Introduction
Pancreatic cancer is a lethal disease that is
notoriously difficult to treat (1). Only a small proportion of cases are
curative through surgical resection and standard chemoradiotherapy
for patients with advanced disease only has modest effects with
substantial toxicity (2,3). Clearly, the continual development of
novel therapeutic agents is required to improve the current
situation.
Several studies on biological approaches targeting
the molecular abnormalities of pancreatic cancer are available
(4–6). One such pathway is the hedgehog (Hh)
signaling pathway, which specifies patterns of cell growth and
differentiation during embryogenesis in a wide range of tissues
(7). In addition to its function
in developmental patterning, the Hh pathway is also important in
maintaining the homeostasis of mature tissues and the number of
somatic cells in various organs. This pathway represents an
attractive target for drug development and has shown promise in
clinical trials of cancer treatments. The specificity of
cyclopamine for the Hh pathway is demonstrated by the absence of
cytotoxicity in cells that lack Hh signaling.
The K-ras oncogene mutation occurs in 75–90% of
pancreatic cancers (8,9). The gene encodes a 21-kDa
membrane-bound guanosine triphosphate-binding protein involved in
growth factor-mediated signal transduction pathways. K-ras is
activated through the overexpression or activation of
ras-activating signaling partners, including the epidermal growth
factor receptor (EGFR) (10). In
the present study, the effects of cyclopamine on pancreatic cancer
radiosensitivity were investigated in vitro using
K-RASwt Colo-357 and K-RASmt SW-1990 human
pancreatic cancer cell lines.
Materials and methods
Cell culture and reagents
Human pancreatic cancer cell lines, Colo-357 and
SW-1990, were purchased from the American Type Culture Collection
(Manassas, VA, USA). Colo-357 cells were maintained in Dulbecco’s
modified Eagle’s medium and SW-1990 cells were seeded onto tissue
culture dishes containing RPMI-1640 medium supplemented with 10%
fetal calf serum, L-glutamine (5 mmol/l), non-essential amino acids
(5 mmol/l), penicillin (100 U/ml) and streptomycin (100 U/ml;
Invitrogen Life Technologies, Carlsbad, CA, USA) at 37°C in a
humidified 5% CO2 atmosphere. Cyclopamine and EGF were
obtained from Cell Signaling Technology, Inc. (Beverly, MA, USA).
The EGFR inhibitor, gefitinib (Iressa), was purchased from
AstraZeneca (Macclesfield, UK).
Inhibitor treatment
Stock solutions of the cyclopamine Hh pathway
inhibitor, gefitinib EGFR inhibitor and EGF were prepared at
appropriate concentrations in dimethyl sulfoxide (DMSO) and then
stored at –70°C. For treatment, inhibitor solutions were diluted
1:1,000 to appropriate working concentrations (20 or 40 μmol/l
cyclopamine, 10 μmol/l gefitinib and 3 μmol/l EGF) in serum-free
medium. Control cultures received medium containing the solvent
DMSO at a concentration of 0.1%. Gefitinib and EGF were
supplemented to the culture media 0.5 h before irradiation and 24 h
of preirradiation treatment with cyclopamine was conducted.
Ionizing radiation
A Siemens 6 MV X-ray linear accelerator (Siemens,
Munich, Germany) was used to deliver a single dose of ionizing
radiation (IR) with a dose rate of 200 cGy/min at room
temperature.
Clonogenic assay
Cells were plated at various cell densities and
irradiated with 0.5, 1, 2, 4 and 6 Gy X-ray 24 h later. Following
12–14 days incubation at 37°C, cells were stained with Giemsa. The
number of colonies per dish was counted and the surviving fractions
were calculated as the ratio of plating efficiencies for irradiated
and unirradiated cells. Plating efficiency is defined as the colony
number divided by the number of cells plated for unirradiated
controls. Experiments were conducted in triplicate and data from
three independent experiments are presented as the means ± SD. All
survival fractions were fitted into the linear quadratic model.
Apoptosis assay
Cells were removed with trypsin and collected into
centrifuge tubes together with the culture medium. Flow cytometry
and Annexin V-fluorescein isothiocyanate (FITC) apoptosis analysis
were performed as previously described (11). Cell cycle distribution and
apoptotic rate were calculated from 1×10−4 cells using
ModFit LT software with the FACS Calibur (both Becton-Dickinson,
San Jose, CA, USA).
Cell cycle assays
Cells were removed with trypsin and collected into
centrifuge tubes together with the culture medium. Detailed methods
for flow cytometry analysis were previously described (12). Cell cycle distribution was
calculated from 1×10−4 cells using ModFit LT software
with the FACS Calibur.
Immunofluorescence assay
Detection of γ-H2AX foci immunofluorescence was
performed to determine residual DNA double-strand breaks (DSBs).
Cells grown on coverslips (Fisher Scientific, Loughborough, UK)
were fixed in ice-cold 4% paraformaldehyde for 30 min, blocked with
3% bovine serum albumin in phosphate buffer solution (PBS) and then
incubated with an antibody against γ-H2AX (ser139; 1:500; Cell
Signaling Technology, Inc.) for 2 h at 4°C. After washing with PBS,
secondary FITC-conjugated antibody was added for 1 h. The slides
were washed with PBS and then mounted with mounting medium
containing 4′,6-diamino-2-phenylindole.
Western blot analysis
Cell lysates were prepared and western blot analysis
was performed as previously described (13). Equal aliquots of total cell protein
(50 μg/lane) were electrophoresed on sodium dodecyl
sulfate-polyacrylamide gels, transferred onto polyvinylidene
fluoride membranes and then probed with β-actin (C-4), DNA-PKcs
(G-4), Ku70 (A-9) (Santa Cruz Biotechnology, Santa Cruz, CA, USA;
1:1,000 dilution), γ-H2AX or p-ATM (Cell Signaling Technology,
Inc.; 1:1,000) primary antibodies, followed by horseradish
peroxidase-labeled goat anti-mouse (GAM-007) and goat anti-rabbit
(SC-2004) IgG secondary antibodies. The protein bands were
visualized using an enhanced chemiluminescence system (Union
Bioscience Corporation, Hangzhou, China) with prestained markers as
molecular size standards. The densitometry of the protein bands was
quantified with Quantity One (Bio-Rad, Hercules, CA, USA) and the
values were expressed relative to β-actin (control for loading and
transfer). At least three independent experiments were performed
for each cell type studied.
Statistical comparisons
Data are presented as the mean ± SD. Experimental
results of the treated and control groups were compared using the
two-tailed Student’s t-test. All statistical tests were performed
using SPSS version 17.0 (SPSS, Inc., Chicago, IL, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
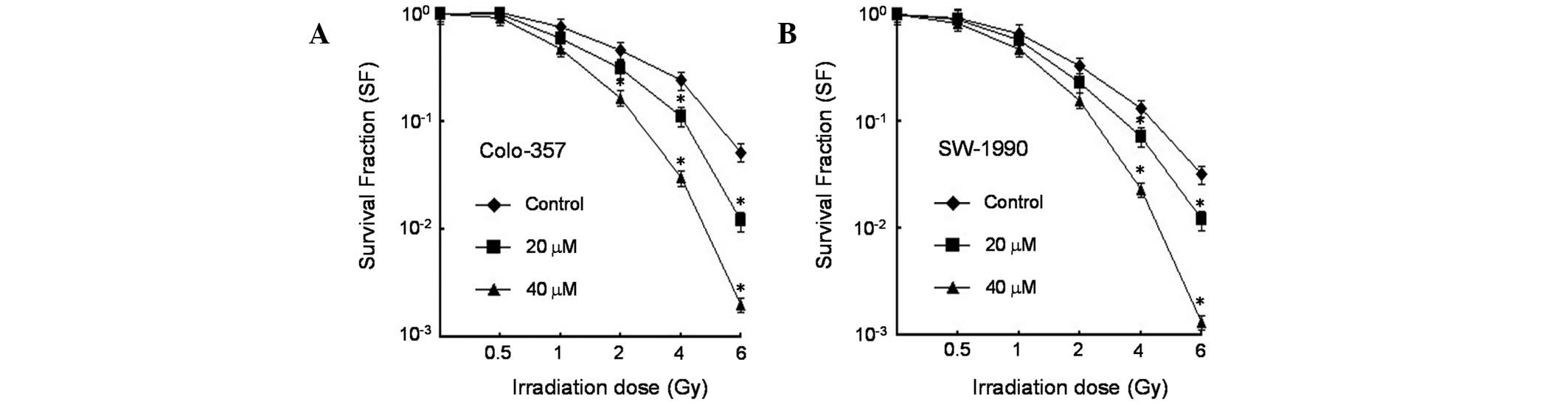
Cyclopamine enhances radiosensitivity of
pancreatic cancer cells
Given that cyclopamine modulates the Hh pathway, the
effect of cyclopamine on the radiation clonogenic survival of
K-RASwt (Colo-357) and K-RASmt (SW-1990) was
analyzed in human pancreatic cancer cell lines. The
radiosensitizing effect of cyclopamine was confirmed by single-dose
irradiation with doses up to 6 Gy. The results revealed that
cyclopamine treatment exerted significant radiosensitization of
Colo-357 and SW-1990 cells to the clinically relevant radiation
dose per fraction of 2 Gy relative to DMSO controls (Fig. 1).
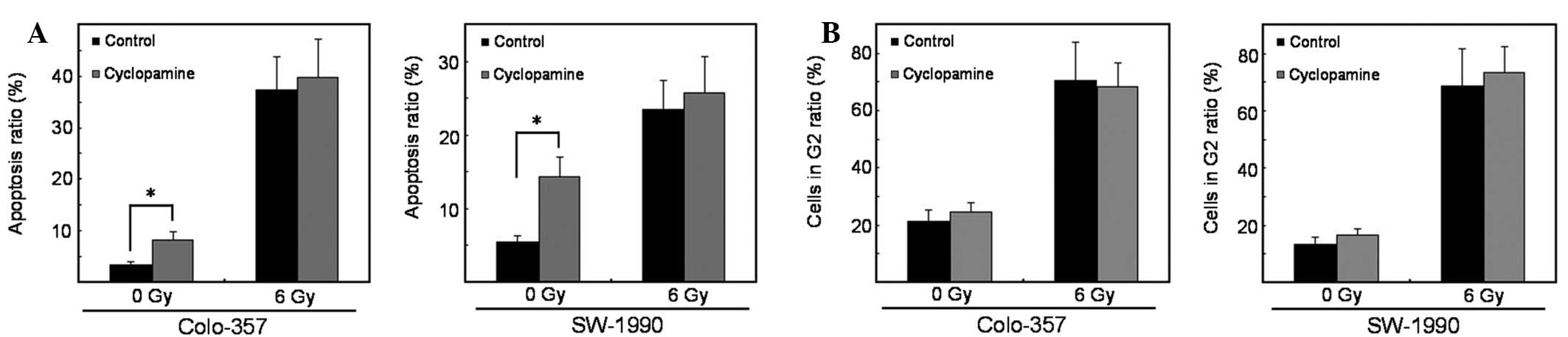
Cyclopamine does not enhance
radiation-induced apoptosis
Previous studies have reported induction of
apoptosis by cyclopamine (14).
Therefore, the rate of apoptosis was examined upon cyclopamine
treatment with irradiation compared with DMSO controls. Cyclopamine
(40 μmol/l) alone significantly induced apoptosis. However, in
combination with irradiation, it failed to induce apoptosis in
K-RASwt and K-RASmt pancreatic cancer cells
(Fig. 2A).
Cyclopamine treatment does not affect
cell cycle redistribution
Cell cycle phases are associated with various
degrees of radiosensitivity. Thus, the percentage of cells in the
radiosensitive G2 cell cycle phase was determined upon treatment
with cyclopamine alone or in combination with irradiation (15). A significant G2 cell cycle arrest
was noted following irradiation. However, cyclopamine (40 μmol/l)
treatment failed to abrogate radiation-induced G2 arrest as
compared with DMSO controls. This effect was observed in
K-RASwt and K-RASmt pancreatic cancer cells
(Fig. 2B).
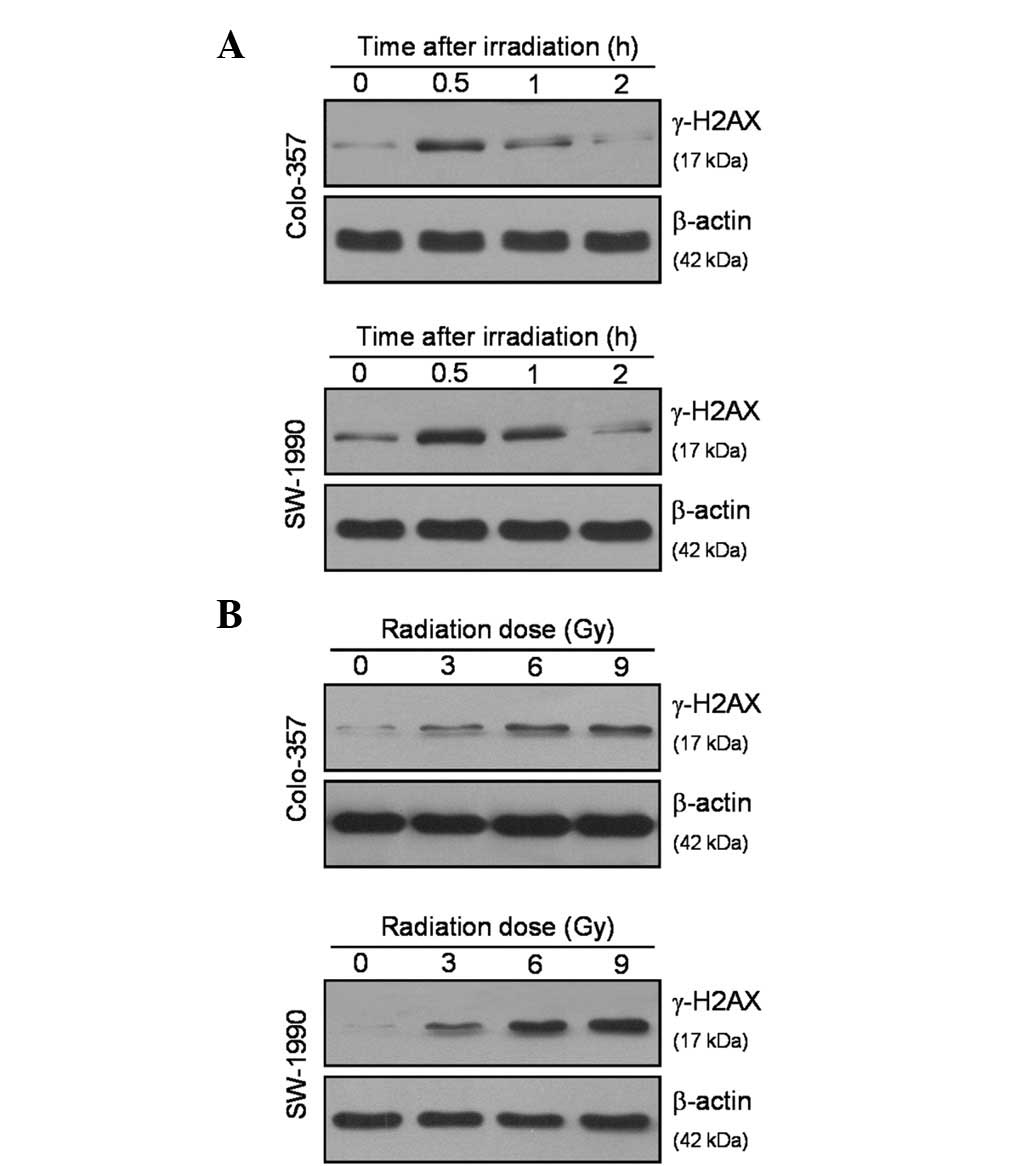
Effects on DNA-DSB repair by
cyclopamine
To analyze how cyclopamine affects radiation-induced
H2AX phosphorylation as an indicator of DNA damage signaling,
Colo-357 and SW-1990 cells were irradiated with a single dose of
ionizing radiation (6 Gy). H2AX phosphorylation at Ser139 reached a
maximum at 0.5 h following ionizing radiation (Fig. 3A). Using western blot analysis, a
dose-dependent increase in γ-H2AX following ionizing radiation was
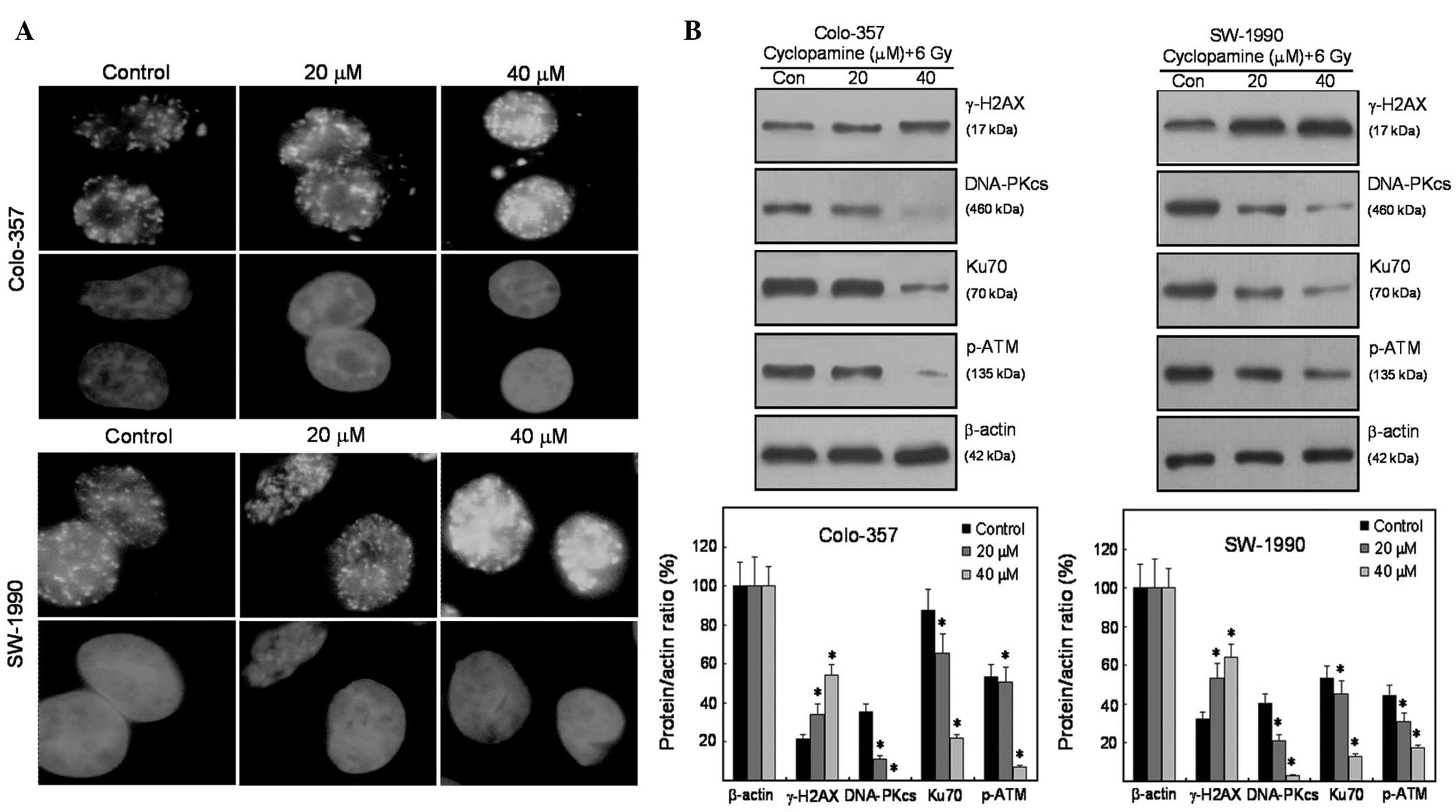
observed, particularly at the 0.5 h time point (Fig. 3B). The formation of γ-H2AX foci was
measured 0.5 h after irradiation of pancreatic cancer cells. This
procedure was conducted in order to recognize the molecular
mechanisms of cyclopamine radiosensitization. Radiation-induced
γ-H2AX foci were significantly increased in cyclopamine-treated
Colo-357 and SW-1990 cells (Fig.
4A), indicative of DNA repair inhibition. Fig. 4B shows the effect of cyclopamine on
the expression of DNA repair-related proteins. For the two cell
lines, H2AX phosphorylation was enhanced following irradiation. In
contrast to γ-H2AX, radiation-induced p-ATM, Ku70 and DNA-PKcs were
all inhibited (Fig. 4B).
Cyclopamine inhibits DNA-DSB repair in an
EGFR-dependent pathway
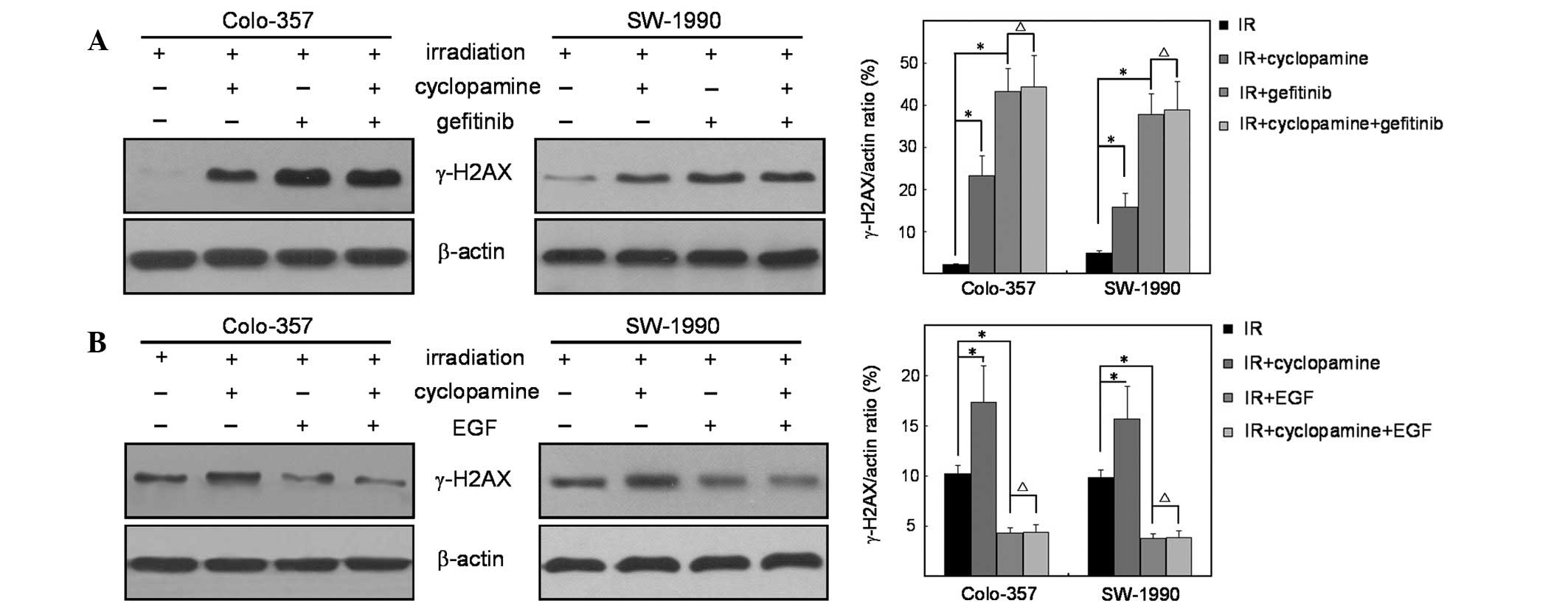
To verify the effect of EGFR in cyclopamine-induced
radiosensitivity, cells were treated with gefitinib prior to
irradiation. The expression levels of γ-H2AX showed that the
inhibitory effect of gefitinib blocked cyclopamine-induced H2AX
phosphorylation, which is consistent with the results discussed
(Fig. 5A). Furthermore, EGF
markedly inhibited cyclopamine-induced phosphorylation of H2AX
following irradiation, indicating the dependence of H2AX
phosphorylation partly through an EGFR-dependent pathway (Fig. 5B).
Discussion
Pancreatic cancer is the fourth leading cause of
cancer-related mortality and is associated with multiple
aberrations in cellular signaling cascades (16). The treatment of pancreatic cancer
is frequently met with poor outcomes due to the development of
resistance to therapy (17).
Currently, an effective treatment for pancreatic cancer is lacking
and conventional therapy has shown limited success in improving
patient survival. Therefore, understanding the mechanisms
regulating the molecular changes that drive the refractoriness to
therapy is a prerequisite for the development of effective
interventions for pancreatic cancer.
In the present study, the effect of cyclopamine on
the radiation clonogenic survival of pancreatic cancer cell lines
was analyzed. Cyclopamine treatment exerted significant
radiosensitization on K-RASwt and K-RASmt
pancreatic cancer cell lines, indicating cyclopamine-induced
radiosensitivity through a K-RAS-independent pathway. Apoptosis and
cell cycle assays showed that cyclopamine failed to affect
radiation-induced apoptosis and cell cycle redistribution. In
addition, phospho-H2AX was analyzed following ionizing radiation to
determine the underlying mechanisms. Radiation-induced γ-H2AX foci
were significantly increased in cyclopamine-treated cells. Western
blot analysis showed that radiation-induced p-ATM, Ku70 and
DNA-PKcs were all inhibited in cyclopamine-treated cells. To verify
the underlying mechanisms in cyclopamine-induced DNA-DSB repair,
cells were treated with gefitinib or EGF prior to irradiation. The
results indicated that the cyclopamine-induced activity of H2AX
occurred, in part, through an EGFR-dependent pathway.
The results of the present study provide convincing
evidence for the function of inhibited Hh pathway in pancreatic
cancer. This study may serve as a basis for clinical studies
identifying the role of cyclopamine in pancreatic cancer
radiotherapy. In conclusion, these observations indicate that the
role of cyclopamine in the radiosensitivity of pancreatic cancer
may be important for translational research on the development of
more effective and targeted therapeutic strategies for pancreatic
cancer.
Acknowledgements
This study was supported by grants from the Hospital
Center Technology Development Fund of Wuxi City (no. YGM1101) and
the Social Development Project of Kunshan City (no. KS1224).
References
|
1
|
Maitra A and Hruban RH: Pancreatic Cancer.
Annu Rev Pathol. 3:157–188. 2008. View Article : Google Scholar
|
|
2
|
Zeng H, Yu H, Lu L, et al: Genetic effects
and modifiers of radiotherapy and chemotherapy on survival in
pancreatic cancer. Pancreas. 40:657–663. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stan SD, Singh SV and Brand RE:
Chemoprevention strategies for pancreatic cancer. Nat Rev
Gastroenterol Hepatol. 7:347–356. 2010.PubMed/NCBI
|
|
4
|
Mahindroo N, Punchihewa C and Fujii N:
Hedgehog-Gli signaling pathway inhibitors as anticancer agents. J
Med Chem. 52:3829–3845. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mazumdar T, Devecchio J, Agyeman A, et al:
Blocking hedgehog survival signaling at the level of the GLI genes
induces DNA damage and extensive cell death in human colon
carcinoma cells. Cancer Res. 71:5904–5914. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Feldmann G, Dhara S, Fendrich V, et al:
Blockade of hedgehog signaling inhibits pancreatic cancer invasion
and metastases: a new paradigm for combination therapy in solid
cancers. Cancer Res. 67:2187–2196. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kumar SK, Roy I, Anchoori RK, et al:
Targeted inhibition of hedgehog signaling by cyclopamine prodrugs
for advanced prostate cancer. Bioorg Med Chem. 16:2764–2768. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bartsch DK, Sina-Frey M, Lang S, et al:
CDKN2A germline mutations in familial pancreatic cancer. Ann Surg.
236:730–737. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Almoguera C, Shibata D, Forrester K, et
al: Most human carcinomas of the exocrine pancreas contain mutant
c-K-ras genes. Cell. 53:549–554. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Toulany M, Kasten-Pisula U, Brammer I, et
al: Blockage of epidermal growth factor
receptor-phosphatidylinositol 3-kinase-AKT signaling increases
radiosensitivity of K-RAS mutated human tumor cells in vitro by
affecting DNA repair. Clin Cancer Res. 12:4119–4126. 2006.
View Article : Google Scholar
|
|
11
|
Jiao Y, Wang HC and Fan SJ: Growth
suppression and radiosensitivity increase by HMGB1 in breast
cancer. Acta Pharmacol Sin. 28:1957–1967. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jiao Y, Ge CM, Meng QH, et al:
Adenovirus-mediated expression of Tob1 sensitizes breast cancer
cells to ionizing radiation. Acta Pharmacol Sin. 28:1628–1636.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiao Y, Sun KK, Zhao L, et al: Suppression
of human lung cancer cell proliferation and metastasis in vitro by
the transducer of ErbB-2.1 (TOB1). Acta Pharmacol Sin. 33:250–260.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Che J, Zhang FZ, Zhao CQ, et al:
Cyclopamine is a novel Hedgehog signaling inhibitor with
significant anti-proliferative, anti-invasive and anti-estrogenic
potency in human breast cancer cells. Oncol Lett. 5:1417–1421.
2013.PubMed/NCBI
|
|
15
|
Ahsan A, Hiniker SM, Davis MA, et al: The
role of cell cycle in epidermal growth factor receptor
inhibitor-mediated radiosensitization. Cancer Res. 69:5108–5114.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cloos CR, Daniels DH, Kalen A, et al:
Mitochondrial DNA depletion induces radioresistance by suppressing
G2 checkpoint activation in human pancreatic cancer cells. Radiat
Res. 171:581–587. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kimple RJ, Vaseva AV, Cox AD, et al:
Radiosensitization of EGFR/HER2 positive pancreatic cancer is
mediated by inhibition of Akt independent of Ras mutational status.
Clin Cancer Res. 16:912–923. 2010. View Article : Google Scholar : PubMed/NCBI
|