Introduction
Epilepsy is a common episodic neurological
condition. In the majority of epilepsy patients, the predominant
cause of the disorder is considered to be genetic rather than
having an extraneous cause (1,2). The
majority of epilepsy phenotypes are the product of interactions
between genetic and environmental factors. A recent study focused
on genetic variations that may affect the aetiology, prognosis and
effects of epilepsy, such as the correlation between the condition
and gene polymorphisms (1). To
date, studies have shown that genetic mutations are correlated with
epilepsy, particularly mutations of the sodium channel (1,3,4).
Voltage-gated sodium channels are crucial for
membrane excitability. Mutations in the genes that code for the
channel components are regarded to be key factors for epilepsy
phenotypes. Voltage-gated sodium ion channels consist of two types
of subunits, α and β subunits. Each α subunit is associated with
one or more β subunit in order to form functional voltage-gated ion
channels. These voltage-gated sodium channels are expressed in
neurons and glia throughout the central and peripheral nervous
system and have been highly conserved during the evolution of
invertebrates and vertebrates (5,6). A
SCN2A mutation has been identified to be associated with seizures,
ataxia and a sensitivity to pain in epilepsy patients and patients
with other neurological disorders (7). In mice, a mutation in the sodium
channel SCN2A gene resulted in seizures and repetitive behaviors
due to a persistent current (8).
Altered sodium channel transcript levels in epilepsy patients
reveal a potential role for sodium channels in the pathophysiology
of epilepsy (9). Although a
mutation in the sodium channel component has been demonstrated to
induce epilepsy, the effect of altered SCN2A expression in epilepsy
requires clarification.
In animals, oxygen radicals are produced as the
byproducts of a normal oxidative metabolism (10). Therefore, in activated cells, more
oxygen radicals were produced. Furthermore, neurons and glia are
produced and reactive oxygen species are released (11). It has long been regarded that a
controlled intracellular redox environment is crucial for proper
cellular function. In order for cells to protect themselves from
constant oxidative toxicity, cells have developed a defense system
that ensures a balance between pro- and antioxidant molecules
(12). Cu-Zn superoxide dismutase
(Cu-Zn SOD) is a key enzyme in the dismutation of superoxide
radicals, which result from the cellular oxidative metabolism
process of cells, converting them into hydrogen peroxide (13). Increasing evidence suggests a role
for oxidative stress in the manifestation of epilepsy (14–17).
Sudha et al(14) reported
that epileptic patients exhibited a low blood antioxidant status
compared with the controls, suggesting that free radicals may be
implicated in epilepsy. Ercegovac et al(18) speculated that the
post-translational modification of existing functional proteins,
particularly alterations to ion channels, may be partially
responsible for the acute early changes in neuronal networks.
However, the evidence that ion channels regulate oxidative stress
action in patients with epilepsy remains limited.
In this study, the levels of SCN2A gene expression
and the concentration of Cu-Zn SOD were investigated in the
cerebral cortexes of patients with primary and secondary temporal
lobe epilepsy and in normal brain cortex tissue. Furthermore, the
changes in the SCN2A gene and Cu-Zn SOD concentration were analyzed
following transfection with the si-SCN2A vector. In addition,
co-transfection with the si-SCN2A vector and the overexpressed
Cu-Zn SOD vector was performed in order to illustrate the effect of
Cu-Zn SOD vector therapy on temporal lobe epilepsy.
Materials and methods
Subjects
In total, 20 patients with temporal lobe epilepsy
(age, 35.12±10.69 years; 10 with primary and 10 with secondary)
were recruited from the outpatient clinics of the Second Xiangya
Hospital, Central South University (Changsha, China). A further 20
healthy controls (age, 40.59±9.77 years) were recruited from the
medical staff of the Second Xiangya Hospital. This study was
approved by the human ethics committee of the Central South
University Xiangya Medical School (Changsha, China). Written
informed consent was obtained from all patients/patients'
families.
Quantitative polymerase chain reaction
(qPCR)
In this study the correlation between SCN2A mRNA and
the temporal lobe was analyzed. All primers and probes were
designed by Applied Biosystems (Carlsbad, CA, USA), and were
hybridized between exons in order to avoid the amplification of
genomic DNA. Total RNA isolation was performed using RNA TRIzol
according to the manufacturer's instructions (Invitrogen Life
Technologies, Carlsbad, CA, USA). By employing the Takara cDNA
Library Construction kit, 1 μg total RNA was used to synthesize
cDNA according to the manufacturer's instructions (Takara Bio,
Inc., Shiga, Japan). The transcription levels for SCN2A and β-actin
(a housekeeping gene) were quantified by the ABI 7500 realtime PCR
system (Applied Biosystems). The amplification conditions were as
follows: 95°C for 10 min, followed 40 cycles of 15 sec at 95°C and
1 min at 60°C, using TaqMan® Universal PCR Master mix
(Applied Biosystems). All results were normalized to the levels of
β-actin RNA (TaqMan probes, Applied Biosystems). The relative
expression level was calculated using the 2−ΔΔCt
method.
Western blot analysis
Samples were separated in 10% SDS-PAGE gels and
transferred to a polyvinylidene fluoride (PVDF) membrane
(Millipore, Billerica, MA, USA). After blocking with 4% non-fat
milk, SCN2A was detected by incubation with monoclonal anti-SCN2A
antibody (SAB5200074, Sigma-Aldrich, St. Louis, MO, USA). β-actin
was detected by the monoclonal anti β-actin antibody (A1978,
Sigma-Aldrich). The anti-mouse IgG secondary antibody was
conjugated to horseradish peroxidase and an enhanced
chemiluminescent substrate (ECL plus, Amersham Pharmacia Biotech,
Piscataway, NJ, USA) for signal development. The image was captured
using a Fuji Film FLA 5000 image reader (Fuji Film, Stamford, CT,
USA).
Cu-Zn SOD detection
Tissues and cells were collected for enzyme
concentration assays. Prior to performing the assays, the samples
were thawed on ice and homogenized in PBS (Polytron PT3100;
Kinematica AG, Littan, Switzerland). Cu-Zn SOD activities were
assayed using a Cu/Zn Superoxide Dismutase ELISA kit (S2147,
Sigma-Aldrich) according to the manufacturer's
instructions.
Single-cell patch clamp technique
The single-cell patch clamp technique was performed
using the procedure as reported previously by Fertig et
al(19). Briefly, before
recordings, cells were transferred to a BX51WI (Olympus, Tokyo,
Japan) upright microscope (equipped with infrared video microscopy)
and incubated in DMEM/F12, 0.075% BSA, Pen/Strep/antimycotic, and
14 mM HEPES, pH 7.4. Individual cells were selected for recordings
based on small round or ovoid cell bodies (diameter, 5–10 μm) and
typically two or more extended processes. For each test, five
patients with primary temporal lobe epilepsy and five with
secondary temporal lobe epilepsy were involved. The methods used
for cell culturing were also used to record the concentration of
electrophysiological intracellular Ca2+. Cells were
differentiated by exposure to the retinoic acid analogue,
4-[(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-napthalenyl), for six
days. Whole cell patch clamp recordings were performed at room
temperature from single cells, which were continually superperfused
(2–3 ml/min) with phosphate-buffered saline (PBS). Ca2+
channel currents were elicited by stepping the membrane potential
from a holding potential of −90 mV to +10 mV for 45 msec every 15
sec. Cells were continually perfused with a solution containing:
NaCl 140 mM, KCl 2 mM, CaCl2 2.5 mM, MgCl2 1
mM, HEPES 10 mM, glucose 10 mM, sucrose 40 mM and bovine serum
albumin 0.05%.
Vector design and transient
transfection
In order to compare the biophysical properties
following the knockdown of SCN2A, the SCN2A interference vector was
constructed following the protocol of a study by Tahiliani et
al(20). To investigate the
effect of Cu-Zn SOD on SH-SY5Y cells following transfection with a
si-SCN2A vector, the overexpressed Cu-Zn SOD vector was constructed
following the method provided by Hu et al(21).
SH-SY5Y cells, grown under standard culture
conditions (5% CO2, 37°C) in Dulbecco's modified Eagle's
medium supplemented with 10% fetal bovine serum, were transiently
transfected with plasmids using Lipofectamine 2000 (Invitrogen Life
Technologies) according to the manufacturer's instructions.
Following 24 h of transfection, the cells were harvested and used
in the following experiments.
Immunofluorescence
SH-SY5Y cells were grown on 24 × 24 mm cover glasses
and subsequently fixed in 4% paraformaldehyde solution in phosphate
buffer for 30 min prior to 30 min incubation with a blocking
reagent [5% fetal bovine serum in PBS]. The primary antibodies of
SCN2A (anti-SCN2A, SAB5200074; Sigma-Aldrich) were incubated
overnight at 4°C prior to being washed with PBS. Immunofluorescence
staining was performed with secondary antibodies conjugated to
fluorescein isothiocyanate (F5262, Sigma-Aldrich). A conventional
fluorescence microscope (Axiovert 100; Carl Zeiss, Oberkochen,
Germany) was used for visualization.
Effects of SCN2A interference on SH-SY5Y
cells
To investigate the biophysical properties of SH-SY5Y
cells following SCN2A knockdown, the cells were planted into
24-well plates. The wells were divided into three groups (8
wells/group): The control group, the empty vector control group and
the si-SCN2A vector group. Following transfection, SCN2A expression
was detected by qPCR, western blotting and immunofluorescence.
Cu-Zn SOD concentrations were assayed. Furthermore, single-cell
patch clamp techniques were used to analyze the
electrophysiological changes of cells in the three groups.
Effect of Cu-Zn SOD on SCN2A knockdown in
SH-SY5Y cells
The effect of Cu-Zn SOD on SH-SY5Y cells following
transfection with a si-SCN2A vector. The wells were divided into
four groups (6 wells/group), including the control group, the empty
vector control group, the si-SCN2A vector group and the si-SCN2A
vector + Cu-Zn SOD vector group. Following 24 h of transfection,
SCN2A expression was analyzed by qPCR, western blotting and
immunofluorescence. Cu-Zn SOD concentrations and cell
electrophysiological changes were also assayed using the
single-cell patch clamp techniques.
Statistical analysis
All values are provided as the mean ± SD. Continuous
variables that did not have a Gaussian distribution underwent log
transformation. Student's t-test was used to compare the
differences between groups. One-way analysis of variance was used
to determine the differences among the groups. P≤0.05 was
considered to indicate a statistically significant difference. If
F-ratios exceeded the critical value (P≤0.05), Newman-Keul's post
hoc test was performed in order to compare the groups.
Results
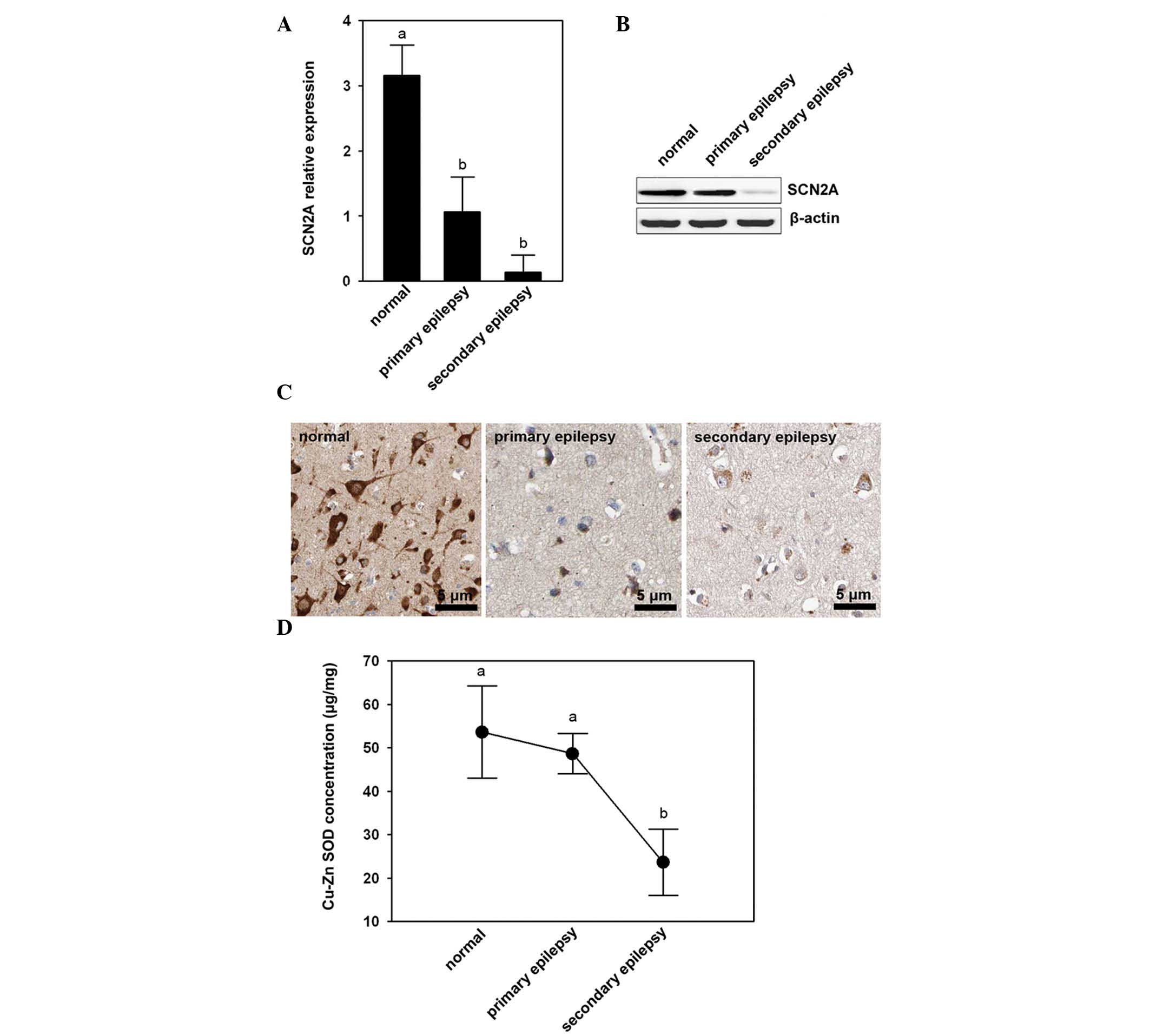
Lower levels of SCN2A expression and low
Cu-Zn SOD content in the cortex of patients with temporal lobe
epilepsy
The differential expression of SCN2A was assessed in
the cerebral cortex of patients with primary and secondary temporal
lobe epilepsy and normal brain cortex tissue. Using qPCR, it was
confirmed that of the three different types of tissues, the normal
brain cortex tissue demonstrated a markedly higher expression of
SCN2A transcripts (P<0.05), while the cerebral cortex of
patients with primary and secondary temporal lobe epilepsy
exhibited lower expression of SCN2A transcripts (Fig. 1A). Furthermore, the cerebral cortex
of patients with secondary temporal lobe epilepsy had weaker
western blotting reactions compared with normal brain cortex tissue
and cerebral cortex of patients with primary temporal lobe epilepsy
(Fig. 1B). Similar to the studies
of mRNA and protein expression levels, immunofluorescence revealed
that the cerebral cortex of patients with primary and secondary
temporal lobe epilepsy had weaker signals (Fig. 1C) when compared with normal brain
cortex tissue.
Cu-Zn SOD concentration in the three
tissue types
The results showed that the cerebral cortexes in
patients with secondary temporal lobe epilepsy had lower Cu-Zn SOD
concentrations than the normal brain cortex tissue (P<0.05) and
cerebral cortex of patients with primary temporal lobe epilepsy
(Fig. 1D).”
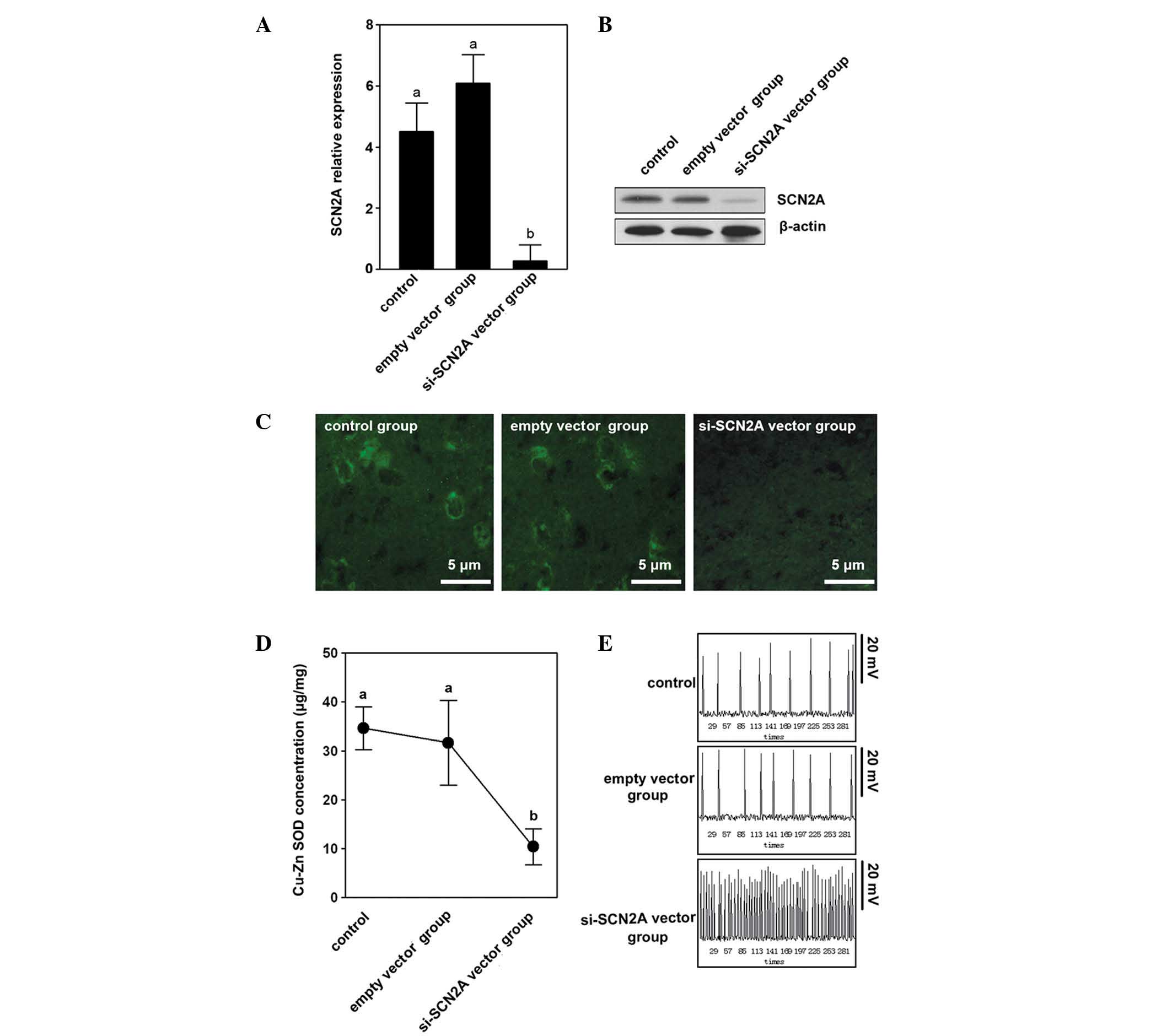
SCN2A knockdown suppresses Cu-Zn SOD
In order to elucidate the regulation of SCN2A by
Cu-Zn SOD, the SCN2A interference vector was used to transfect
SH-SY5Y cells and detected the concentration of Cu-Zn SOD in the
different groups. SCN2A expression was determined by qPCR, western
blotting and immunofluorescence. The control and empty vector
control groups demonstrated higher mRNA and protein expression
levels compared with the si-SCN2A vector group (Fig. 2A and B). The immunofluorescence
result indicated that the si-SCN2A vector group had weaker signals
compared with the control and the empty vector control groups
(Fig. 2C). A notable difference
was identified in the si-SCN2A vector group among the three groups
that were studied.
The Cu-Zn SOD concentration in the si-SCN2A vector
group was 11.23±4.52 μg/mg, which was significantly lower than the
control and empty vector control groups (P<0.05; Fig. 2D).
The control and empty vector control groups showed a
stable resting membrane electric potential (53.34±3.25 mV and
52.68±3.48 mV) with a discharge frequency of approximately once
every minute. However, the si-SCN2A vector group showed repeated
discharge, which was similar to the phenomenon of epilepsy. The
volatility (between 120–150 mV) and frequency (6–10 times/min) were
regular (Fig. 2E).
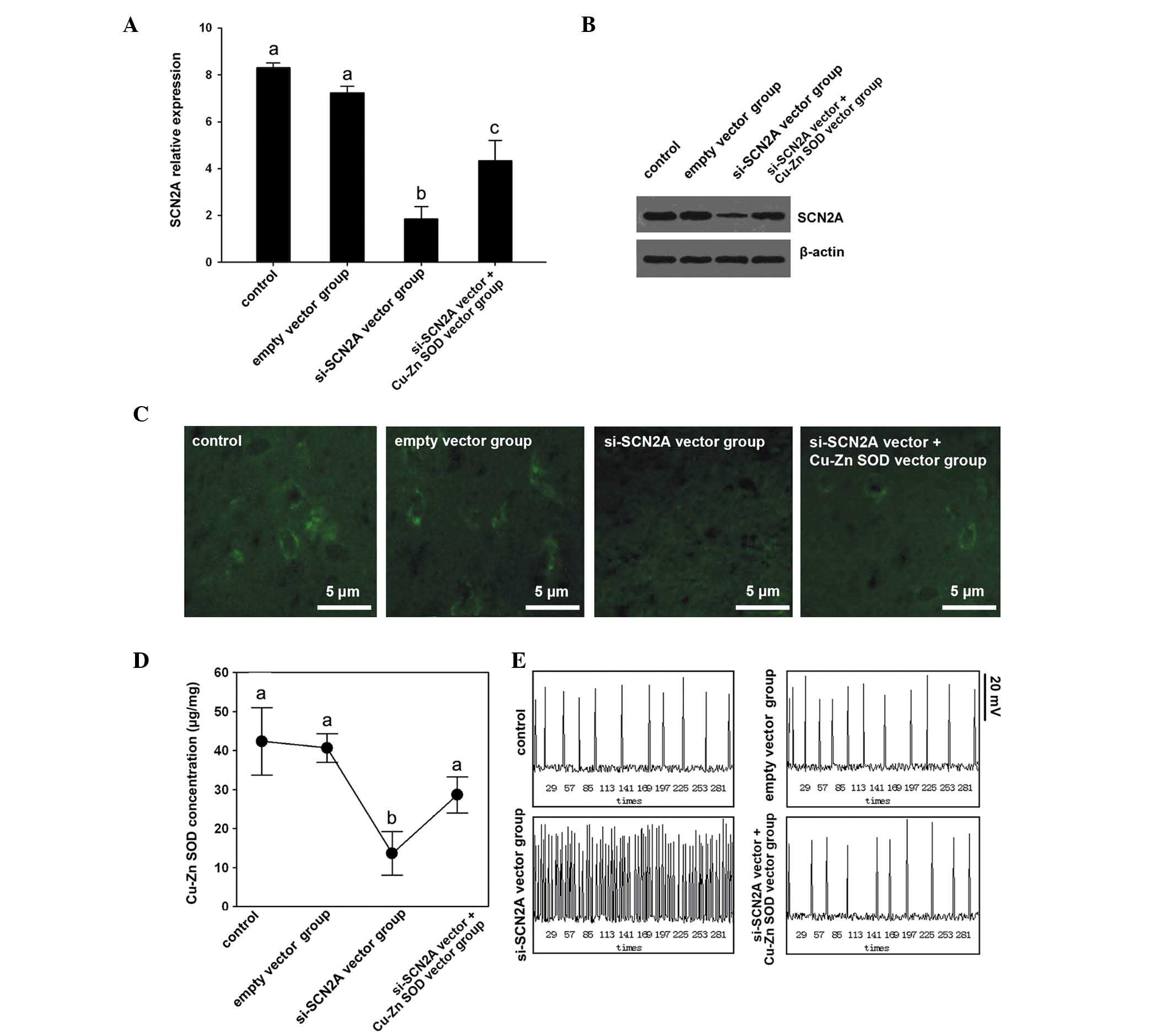
Poor efficacy of Cu-Zn SOD in SCN2A
knockdown SH-SY5Y cells
The response of SH-SY5Y cells following the
knockdown of SCN2A was also evaluated in order to assess the
efficacy of Cu-Zn SOD with a deficiency of SCN2A. Four groups were
employed in this study, including the control, the empty vector
control, the si-SCN2A vector and the si-SCN2A vector + Cu-Zn SOD
vector groups. The expression levels of SCN2A were assayed in order
to confirm the silencing efficiency. mRNA and protein levels showed
a significant decrease following si-SCN2A vector transfection
(Fig. 3A and B). Notably, SCN2A
mRNA expression levels were significantly lower in the si-SCN2A
vector group compared with the si-SCN2A vector + Cu-Zn SOD vector
group (Fig. 3A and B).
Furthermore, the distribution results of SCN2A (Fig. 3C) indicated that the si-SCN2A
vector group and the si-SCN2A vector + Cu-Zn SOD vector group had
weaker signals compared with the other two groups.
Cu-Zn SOD concentrations of the
groups
The si-SCN2A vector group exhibited the lowest
concentration of Cu-Zn SOD compared with the si-SCN2A vector +
Cu-Zn SOD vector group, the control group and empty vector group
(Fig. 3D). The empty vector
control group had the highest concentration of Cu-Zn SOD while the
lowest concentration of Cu-Zn SOD was observed in the si-SCN2A
vector group (Fig. 3D).
The resting membrane electric potential of the
si-SCN2A vector group was similar to that identified in a previous
study (8). Notably, the
overexpression of Cu-Zn SOD rescued the repeated discharge caused
by si-SCN2A, which demonstrated that Cu-Zn SOD is beneficial for
the low expression of SCN2A in SH-SY5Y cells (Fig. 3E).
Discussion
The correlation between voltage-gated sodium
channels and epilepsy has been widely reported (22). The Scn2aQ54 transgenic line may be
capable of inducing sodium-channel-dependent epilepsy in a mouse
model (8). In this study, Scn2aQ54
heterozygotes exhibited adult-onset, progressive epilepsy that
begins with partial seizures of short duration of hippocampal
origin. The causal mutation, GAL879–881QQQ, in the cytoplasmic
S4–S5 linker of domain 2 results in delayed channel inactivation
and increased persistent current. Thus, the genome mutation has
been revealed. However, the SCN2A expression changes in epilepsy
have not been discussed, particularly with reference to the
cerebral cortexes of patients with primary or secondary temporal
lobe epilepsy and normal brain cortex tissues. In the present
study, in terms of RNA levels and protein levels, normal brain
cortex tissues exhibited a higher expression of SCN2A than the
cerebral cortexes of patients with primary or secondary temporal
lobe epilepsy. This result suggested that in epileptic tissues,
there is a lower level of expression of SCN2A. In organisms,
voltage-gated calcium channels allow for the conversion of
electrical signals to chemical signals, which, due to the input of
calcium ions (Ca2+) then induce the release of
neurotransmitters from synaptic vesicles (23). SCN2A encodes the α-subunit of the
voltage-gated sodium channel (24). Thus, the lower levels of SCN2A
expression may contribute to disorder in the transmission of
electrical signals to chemical signals in the cortex, which induces
pathological changes. Similar results were observed in a study of
the Cu-Zn SOD concentration that was higher in epileptic tissues
than normal brain cortex tissue (25). Cu-Zn SOD has been regarded as the
first-line of defense against free radical damage (26). Abnormal concentrations of Cu-Zn SOD
may induce neurotoxicity, which results in exacerbations (27). Subsequently, the Cu-Zn SOD
concentrations were investigated following SCN2A knockdown in
SH-SY5Y cells in order to understand the correlation between Cu-Zn
SOD and SCN2A.
Following SCN2A knockdown with transfection of the
si-SCN2A vector, SCN2A expression decreased significantly. These
results demonstrated that the SCN2A knockdown was efficient and
successful. Furthermore, the result also demonstrated that the
concentration of Cu-Zn SOD was downregulated following a reduction
in the levels of SCN2A. To date, little information has been
provided regarding the effect of SCN2A expression on Cu-Zn SOD in
organisms or cells. Therefore, the present study provided a novel
insight in to Cu-Zn SOD and its regulation by SCN2A in epilepsy;
the decrease in SCN2A expression may reduce the concentration of
Cu-Zn SOD in the brain cortex, which contributes to serious trauma
to the cortex. The resting membrane electric potential was also
analyzed by single-cell patch clamp. The cells, which had been
transfected with the si-SCN2A vector, exhibited a repeated
discharge, similar to that identified in epilepsy. The result
suggested that the low expression of SCN2A in the cerebral cortexes
of patients with primary and secondary temporal lobe epilepsy
induced the repeated discharge. Thus, the upregulation of SCN2A
expression in the cortex may be an effective treatment for the
repeated discharge that occurs in epilepsy.
It has been reported that Cu-Zn SOD is crucial in
preventing oxidative damage to cells (28). Thus, a decrease in Cu-Zn SOD levels
in the cortex induces damage and a Cu-Zn SOD supplement may repair
the damage caused. In addition, it was observed that in cortex
tissue of epileptics, the SCN2A expression was abnormal, with a
lower expression than that in normal cortex tissue. Subsequently,
the protective effect of Cu-Zn SOD on SH-SY5Y cells was analyzed
following SCN2A knockdown. Notably, the Cu-Zn SOD supplement group
demonstrated a higher level of SCN2A expression compared with the
group that received no treatment, which indicated that the addition
of Cu-Zn SOD may be capable of reducing the depression induced by
SCN2A knockdown. As demonstrated, the low expression of SCN2A
induced repeated discharge. Subsequently, the effect of Cu-Zn SOD
overexpression on si-SCN2A-induced repeated discharge was observed.
Notably, the overexpression of Cu-Zn SOD prevented the repeated
discharge due to si-SCN2A. Thus, it was presumed that Cu-Zn SOD may
be capable of treating the repeated discharge, which may be a
feasible treatment strategy for epilepsy.
In the present study, it was confirmed that SCN2A
expression was lower in epileptic cerebral cortexes. This change
may induce pathological changes in the cortex tissue. In SH-SY5Y
cells, SCN2A expression was downregulated artificially by knockdown
technology. With the low expression of SCN2A in the cell lines the
concentration of Cu-Zn SOD also decreased. No information has been
reported regarding the regulation of Cu-Zn SOD in the cerebral
cortex by SCN2A thus far. SCN2A variation in the genome or
expression levels leads to tissue injury, due to the abnormal
release of neurotransmitters from synaptic vesicles. Thus, in the
cerebral cortex, tissue injury is caused by SCN2A disorder and the
downregulation of Cu-Zn SOD concentration. However, whether SCN2A
may be capable of regulating Cu-Zn SOD directly requires further
study. Conversely, Cu-Zn SOD was capable of rescuing SCN2A in SCN2A
knockdown cell lines. This result indicates that a Cu-Zn SOD
supplement may be an effective therapy for epilepsy but the
mechanism underlying this requires further study. Furthermore, it
was demonstrated that Cu-Zn SOD may prevent the repeated discharge
that is caused by the low expression of SCN2A.
In conclusion, a significant decline in the
expression of SCN2A and the concentration of Cu-Zn SOD was observed
in the cerebral cortex of epileptic patients and the decrease in
SCN2A expression may be restored by increasing the Cu-Zn SOD.
However, further investigation and a larger cohort are required to
investigate the underlying molecular mechanism.
References
|
1
|
Lakhan R, Kumari R, Misra UK, Kalita J,
Pradhan S and Mittal B: Differential role of sodium channels SCN1A
and SCN2A gene polymorphisms with epilepsy and multiple drug
resistance in the North Indian population. Br J Clin Pharmacol.
68:214–220. 2009. View Article : Google Scholar
|
|
2
|
Clary HM: Promising New Directions in
Epilepsy Management. Neurology. 21:21–25. 2012.
|
|
3
|
Estacion M, Gasser A, Dib-Hajj SD and
Waxman SG: A sodium channel mutation linked to epilepsy increases
ramp and persistent current of Nav1.3 and induces hyperexcitability
in hippocampal neurons. Exp Neurol. 224:362–368. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liao Y, Anttonen AK, Liukkonen E, et al:
SCN2A mutation associated with neonatal epilepsy, late-onset
episodic ataxia, myoclonus, and pain. Neurology. 75:1454–1458.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Felts P, Yokoyama S, Dib-Hajj S, Black J
and Waxman SG: Sodium channel α-subunit mRNAs I, II, III, NaG, Na6
and hNE (PN1): different expression patterns in developing rat
nervous system. Brain Res Mol Brain Res. 45:71–82. 1997.
|
|
6
|
Whitaker WR, Clare JJ, Powell AJ, Chen YH,
Faull RL and Emson PC: Distribution of voltage-gated sodium channel
α-subunit and β-subunit mRNAs in human hippocampal formation,
cortex and cerebellum. J Comp Neurol. 422:123–139. 2000.
|
|
7
|
Meisler MH and Kearney JA: Sodium channel
mutations in epilepsy and other neurological disorders. J Clin
Invest. 115:2010–2017. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kearney JA, Plummer NW, Smith MR, et al: A
gain-of-function mutation in the sodium channel gene Scn2a results
in seizures and behavioral abnormalities. Neuroscience.
102:307–317. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lombardo AJ, Kuzniecky R, Powers RE and
Brown GB: Altered brain sodium channel transcript levels in human
epilepsy. Brain Res Mol Brain Res. 35:84–90. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Malmström BG: Enzymology of oxygen. Annu
Rev Biochem. 51:21–59. 1982.
|
|
11
|
Dugan LL, Sensi SL, Canzoniero LM, et al:
Mitochondrial production of reactive oxygen species in cortical
neurons following exposure to N-methyl-D-aspartate. J Neurosci.
15:6377–6388. 1995.PubMed/NCBI
|
|
12
|
Forman HJ and Torres M: Redox signaling in
macrophages. Mol Aspects Med. 22:189–216. 2001. View Article : Google Scholar
|
|
13
|
Fridovich I: The biology of oxygen
radicals. Science. 201:875–880. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sudha K, Rao AV and Rao A: Oxidative
stress and antioxidants in epilepsy. Clin Chim Acta. 303:19–24.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mori A, Hiramatsu M, Yokoi I and Edamatsu
R: Biochemical pathogenesis of post-traumatic epilepsy. Integr
Physiol Behav Sci. 25:54–62. 1990.
|
|
16
|
Nazıroglu M: Role of selenium on calcium
signaling and oxidative stress-induced molecular pathways in
epilepsy. Neurochem Res. 34:2181–2191. 2009.PubMed/NCBI
|
|
17
|
Waldbaum S and Patel M: Mitochondria,
oxidative stress, and temporal lobe epilepsy. Epilepsy Res.
88:23–45. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ercegovac M, Jovic N, Simic T, et al:
Byproducts of protein, lipid and DNA oxidative damage and
antioxidant enzyme activities in seizure. Seizure. 19:205–210.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fertig N, Blick RH and Behrends JC: Whole
cell patch clamp recording performed on a planar glass chip.
Biophys J. 82:3056–3062. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tahiliani M, Mei P, Fang R, et al: The
histone H3K4 demethylase SMCX links REST target genes to X-linked
mental retardation. Nature. 447:601–605. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hu Y, Rosen DG, Zhou Y, et al:
Mitochondrial manganese-superoxide dismutase expression in ovarian
cancer: role in cell proliferation and response to oxidative
stress. J Biol Chem. 280:39485–39492. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Scheffer IE and Berkovic SF: The genetics
of human epilepsy. Trends Pharmacol Sci. 24:428–433. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Michaelis EK: Molecular biology of
glutamate receptors in the central nervous system and their role in
excitotoxicity, oxidative stress and aging. Prog Neurobiol.
54:369–415. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Goldin AL: Evolution of voltage-gated
Na+channels. J Exp Biol. 205:575–584. 2002.PubMed/NCBI
|
|
25
|
Akbas SH, Yegin A and Ozben T: Effect of
pentylenetetrazol-induced epileptic seizure on the antioxidant
enzyme activities, glutathione and lipid peroxidation levels in rat
erythrocytes and liver tissues. Clin Biochem. 38:1009–1014. 2005.
View Article : Google Scholar
|
|
26
|
Ansenberger-Fricano K, Ganini DS, Mao M,
et al: The peroxidase activity of mitochondrial superoxide
dismutase. Free Radic Biol Med. 54:116–124. 2012. View Article : Google Scholar
|
|
27
|
Lobo V, Patil A, Phatak A and Chandra N:
Free radicals, antioxidants and functional foods: Impact on human
health. Pharmacogn Rev. 4:118–126. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Simonian NA and Coyle JT: Oxidative stress
in neurodegenerative diseases. Annu Rev Pharmacol Toxicol.
36:83–106. 1996. View Article : Google Scholar : PubMed/NCBI
|