLL patients
and ~1/3 of the CLL cases with del11q23 (19). ATM functions in regulating cell
cycle progression and controlling the integrity of DNA repair. CLL
patients with this deletion or mutation have a significantly
shorter treatment-free interval (23.5 months) compared with those
without such abnormalities (64.2 months) (20). A combination of an 11q deletion and
an ATM mutation in CLL resulted in a shorter overall survival (OS)
and progression-free survival (PFS) time following first-line
therapy with alkylating agents and purine analogs (20). ATM is important in the regulation
of miRNA biogenesis. It directly phosphorylates KH-type splicing
regulatory protein (KSRP, a key component in Drosha and Dicer
miRNA-processing complexes) upon DNA damage, facilitating the
interaction between KSRP and pri-miRNAs and promoting miRNA
maturation (21). Wang et
al(22) demonstrated that
miR-181 was directly targeted by ATM in breast cancer. Moreover,
miR-34b and miR-34c are located in the 11q23 region, thus deletion
of this region led to the downregulation of these two miRNAs
(23).
Deletion of 17p13 was observed in ~5–10% of CLL
cases at first diagnosis or treatment (9). Over 80% of CLL cases with 17p
deletion carried TP53 (a tumor suppressor involved in cell cycling
and cell death) mutations in the remaining allele. Inactivation of
the second TP53 allele by point mutation was identified in numerous
CLL cases and was correlated with chemorefractiveness and reduced
survival (24). Expression of
miR-34a, miR-29c and miR-17-5p were significantly downregulated in
CLL with TP53 abnormalities (25).
miRNAs involved in B cell
development
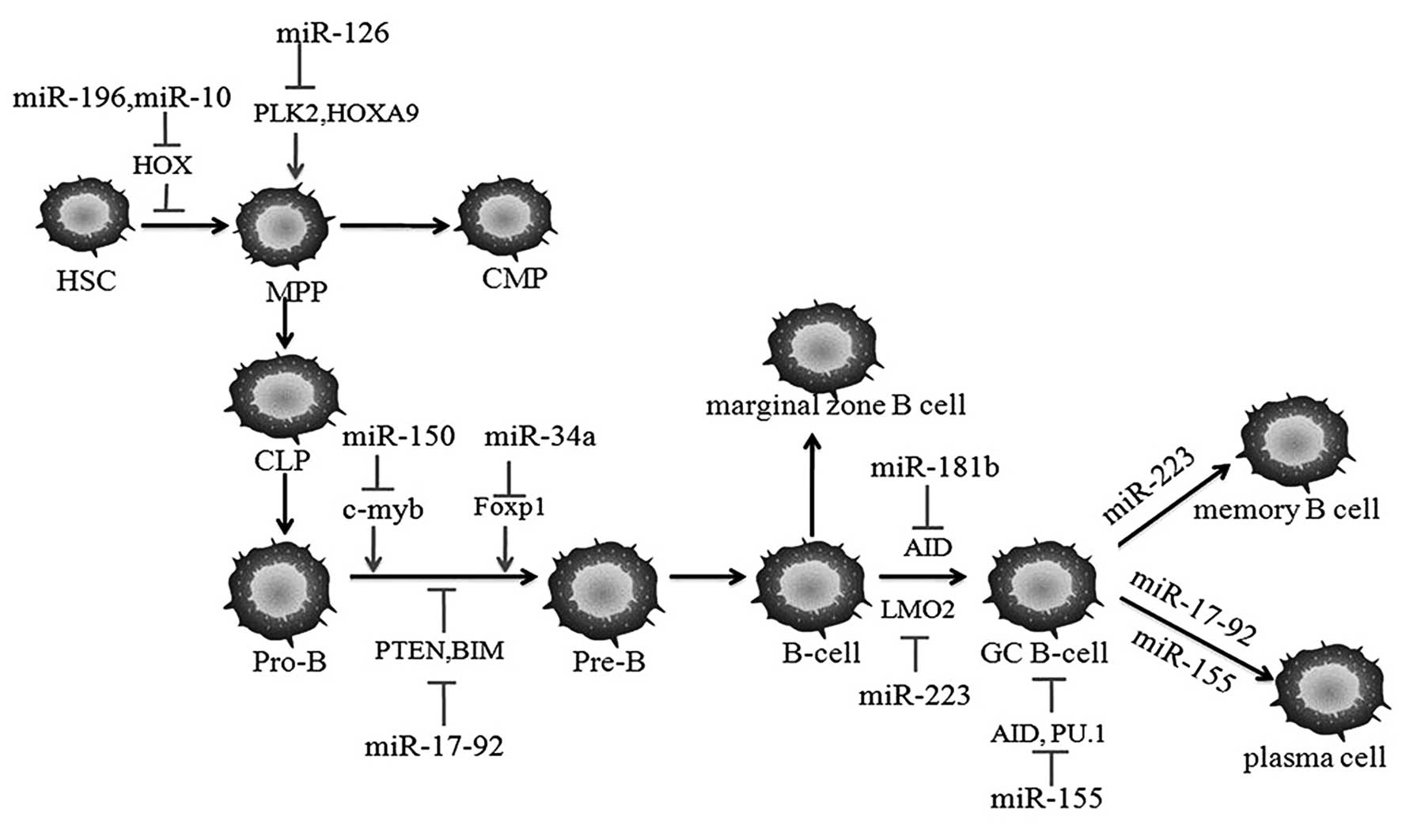
All blood cell lineages are generated from the
hematopoietic stem cells (HSCs) in the bone marrow. miRNAs
contribute to B cell development by targeting different
transcription factors. Developmental arrest at the progenitor (pro)
B cell to precursor (pre) B cell transition and an influence on
antibody diversity is induced by knocking out Dicer in early B
cells (26). The involvement of
miRNAs during B cell development are shown in Fig. 2. Homeobox (HOX) family members,
which are essential in modulating HSC homeostasis, are directly
repressed by the miR-196b and miR-10 families, leading to the
promotion of HSC differentiation. The development of progenitor
cells is also blocked by miR-126, through the direct inhibition of
the mRNAs encoding polo-like kinase 2 and HOXA9 (27). Constitutive expression of miR-150,
a hematopoietic-specific miRNA, blocks the transition from pro-B
cell to pre-B cell by inhibiting the expression of the translation
factor, c-myb (28,29). It has been demonstrated that the
miR-17-92 cluster (miR-17-5p, miR-18a, miR-19a, miR-19b-1, miR-20a
and miR-92) suppressed the expression of tumor suppressor
phosphatase and tensin homolog (PTEN), which is often mutated in
human lymphomas, and the pro-apoptosis protein BCL2-interacting
mediator of cell death (30). This
cluster is required for B cell development in normal hematopoiesis
and increased expression of these genes was demonstrated to be
oncogenic promoting the transition from pro-B cell to pre-B cell
during B cell development (31).
There are multiple putative binding sequences for miR-181b in the
3′UTR of activation-induced cytidine deaminase (AID) and these
sequences are directly inhibited by miR-181b (32). miR-223 inhibits LMO2 in the
transition from naïve B-cell to germinal center (GC) B-cell and GC
B-cell to memory B-cell (33).
Expression of miR-155 was observed to be at a moderate level in
HSC, a high level in the germinal center and relatively lower level
in mature B cells during normal lymphopoiesis (34). Upregulation of miR-155 has been
observed in various human B-cell neoplasms, including diffuse large
B cell lymphoma, Hodgkin’s lymphoma and CLL. Numerous studies have
demonstrated that miR-155 is important in the immune system and in
homeostasis. Mice lacking miR-155 exhibited a normal steady
condition of immune cell populations; however, showed a defective
humoral response when immunized (35). miR-155 is upregulated subsequent to
B cell activation in the GC, defective antibody class switching and
reductive differentiation into plasma cells occurs in miR-155
deficient B cells (36). PU.1 and
AID are two targets through which miR-155 functions. However,
miR-155 and miR-181b do not exhibit overlapping functions in
regulating AID expression as miR-181b prevents premature AID
activity in the early stages of activation and miR-155 functions at
a relatively later activation time. High levels of miR-34a also
restricts the transition from pro-B to pre-B cell through
inhibiting forkhead box transcription factor 1 (37).
miRNAs in the canonical Wnt signaling
pathway
Numerous studies have demonstrated that the Wnt
signaling pathway is significant in embryonic development and
fundamental cellular functions throughout life. It has recently
been demonstrated that the Wnt/β-catenin signaling pathway is
significant in tumorigenesis and angiogenesis (38). Constitutive activation of β-catenin
inhibits multilineage differentiation from HSCs, including the
development of B cells (39). Lu
et al(40) observed the
activation of the Wnt signaling pathway in CLL. Significantly
higher expression levels of Wnt3, Wnt5b, Wnt6, Wnt10a, Wnt14,
Wnt16, Fzd3 and LRP5/6, as well as LEF1 were observed in CLL cells
compared with that in normal B cells. Survival of CLL lymphocytes
was enhanced by SB-216763, an inhibitor of GSK3β. Two small
molecule inhibitors of the LEF-1/β-catenin complex, CGP049090 and
PKF115-584, were observed to efficiently induce the apoptosis of
CLL cells in vitro and in vivo, whereas the normal B
cells were not significantly affected (41). The Wnt canonical signaling pathway
may be significant in the survival of CLL cells.
Several studies demonstrated the correlation between
miRNAs and the canonical Wnt/β-catenin signaling pathway. Valastyan
et al(42) observed that
Fzd3 was directly targeted by miR-31 in breast cancer and may have
been a mechanism causing miR-31 to oppose metastasis. Analyses of
the mRNA and protein levels determined that Wnt1 is
transcriptionally inhibited by miR-21. Differentiation of
monocyte-derived dendritic cells is inhibited by the antagonism of
miR-21, either by adding exogenous Wnt1 or transfected with miR-21
inhibitors (43). PTEN is an
important tumor suppressor and the loss of PTEN results in the
upregulation of anti-apoptotic factor myeloid cell leukemia 1
(MCL1) in HSC. PTEN was also shown to be targeted by miR-21 in
hepatocellular cancer and epithelial ovarian cancer (44,45).
In addition, Rossi et al(46) suggested the 21FK (miR-21
expression, fluorescence in situ hybridization and
karyotype) score, a three-category score model (scores of 0, 1 or
2) to stratify the CLL patients according to OS. The lower the
score, the longer the OS time in CLL patients. One point was
allocated for miR-21 expression with a 17p deletion, and no points
were allocated for low miR-21 expression or a normal karyotype.
Patients with a score of zero exhibited a significantly higher
survival rate than those with a score of 1 or 2. An miR-29a/Wnt
positive feedback loop was discovered in osteoblastic
differentiation. Transcription of miR-29 was induced by the
canonical Wnt signaling pathway. Dikkopf-1, Kremen2 and secreted
frizzled related protein 2, which are negative regulators of the
canonical Wnt signaling pathway were directly inhibited by miR-29a.
The expression levels of these Wnt signaling inhibitors were
increased in miR-29a antagonist transfected cells (47). Furthermore, miR-315 and miR-135a/b
directly targeted Axin and APC, respectively, in the canonical Wnt
pathway (48). Few studies are
available concerning miRNAs in the Wnt/β-catenin signaling of CLL.
miRNAs are involved in several components of the canonical Wnt
signaling pathway; however, the majority of the specific mechanisms
remain unknown possibly due to the crosstalk between Wnt/β-catenin
signaling and other pathways.
3. miRNAs as hallmarks of CLL prognosis
miRNAs in CLL with ZAP-70+
and/or unmutated IgVH
Patients with aggressive CLL usually express the
unmutated immunoglobulin heavy-chain variable-region gene
(IgVH) and a high level of the 70-kDa ζ-associated
protein (ZAP-70), whereas patients with mutated IgVH and
low ZAP-70 expression present with indolent CLL (49). Through an miRNA microarray composed
of 190 human genes, Calin et al(50) identified a significant correlation
between miRNAs and CLL with ZAP-70 expression and unmutated
IgVH. A signature of 13 miRNAs was identified, with
upregulation of miR-15a, miR-16-1, miR-16-2, miR-23b, miR-24-1,
miR-146, miR-155, miR-195 and miR-221, and downregulation of
miR-223, miR-29a-2, miR-29b-2 and miR-29c in CLL with high ZAP-70
expression and unmutated IgVH. Therefore, the expression
of certain miRNAs may act as prognostic markers of CLL. The
predictive value of circulating miRNAs in CLL disease
stratification has been demonstrated (51). Expression levels of certain
circulating miRNAs present in CLL patient plasma, such as miR-150
and miR-135, were significantly different from that in control
groups. Circulating levels of certain miRNAs in CLL with
ZAP-70+ were also different from those in
ZAP-70− CLL patients. The level of miR-150 in the
ZAP-70− CLL plasma was increased, which was thought to
be correlated with the disease stage (51). Bomben et al(52) observed a significantly higher
expression level of miR-17 (the prototypic member of the miR-17-92
family) in IgVH unmutated CLL cells with high ZAP-70
compared with that in CLL cells with mutated IgVH and a
low ZAP-70 phenotype.
miRNAs and TP53 in CLL
The prognostic value of TP53 mutations in CLL has
been shown in numerous studies. Zenz et al(53) demonstrated that the TP53 mutation
acted as a central prognostic factor for PFS and OS in CLL
independent of 17p deletions. TP53 mutation and/or 17p deletions
were identified in ~29% of CLL patients refractory to fludarabin in
first-line treatment settings. Notably, the correlation between
TP53 mutations and certain other factors, such as IGHV gene
mutations, CD38 and ZAP-70 expression, or any chromosomal
abnormality (other than 17p deletion) was of no significance
(24). Therefore, TP53 mutations
may be beneficial if added to the genetic tests for CLL.
microRNA/TP53 feedback circuitry was demonstrated to be correlated
with CLL pathogenesis and outcome (54). In this feedback circuit, TP53 acts
as a molecular linker between miR-15a/miR-16-1 and miR-34b/miR-34c
clusters, which are located on chromosome 13q and chromosome 11q,
respectively. miR-15a and miR-16-1 directly bind to the 3′UTR of
TP53 and inhibit its expression. TP53 binds directly to a TP53
binding site on pre-miR-34b/miR-34c to promote their translation
and the downstream signal is activated. In CLL cases with a 13q
deletion only, downregulated or absent miR-15a/miR-16-1 resulted in
increased levels of BCL, MCL1 and TP53, further activated the
expression of miR-34a, miR-34b and miR-34c and induced ZAP-70
inhibition. Thus, in CLL patients with a 13q deletion, the number
of apoptotic cells may be decreased due to the increased levels of
anti-apoptotic proteins; however, the increase in tumor burden is
relatively low due to the enhancement of the TP53 tumor suppressor
pathway (54). This feedback
circuitry provides a novel mechanism for the involvement of miRNAs
in CLL with 13q deletions and their interaction with TP53. Mraz
et al(55) demonstrated the
involvement of miRNAs in cases of CLL with TP53 mutation.
Thirty-five miRNAs were detected in CLL samples with TP53 mutations
(n=12) and wild type TP53 samples (n=18), as a consequence,
miR-34a, miR-29 and miR-17-5p were significantly downregulated in
CLL with mutated TP53. miR-34a, which is located in 1p36 and is
frequently deleted in numerous types of tumors, was the most
differentially expressed miRNA with an ~9-fold decrease in CLL with
mutated TP53. miR-29 was reported to directly downregulate the
expression levels of TCL1 and MCL1 in CLL. As a member of the
miR-17-92 family, miR-17-5-p directly targeted E2F1, p21 and cyclin
D1, and other genes involved in the G1/S-phase cell-cycle
transition.
miRNAs involved in Notch signaling
Leong and Karsan (56) observed the activation of NOTCH1 in
leukemia through the study of the chromosomal translocation
t(7;9)(q34;q34.3) in T cell acute lymphoblastic cases (56). Activating mutations of NOTCH1 were
observed in 8.3% of CLL at diagnosis, 31% in CLL transformation to
Richter’s syndrome and 20% in CLL chemorefraction (57). CLL patients with a NOTCH1 mutation
presented with a more advanced clinical stage at diagnosis and a
shorter 10-year OS (21%) compared with those without NOTCH1
mutations (56%) (58). NOTCH1
mutation in CLL generates a premature stop codon and leads to a
constitutively activated and more stable form of the NOTCH1
protein, which lacks the C-terminal domain which contains a PEST
sequence. Mutations of NOTCH1 act as an independent prognostic
marker of CLL, which is prone to be mutually exclusive with TP53
mutations and a poor prognosis (59). Microarray profiling of miRNAs in
T-ALL cells was performed prior to and following inhibition of
Notch signaling with γ-secretase inhibitor (GSI). The expression
level of miR-223 significantly increased ~1.5-fold when Notch
signaling was inhibited by GSI. miR-223 targeted the 3′UTR of the
insulin-like growth factor 1 receptor (IGF1R) and downregulated its
expression. Thus, Notch signaling inhibits the miR-223 expression
level in T-ALL cells and the total IGF1R protein levels are
negatively regulated by miR-223 (18). miR-181a and miR-181-b-1, miRNAs
related to NOTCH1, are also observed in T-ALL. Deletion of
miR-181a-1/miR-181b-1 significantly inhibits the NOTCH1
oncogene-induced T-ALL by downregulating the negative regulators
downstream of the NOTCH and pre-TCR signaling pathway (17). In addition, miR-181a/miR-181-b
promoted the development of NK cells from CD34+
hematopoietic progenitor through the suppression of nemo-like
kinase (NLK), a protein kinase that negatively regulates Notch
signaling by inhibiting the formation of the Notch active
transcriptional complex. Transcription levels of miR-181a/b
increased during NK cell development from NK progenitor cells to
final CD56+ NK cells. Moreover, miR-181a/miR-181b also
increased IFN-γ production in primary CD56+ NK cells,
possibly by regulating the Notch signaling pathway (60). In addition, expression of miR-451
and miR-709 were repressed by intracellular NOTCH1 in a T-ALL mouse
model through induction of the degradation of the E2A tumor
suppressor (61).
In conclusion, the results of these studies suggest
that miRNAs involved in the NOTCH1 mutation are important in
several diseases, particularly T-ALL. A previous study demonstrated
that NOTCH1 may be added to the hierarchical prognostic
classification in CLL (62).
However, few studies have been conducted concerning the correlation
between miRNAs and NOTCH1 mutation in CLL at present. Thus further
studies regarding the mechanism of miRNAs and NOTCH1 in CLL may aid
in a more accurate prognosis for CLL patients.
4. miRNAs in CLL treatment and
chemoresistance
Current therapy for the clinical treatment of CLL
consists of chemotherapy with agents such as fludarabine,
cyclophosphamide, bendamustine and chlorambucil, and
immunotherapeutic agents such as rituximab and alemtuzumab
(63). However, resistance to
chemotherapeutic drugs is emerging as a challenging issue in the
effective management of CLL patients. The p53 pathway has been
demonstrated to be involved in the chemoresistance of CLL (64). Moreover, a model of the p53/miR-34a
network exists in the DNA-damage response pathway in CLL,
disturbance of this network results in chemoresistance. DNA damage
leads to the induction of p53 through the activation of ATM. Arrest
of the cell cycle and cell apoptosis are induced by p53 via direct
targeting of p21, Puma, Rax and miR-34a. Therefore, a p53-dependent
upregulation of miR-34a following DNA damage exists in CLL. An
increase in miR-34a affects cell death through targeting CDK4,
CDK6, CCND1, MYCN, BCL2 and Sirtuin 1. With TP53 mutation/17p
deletion, ATM mutation/11q deletion or miR-34a downregulation, the
DNA damage response pathway is reduced and the effect of cell
apoptosis and cell cycle arrest is weakened resulting in
chemoresistance (65). The
expression of miR-34a is not only related to 17p deletion but also
to fludarabine-refractory disease (in the absence of 17p deletion),
mono-allelic TP53 mutation and an impaired DNA damage response.
Expression of miR-34a is correlated with murine double minute 2
(MDM2) single nucleotide polymorphism 309 (SNP309) polymorphism
(66). SNP309 leads to a T to G
mutation in the first intron of MDM2 gene promoter and an
enhancement in the MDM2 protein and further inhibits the function
of p53 (67). Expression levels of
miR-34a in CLL patients with SNP309 G/G genotype were significantly
lower than in those with the TT genotype (66).
One of the most effective single therapeutic agents
for CLL treatment is fludarabine. An investigation of miRNA
expressions in fludarabine-sensitive and resistant cells
demonstrated that miR-181a and miR-221 were significantly
upregulated in resistant cases, and miR-29a was downregulated in
resistant cases in vitro(68). Additionally, miR-181a and miR-181b
significantly enhanced drug sensitivity in CLL cells by targeting
the 3′UTR of BCL2, MCL1 and X-linked inhibitor of apoptosis protein
(69). Ferracin et
al(70) investigated the
changes in the expression of 723 miRNAs in 17 CLL patients prior to
and following 5-day fludarabine monotherapy. Differential
expression of 37 miRNAs were identified prior to and following
fludarabine treatment. Among them, significant upregulation of
miR-21, miR-222 and miR-148a were observed in non-responsive
patients compared with patients sensitive to fludarabine treatment.
Moreover, a significant increase in caspase activity of
fludarabine-treated p53-mutant MEG-01 cells was induced by
anti-miR-21 and anti-miR-222 oligonucleotides, suggesting the
involvement of these two miRNAs in fludarabine resistance. These
results indicated that miR-222 and miR-21 are involved in CLL
fludarabine resistance independently of the p53 pathway. The
studies demonstrated that miRNAs may be beneficial in the
therapeutic approach to CLL by interfering with chemoresistance or
as novel target therapy drugs. In the future, miRNAs may be used
singly or coupled with other factors, or as target drugs.
5. Conclusion and future perspectives
In conclusion, miRNAs are extensively involved in
the pathogenesis and the chemoresistant mechanisms of CLL, and may
be used as prognostic markers. Cluster of miR-15a and miR-16-1 in
CLL with del13q14 acts as a model of miRNA involvement in CLL
pathogenesis. The development of the 21FK score suggests the
beneficial implication of a single miRNA or an miRNA in combination
with other prognostic markers in the accurate stratification of
CLL. Due to the significant involvement of miRNAs in
chemoresistance, novel therapeutic strategies and personalized
therapy approaches may be developed. However, further studies are
required to clarify the detailed mechanisms of specific miRNAs on
their various targets and the association between miRNAs and bone
marrow stromal cells prior to the use of miRNAs for clinical
treatment of CLL.
Acknowledgements
This study was supported by the grants from the
National Natural Science Foundation (grant no. 81270598), the
Natural Science Foundation of Shandong Province (grant nos.
Y2007C053 and 2009ZRB14176) and the Technology Development Project
of Shandong Province (grant nos. 2007GG10 and 2010 GSF10250).
Abbreviations:
|
HSC
|
hematopoietic stem cell
|
|
GC B cell
|
germinal center B cell
|
References
|
1
|
Dores GM, Anderson WF, Curtis RE, et al:
Chronic lymphocytic leukaemia and small lymphocytic lymphoma:
overview of the descriptive epidemiology. Br J Haematol.
139:809–819. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tsimberidou AM and Keating MJ: Richter
syndrome: biology, incidence, and therapeutic strategies. Cancer.
103:216–228. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lee RC, Feinbaum RL and Ambros V: The
C. elegans heterochronic gene lin-4 encodes small RNAs with
antisense complementarity to lin-14. Cell. 75:843–854. 1993.
|
|
4
|
Carthew RW and Sontheimer EJ: Origins and
mechanisms of miRNAs and siRNAs. Cell. 136:642–655. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim VN, Han J and Siomi MC: Biogenesis of
small RNAs in animals. Nat Rev Mol Cell Biol. 10:126–139. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hutvágner G and Zamore PD: A microRNA in a
multiple-turnover RNAi enzyme complex. Science. 297:2056–2060.
2002.PubMed/NCBI
|
|
7
|
Bartels CL and Tsongalis GJ: MicroRNAs:
novel biomarkers for human cancer. Clin Chem. 55:623–631. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Joshi D, Gosh K and Vundinti BR: MicroRNAs
in hematological malignancies: a novel approach to targeted
therapy. Hematology. 17:170–175. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dohner H, Stilgenbauer S, Benner A, et al:
Genomic aberrations and survival in chronic lymphocytic leukemia. N
Engl J Med. 343:1910–1916. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Calin GA, Dumitru CD, Shimizu M, et al:
Frequent deletions and down-regulation of micro-RNA genes miR15 and
miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci
USA. 99:15524–15529. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lagos-Quintana M, Rauhut R, Lendeckel W
and Tuschl T: Identification of novel genes coding for small
expressed RNAs. Science. 294:853–858. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lia M, Carette A, Tang H, et al:
Functional dissection of the chromosome 13q14 tumor-suppressor
locus using transgenic mouse lines. Blood. 119:2981–2990. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cimmino A, Calin GA, Fabbri M, et al:
miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl
Acad Sci USA. 102:13944–13949. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu Q, Fu H, Sun F, et al: miR-16 family
induces cell cycle arrest by regulating multiple cell cycle genes.
Nucleic Acids Res. 36:5391–5404. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Balatti V, Bottoni A, Palamarchuk A, et
al: NOTCH1 mutations in CLL associated with trisomy 12. Blood.
119:329–331. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
López C, Delgado J, Costa D, et al:
Different distribution of NOTCH1 mutations in chronic lymphocytic
leukemia with isolated trisomy 12 or associated with other
chromosomal alterations. Genes Chromosomes Cancer. 51:881–889.
2012.PubMed/NCBI
|
|
17
|
Fragoso R, Mao T, Wang S, et al:
Modulating the strength and threshold of NOTCH oncogenic signals by
mir-181a-1/b-1. PLoS Genet. 8:e10028552012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gusscott S, Kuchenbauer F, Humphries RK
and Weng AP: Notch-mediated repression of miR-223 contributes to
IGF1R regulation in T-ALL. Leuk Res. 36:905–911. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Austen B, Powell JE, Alvi A, et al:
Mutations in the ATM gene lead to impaired overall and
treatment-free survival that is independent of IGVH mutation status
in patients with B-CLL. Blood. 106:3175–3182. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Skowronska A, Parker A, Ahmed G, et al:
Biallelic ATM inactivation significantly reduces survival in
patients treated on the United Kingdom Leukemia Research Fund
Chronic Lymphocytic Leukemia 4 trial. J Clin Oncol. 30:4524–4532.
2012. View Article : Google Scholar
|
|
21
|
Zhang X, Wan G, Berger FG, He X and Lu X:
The ATM kinase induces microRNA biogenesis in the DNA damage
response. Mol Cell. 41:371–383. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang Y, Yu Y, Tsuyada A, et al:
Transforming growth factor-β regulates the sphere-initiating stem
cell-like feature in breast cancer through miRNA-181 and ATM.
Oncogene. 30:1470–1480. 2011.
|
|
23
|
Auer RL, Riaz S and Cotter FE: The 13q and
11q B-cell chronic lymphocytic leukaemia-associated regions derive
from a common ancestral region in the zebrafish. Br J Haematol.
137:443–453. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gonzalez D, Martinez P, Wade R, et al:
Mutational status of the TP53 gene as a predictor of response and
survival in patients with chronic lymphocytic leukemia: results
from the LRF CLL4 trial. J Clin Oncol. 29:2223–2229. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mraz M, Pospisilova S, Malinova K, Slapak
I and Mayer J: MicroRNAs in chronic lymphocytic leukemia
pathogenesis and disease subtypes. Leuk Lymphoma. 50:506–509. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Koralov SB, Muljo SA, Galler GR, et al:
Dicer ablation affects antibody diversity and cell survival in the
B lymphocyte lineage. Cell. 132:860–874. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
O’Connell RM, Rao DS, Chaudhuri AA and
Baltimore D: Physiological and pathological roles for microRNAs in
the immune system. Nat Rev Immunol. 10:111–122. 2010.PubMed/NCBI
|
|
28
|
Zhou B, Wang S, Mayr C, Bartel DP and
Lodish HF: miR-150, a microRNA expressed in mature B and T cells,
blocks early B cell development when expressed prematurely. Proc
Natl Acad Sci USA. 104:7080–7085. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xiao C, Calado DP, Galler G, et al:
MiR-150 controls B cell differentiation by targeting the
transcription factor c-Myb. Cell. 131:146–159. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xiao C, Srinivasan L, Calado DP, et al:
Lymphoproliferative disease and autoimmunity in mice with increased
miR-17-92 expression in lymphocytes. Nat Immunol. 9:405–414. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ventura A, Young AG, Winslow MM, et al:
Targeted deletion reveals essential and overlapping functions of
the miR-17 through 92 family of miRNA clusters. Cell. 132:875–886.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
de Yébenes VG, Belver L, Pisano DG, et al:
miR-181b negatively regulates activation-induced cytidine deaminase
in B cells. J Exp Med. 205:2199–2206. 2008.PubMed/NCBI
|
|
33
|
Zhang J, Jima DD, Jacobs C, et al:
Patterns of microRNA expression characterize stages of human B-cell
differentiation. Blood. 113:4586–4594. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fernando TR, Rodriguez-Malave NI and Rao
DS: MicroRNAs in B cell development and malignancy. J Hematol
Oncol. 5:72012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rodriguez A, Vigorito E, Clare S, et al:
Requirement of bic/microRNA-155 for normal immune function.
Science. 316:608–611. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Thai TH, Calado DP, Casola S, et al:
Regulation of the germinal center response by microRNA-155.
Science. 316:604–608. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rao DS, O’Connell RM, Chaudhuri AA,
Garcia-Flores Y, Geiger TL and Baltimore D: MicroRNA-34a perturbs B
lymphocyte development by repressing the forkhead box transcription
factor Foxp1. Immunity. 33:48–59. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ge X and Wang X: Role of Wnt canonical
pathway in hematological malignancies. J Hematol Oncol. 3:332010.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Seke Etet PF, Vecchio L and Nwabo Kamdje
AH: Interactions between bone marrow stromal microenvironment and
B-chronic lymphocytic leukemia cells: any role for Notch, Wnt and
Hh signaling pathways? Cell Signal. 24:1433–1443. 2012.PubMed/NCBI
|
|
40
|
Lu D, Zhao Y, Tawatao R, et al: Activation
of the Wnt signaling pathway in chronic lymphocytic leukemia. Proc
Natl Acad Sci USA. 101:3118–3123. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gandhirajan RK, Staib PA, Minke K, et al:
Small molecule inhibitors of Wnt/beta-catenin/lef-1 signaling
induces apoptosis in chronic lymphocytic leukemia cells in vitro
and in vivo. Neoplasia. 12:326–335. 2010.PubMed/NCBI
|
|
42
|
Valastyan S, Reinhardt F, Benaich N, et
al: A pleiotropically acting microRNA, miR-31, inhibits breast
cancer metastasis. Cell. 137:1032–1046. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hashimi ST, Fulcher JA, Chang MH, Gov L,
Wang S and Lee B: MicroRNA profiling identifies miR-34a and miR-21
and their target genes JAG1 and WNT1 in the coordinate regulation
of dendritic cell differentiation. Blood. 114:404–414. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Meng F, Henson R, Wehbe-Janek H, Ghoshal
K, Jacob ST and Patel T: MicroRNA-21 regulates expression of the
PTEN tumor suppressor gene in human hepatocellular cancer.
Gastroenterology. 133:647–658. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lou Y, Yang X, Wang F, Cui Z and Huang Y:
MicroRNA-21 promotes the cell proliferation, invasion and migration
abilities in ovarian epithelial carcinomas through inhibiting the
expression of PTEN protein. Int J Mol Med. 26:819–827.
2010.PubMed/NCBI
|
|
46
|
Rossi S, Shimizu M, Barbarotto E, et al:
microRNA fingerprinting of CLL patients with chromosome 17p
deletion identify a miR-21 score that stratifies early survival.
Blood. 116:945–952. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kapinas K, Kessler C, Ricks T, Gronowicz G
and Delany AM: miR-29 modulates Wnt signaling in human osteoblasts
through a positive feedback loop. J Biol Chem. 285:25221–25231.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Huang K, Zhang JX, Han L, You YP, Jiang T,
Pu PY and Kang CS: MicroRNA roles in beta-catenin pathway. Mol
Cancer. 9:2522010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Rassenti LZ, Huynh L, Toy TL, et al:
ZAP-70 compared with immunoglobulin heavy-chain gene mutation
status as a predictor of disease progression in chronic lymphocytic
leukemia. N Engl J Med. 351:893–901. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Calin GA, Ferracin M, Cimmino A, et al: A
MicroRNA signature associated with prognosis and progression in
chronic lymphocytic leukemia. N Engl J Med. 353:1793–1801. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Moussay E, Wang K, Cho JH, et al: MicroRNA
as biomarkers and regulators in B-cell chronic lymphocytic
leukemia. Proc Natl Acad Sci USA. 108:6573–6578. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bomben R, Gobessi S, Dal Bo M, et al: The
miR-17~92 family regulates the response to Toll-like receptor 9
triggering of CLL cells with unmutated IGHV genes. Leukemia.
26:1584–1593. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zenz T, Eichhorst B, Busch R, et al: TP53
mutation and survival in chronic lymphocytic leukemia. J Clin
Oncol. 28:4473–4479. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Fabbri M, Bottoni A, Shimizu M, et al:
Association of a microRNA/TP53 feedback circuitry with pathogenesis
and outcome of B-cell chronic lymphocytic leukemia. JAMA.
305:59–67. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Mraz M, Malinova K, Kotaskova J, et al:
miR-34a, miR-29c and miR-17-5p are downregulated in CLL patients
with TP53 abnormalities. Leukemia. 23:1159–1163. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Leong KG and Karsan A: Recent insights
into the role of Notch signaling in tumorigenesis. Blood.
107:2223–2233. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Fabbri G, Rasi S, Rossi D, et al: Analysis
of the chronic lymphocytic leukemia coding genome: role of NOTCH1
mutational activation. J Exp Med. 208:1389–1401. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Puente XS, Pinyol M, Quesada V, et al:
Whole-genome sequencing identifies recurrent mutations in chronic
lymphocytic leukaemia. Nature. 475:101–105. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Rossi D, Rasi S, Fabbri G, et al:
Mutations of NOTCH1 are an independent predictor of survival in
chronic lymphocytic leukemia. Blood. 119:521–529. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Cichocki F, Felices M, McCullar V,
Presnell SR, Al-Attar A, Lutz CT and Miller JS: Cutting edge:
microRNA-181 promotes human NK cell development by regulating Notch
signaling. J Immunol. 187:6171–6175. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Li X, Sanda T, Look AT, Novina CD and von
Boehmer H: Repression of tumor suppressor miR-451 is essential for
NOTCH1-induced oncogenesis in T-ALL. J Exp Med. 208:663–675. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Mansouri L, Cahill N, Gunnarsson R, et al:
NOTCH1 and SF3B1 mutations can be added to the hierarchical
prognostic classification in chronic lymphocytic leukemia.
Leukemia. 27:512–514. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Hallek M, Cheson BD, Catovsky D, et al:
Guidelines for the diagnosis and treatment of chronic lymphocytic
leukemia: a report from the International Workshop on Chronic
Lymphocytic Leukemia updating the National Cancer Institute-Working
Group 1996 guidelines. Blood. 111:5446–5456. 2008. View Article : Google Scholar
|
|
64
|
Zenz T, Mohr J, Edelmann J, et al:
Treatment resistance in chronic lymphocytic leukemia: the role of
the p53 pathway. Leuk Lymphoma. 50:510–513. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zenz T, Mohr J, Eldering E, et al: miR-34a
as part of the resistance network in chronic lymphocytic leukemia.
Blood. 113:3801–3808. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Asslaber D, Piñón JD, Seyfried I, et al:
microRNA-34a expression correlates with MDM2 SNP309 polymorphism
and treatment-free survival in chronic lymphocytic leukemia. Blood.
115:4191–4197. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bond GL, Hu W, Bond EE, et al: A single
nucleotide polymorphism in the MDM2 promoter attenuates the p53
tumor suppressor pathway and accelerates tumor formation in humans.
Cell. 119:591–602. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Moussay E, Palissot V, Vallar L, et al:
Determination of genes and microRNAs involved in the resistance to
fludarabine in vivo in chronic lymphocytic leukemia. Mol Cancer.
9:1152010. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhu DX, Zhu W, Fang C, et al: miR-181a/b
significantly enhances drug sensitivity in chronic lymphocytic
leukemia cells via targeting multiple anti-apoptosis genes.
Carcinogenesis. 33:1294–1301. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Ferracin M, Zagatti B, Rizzotto L, et al:
MicroRNAs involvement in fludarabine refractory chronic lymphocytic
leukemia. Mol Cancer. 9:1232010. View Article : Google Scholar : PubMed/NCBI
|