Introduction
Hemophilia A (HA) is an X-linked recessive
congenital coagulation disorder caused by a factor VIII (FVIII)
deficiency, which results from mutations in the FVIII gene (F8). HA
typically occurs in males with an incidence of ~1 in 5000 (1). Female HA patients have rarely been
reported, although they may carry a defective allele. Human F8 maps
to the long arm of the Xq28 region of the X chromosome and consists
of 26 exons and 25 introns. The protein product of F8 possesses no
enzyme activity. The 19 amino acid signal peptide at the
N-terminus, encoded by an open reading frame (ORF), leads to the
passage of FVIII through hepatocytes to blood vessels (2). The matured FVIII protein is composed
of 2332 amino acids and comprises three homologous A domains, two
homologous C domains and a single B domain. These domains, arranged
as A1-A2-B-A3-C1-C2 from the N- to the C-terminus, are important
for FVIII function. FVIII synthesized in hepatocytes is secreted
into the plasma in the form of an inactive pro-cofactor (3). FVIII, an essential cofactor for
coagulation factor IX, readily binds to von Willebrand factor
(vWF), forming a tight non-covalent complex that protects against
degradation. Furthermore, FVIII may also interact with a number of
proteins, such as thrombin and factor X. These interactions are
indispensable for effective coagulation. Once FVIII is cleaved by
thrombin, activated FVIII dissociates from vWF and participates in
the coagulation cascade (4).
Genetic abnormalities in F8 may result in
qualitative or quantitative defects of the FVIII protein. The most
common F8 abnormality to cause HA is an inversion of intron 1 or 22
(5,6). The remaining HA cases are caused by
numerous point mutations spread throughout the gene, including
missense, nonsense, splice site, frameshift mutations and gross
deletions (7,8). To date, >1400 F8 mutations within
the FVIII coding and non-coding regions have been identified in the
Haemophilia A Mutation, Search, Test and Resource Site (HAMSTeRS)
database (http://hadb.org.uk/).
In this study, we report the case of a female HA
patient with F8 compound heterozygote mutations. Diagnosis and
differential diagnosis were complex. Furthermore, F8 compound
heterozygote mutations (c.1505T>A, p.Val502Asp and c.3279G>A,
p.Met1093Ile) were identified using multiplex PCR and DNA
sequencing.
Case report
Patient history
A 37-year-old female patient presented with an
8-month history of severe uterine abnormal bleeding following
surgery for the removal of an intrauterine device. After a short
time, the patient developed profound anemia (hemoglobin level of 58
g/l). Prior to being admitted to our hospital, the patient had
received a blood transfusion (1200 ml) for hemorrhagic anemia in
the Second Hospital of Hebei Medical University. Furthermore, the
patient complained of repeated skin purpura since childhood. The
patient had no other history of bleeding, such as nosebleeds,
gingival bleeding, hematemesis or melena. There was no personal or
family history of bleeding disorders. The patient denied any intake
of medicines or herbs. The biochemical and hematological data of
the patient are shown in Table
I.
 | Table IInitial biochemistry and hematology
results. |
Table I
Initial biochemistry and hematology
results.
| Variable | Value | Reference range |
|---|
| Whole blood |
| White blood
cell |
5.8×109/l |
type4/xref>–10)x109/l |
| Hemoglobin | 92 g/l | 110–155 g/l |
| Platelet |
216×109/l |
(100–300)×109/l |
| Coagulation
panel |
| PT | 14.6 sec | 12–16 sec |
| INR | 1.12 | 0.8–1.2 |
| APTT | 46.5 sec | 24–37sec |
| APTT-R | 1.62 | 0.8–1.2 |
| Fib | 242 mg/dl | 200–400 mg/dl |
| BT | 11 min | 4.8–9 min |
| FVIII:C | 4.7% | 50–150% |
| vWF-Ag | 128% | 60–150% |
| LA | 29.5 sec | 27–41 sec |
| RIPA (1.2
mg/ml) | 91% | 87–102% |
| Factor VIII
antibody | Negative | Negative |
White blood cell and platelet levels were within the
normal ranges. Hemoglobin was only marginally reduced (92 g/l), due
to the blood transfusion. The prothrombin time (PT) and
international normalized ratio (INR) were normal, but the activated
partial thromboplastin time (APTT) and bleeding time (BT) were
noticeably prolonged, which may be corrected easily by a plasma
transfusion. Plasma FVIII levels were significantly decreased
(4.7%). vWF antigen levels were within the normal range (128%).
Autoimmune markers, such as the antinuclear, anticardiolipin,
antidouble stranded DNA and extractable nuclear antigen (ENA)
polypeptide antibodies were all negative. Other coagulation tests
were also normal, including platelet count and function, and
fibrinogen levels.
Diagnosis and differential diagnosis
Based on the clinical manifestations and laboratory
tests, the following diagnoses were considered, in order of
decreasing probability: i) Acquired HA (AHA); ii) von Willebrand
disease type 2N (vWD-Type 2N); and iii) HA. There is a certain
degree of overlap in the clinical manifestations and laboratory
results among these three disorders. Although female HA is
extremely rare, it could not be ruled out as a possibility. The key
points of diagnosis and differential diagnosis for these three
diseases are discussed in the following paragraphs.
AHA is an autoimmune disorder caused by
autoantibodies against FVIII, neutralizing its coagulation
functions and resulting in severe, often life-threatening bleeding.
It is characterized by severe, spontaneous hemorrhaging at sites,
such as the skin, muscle and soft tissues, or by excessive bleeding
during surgery. Hemarthrosis, the hallmark of severe congenital HA,
seldom occurs in AHA patients (9).
AHA may be associated with several clinical conditions, including
pregnancy, autoimmune diseases, malignancies, infections or drugs.
Approximately 50% of patients are idiopathic with no known
underlying disease association (10). If a patient presents with
spontaneous hemorrhaging, then previous personal or family history
of bleeding must be ruled out. Once laboratory results reveal the
isolated prolongation of APTT and reduced FVIII levels, which fail
to normalize following a normal plasma transfusion, AHA should be
suspected. Following confirmation that FVIII antibodies are absent
and that FVIII levels may be improved by transfusing normal plasma,
a diagnosis of AHA was definitively ruled out.
vWD is the most common inherited bleeding disorder
and is caused by deficiency or dysfunction of vWF. vWD is divided
into three types: Type 1 (partial quantitative deficiency); Type 2,
with four subtypes 2A, 2B, 2M and 2N (qualitative deficiency); and
Type 3 (complete quantitative deficiency) (11). Type 2N vWD show a marked decrease
in the vWF binding affinity for FVIII. Its clinical manifestations
are similar to that of mild HA: FVIII levels are decreased, but vWF
antigen (vWF:Ag) levels are within the normal range (12). These patients are usually
misdiagnosed as HA. When the laboratory results were returned, it
was observed that the vWF binding affinity for FVIII and the
ristocetin-induced platelet agglutination (RIPA) were normal in our
patient. Therefore, a diagnosis of vWD-Type 2N was ruled out. Thus,
a diagnosis of HA was considered for the patient. The differential
diagnoses of HA, VWD-Type 2N and AHA are outlined in Table II.
| Table IIDifferential diagnosis of HA, vWD-Type
2N and AHA. |
Table II
Differential diagnosis of HA, vWD-Type
2N and AHA.
| Variable | HA | vWD-Type 2N | AHA |
|---|
| BT | Normal | Normal or (↑) | Normal |
| APTT | ↑ | ↑ | ↑ |
| FVIII:C | ↓ | ↓ | ↓ |
| vWF-Ag | Normal | (↓) or Normal | Normal |
| FVIII inhibitor | Negative | Negative | Positive |
| vWF-FVIII binding
affinity | Normal | ↓ | Normal |
| vWF:Rcof | Normal | (↓) or Normal | Normal |
Mutational screening
In order to confirm the cause of HA in the patient,
a mutational screening of F8 was performed. Genomic DNA was
obtained from EDTA-anticoagulated blood from the patient.
Inversions in introns 1 and 22 were first screened using
long-distance PCR (13). All exons
and their flanking regions of F8 were amplified by PCR using
specific primers, as described previously (14). PCR products were purified and
sequenced.
DNA sequencing
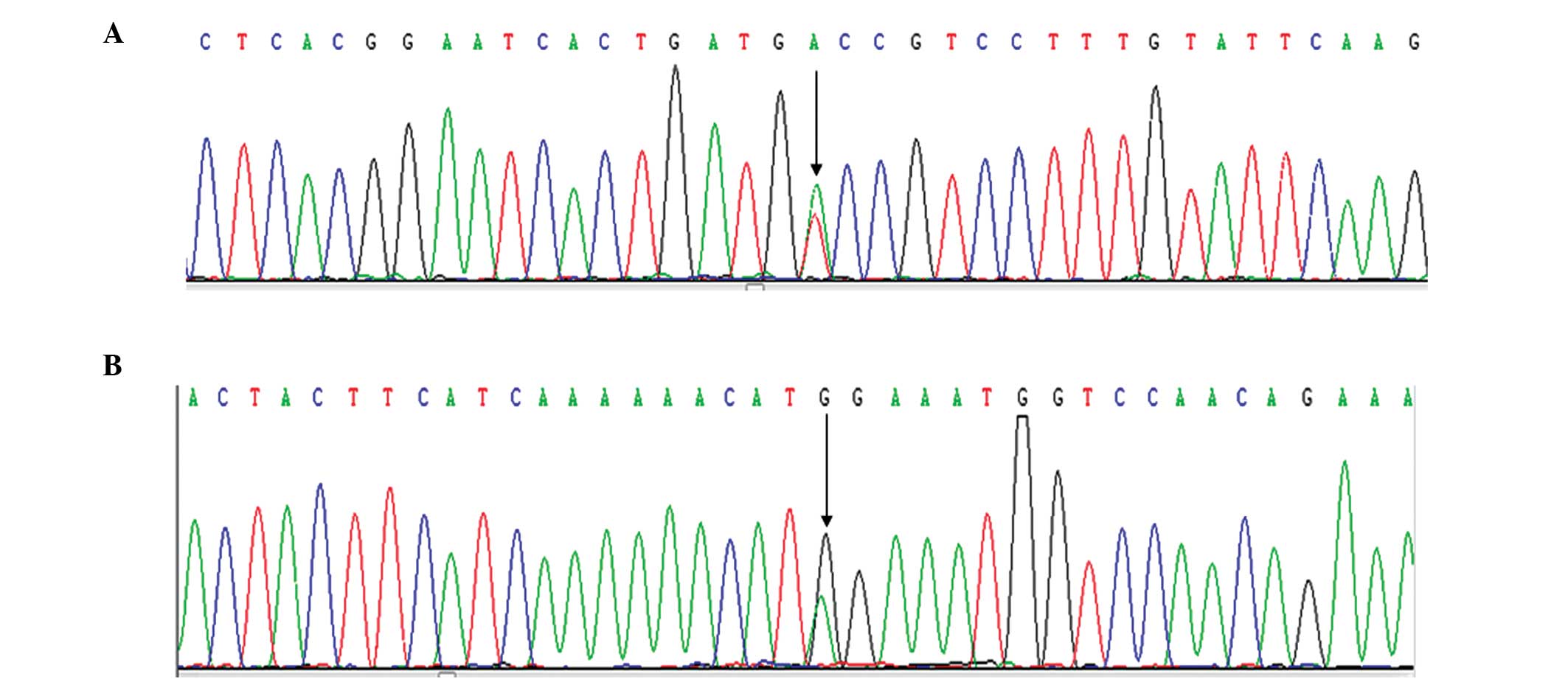
Introns 1 and 22 were not inverted. A previously
reported mutation (15) was
detected (exon 10, c.1505T>A, p.Val502Asp), and a novel
unreported missense point mutation (exon 14, c.3279G>A,
p.Met1093Ile) (Fig. 1).
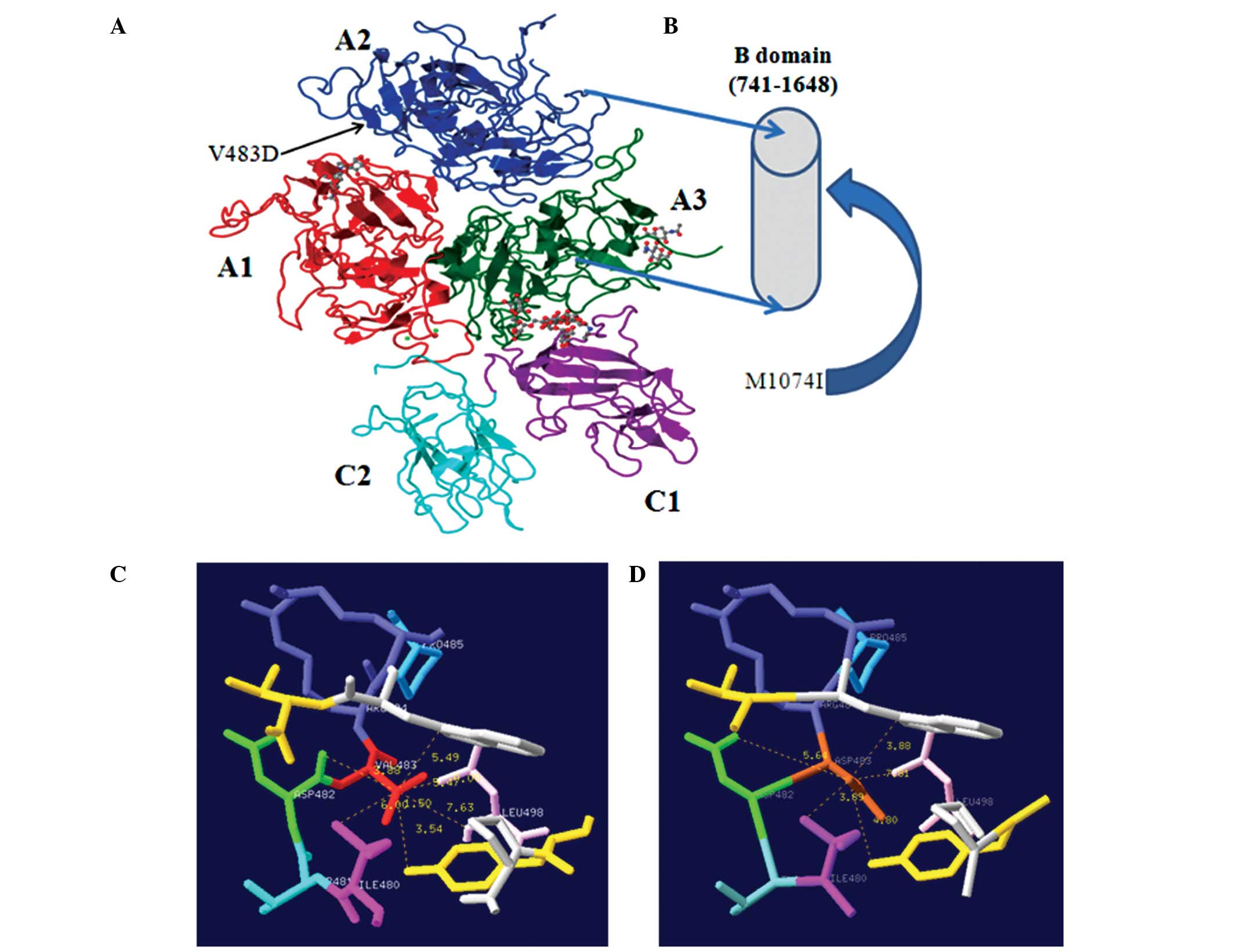
Molecular modeling
Mutated FVIII proteins were modeled using the
crystal structure of activated recombinant FVIII (16,17).
Structural images were generated using the Swiss-Pdb Viewer (Swiss
Institute of Bioinformatics, Geneva, Switzerland) (18) (Fig.
2).
Conserved domains analysis
A conserved domains analysis was conducted for
p.Val502 and p.Met1093 by using the Multiple Alignment Tool
(http://www.ncbi.nlm.nih.gov/tools/cobalt/cobalt.cgi?link_loc=BlastHomeLink).
It was observed that p.Val502 of the A2 domains in F8 is highly
conserved among mammals (Table
III). By contrast, p.Met1093 in the B domain shares little
amino acid homology, indicating that it is not conserved among
different species.
| Table IIIConserved domains analysis in F8 for
p.V483(502). |
Table III
Conserved domains analysis in F8 for
p.V483(502).
| Species | Domain | Nucleotide
number |
|---|
| Hs |
PLLYGEVGDTLLIIFKNQASRPYNIYPHGITDVRPLYSRRLPKGVKHLKDFPILGEIFKYKWTVTVEDGPT | 541 |
| Mm |
PLLYGEVGDTLLIIFKNQASRPYNIYPHGITDVSPLHARRLPRGIKHVKDLPIHPGEIFKYKWTVTVEDGPT | 541 |
| Rn |
PLLYGEVGDSLLIVFKNRASRAYNIHPHGIRDVGAVHAGRLPRGVKHVKDLPIRPGETFKYRWTLTAEDGPA | 529 |
| Cf |
PLLYGEVGDTLLIIFKKQASRPYNIYPHGINYVTPLHTGRLPKGVKHLKDMPILPGEIFKYKWTVTVEDGPT | 535 |
| Bt |
PLLYGEVGDTLLIIFKNQASRPYNIYPHGITDVSPLHSGRFPKGVKHLKDMPILGEVFKYKWTVTVEDGPT | 536 |
| Oc |
PLLYGEVGDTLLIIFKNQASRPYNIYPHGITDVSPLHSGRLSKGMKHLKDLPILPGEIFYKWKVTVEDGPT | 537 |
| Gg |
PVLKGEVGDQFKIVFRNLASRPYNIYPHGLTSVNPYHAMKPSQGKKDVKDIPAPGQSFTYRWSITTEDGPT | 529 |
| Oa |
PLLYGEVGDTLLIIFKNQASRPYNIYPHGITDVSPLHSGRFPKGVKHLKDMPILPGEVFKYKWTVTVEDGPT | 536 |
| Ss |
PLLYGEVGDTLLIIFKNKASRPYNIYPHGITDVSALHPGRLLKGWKHLKDMPILPGETFKYKWTVTVEDGPT | 541 |
| Tr |
PLLKGKVGDQIHIMLKNTASRPFNIYPNGLSSIRPMKRSKNAS-EKDLRTMGVGPNETFGYMWELTANDRPL | 546 |
| Sh |
PLLYGEVGDMLLITFKNLASRPYNIYPHGLTSVSPLHSGRLPKGVKDVKDMPIMPGQTFKYKWEVTMEDGPT | 541 |
| La |
PLLYGEVGDTLLIIFKNQASRPYNIYPHGITNVSPLHSGRLSKGVKHLKDLQIMPGEIFKYKWTVTLEDGPT | 542 |
| Dr |
PELRGEVGDKFQIVFKNMASRPFNIYPNGLTSVQPLKTTNKDK-QVDLRSLAVPPGEIMTYLWKLTAEGDPT | 530 |
Discussion
Due to the X-linked recessive mode of inheritance,
HA usually affects males, and females usually are carriers who may
pass the disease on to their progeny (19). Thus, female HA cases are rarely
observed. However, in certain cases, there are a variety of
potential genetic mechanisms leading to HA in females: i)
Non-random inactivation of the normal X-chromosome in a female HA
carriers (the most common cause of female HA (20,21);
ii) homozygous F8 mutations (mostly reported in India where
consanguineous marriages are more common) (22,23);
iii) compound heterozygous mutations affecting both F8 alleles
(21); and iv) X-chromosome
monosomy or gross structural defects, such as a deletion or
translocation (24–26).
Disease severity in HA patients is classified
according to residual plasma FVIII activity (FVIII:C): severe
(<1%), moderate (1–5%) and mild (5–35%) (27). In the severe phenotype, the most
prevalent mutations are inversions of introns 1 or 22, accounting
for 5% and 40–50% of patients, respectively (5,28).
Moderate or mild HA are usually caused by missense mutations
(29).
In this study, we presented the case of a female HA
patient with a moderately clinical phenotype (FVIII: C=4.8%)
resulting from F8 compound heterozygous mutations. The p.Val502Asp
mutation located in A2 domains was first reported to be associated
with mild HA by Fernández-López et al (15) in 2005. As the original amino acid
Val502 in F8 is highly conserved among all known mammals, it was
predicted that Val502 is involved in an A2-specific functional
role. The non-polar, hydrophobic amino acid Val is replaced by the
polar and acidic amino acid Asp, resulting in altered stability and
protein folding. The p.Met1093Ile mutation is located in the B
domain of F8, which is encoded by exon 14 (amino acids 741–1648).
Contrary to the A and C domains, there are no available molecular
models or crystal structures that elucidate the detailed structural
information of the B domain. The FVIII B domain shares little
sequence homology with other known mammalian species. Despite the
fact that the B domain is not directly required for FVIII
coagulation activity, it has been shown to exhibit a major role in
the intracellular interactions that regulate quality control and
secretion, as well as potential regulatory roles within plasma
during activation, platelet binding, inactivation and clearance
(30). Therefore, the p.Met1093Ile
mutation in the B domain also affects FVIII coagulation activity.
Furthermore, missense mutations within the F8 B domain have often
been reported in HA patients (31,32).
p.Met1093Ile is a novel missense mutation unreported in the
HAMSTeRS database. The mutation was submitted to the HAMSTeRS
database and was accepted (unpublished). It was estimated that the
compound heterozygous mutations (p.Val502Asp and p.Met1093Ile) are
causative gene defects, which resulted in a moderate HA phenotype
in this female patient.
In conclusion, female HA patients, particularly
those without a personal or family history of bleeding disorders,
are often misdiagnosed as AHA or vWD-type 2N. A diagnosis of HA
should be made in a female patient when isolated APTT is prolonged,
FVIII binding capacity for vWF is normal, FVIII levels are
decreased and FVIII auto-antibodies are absent. Genetic analysis of
F8 using multiplex PCR and DNA sequencing is an essential tool in
elucidating the nature of the various molecular mechanisms
resulting in HA in females.
Acknowledgements
The authors are grateful to Dr Ye-ling Lu (Ruijin
Hospital, Shanghai Jiaotong University School of Medicine,
Shanghai, China) for his technical assistance in mutational
analysis and to Professor Jing-yu Zhang (The Second Hospital of
Hebei Medical University, Shijiazhuang, Hebei, China) for measuring
the vWF binding affinity for FVIII.
References
|
1
|
Klinge J, Ananyeva NM, Hauser CA and
Saenko EL: Hemophilia A - from basic science to clinical practice.
Semin Thromb Hemost. 28:309–322. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Saenko EL, Ananyeva NM, Tuddenham EG and
Kemball-Cook G: Factor VIII - novel insights into form and
function. Br J Haematol. 119:323–331. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lenting PJ, van Mourik JA and Mertens K:
The life cycle of coagulation factor VIII in view of its structure
and function. Blood. 92:3983–3996. 1998.PubMed/NCBI
|
|
4
|
Lenting PJ, Pegon JN, Christophe OD and
Denis CV: Factor VIII and von Willebrand factor - too sweet for
their own good. Haemophilia. 16:194–199. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bagnall RD, Waseem N, Green PM and
Giannelli F: Recurrent inversion breaking inversion intron 1 of the
factor VIII gene is a frequent cause of severe hemophilia A. Blood.
99:168–174. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oldenburg J, Brackmann HH, Hanfland P and
Schwaab R: Molecular genetics in haemophilia A. Vox Sang. 78:33–38.
2000.
|
|
7
|
Castaldo G, D’Argenio V, Nardiello P, et
al: Haemophilia A: molecular insights. Clin Chem Lab Med.
45:450–461. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Oldenburg J: Mutation profiling in
haemophilia A. Thromb Haemost. 85:577–579. 2001.PubMed/NCBI
|
|
9
|
Shetty S, Bhave M and Ghosh K: Acquired
hemophilia A: diagnosis, aetiology, clinical spectrum and treatment
options. Autoimmun Rev. 10:311–316. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Franchini M, Targher G, Montagnana M and
Lippi G: Laboratory, clinical and therapeutic aspects of acquired
hemophilia A. Clin Chim Acta. 395:14–18. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sadler JE, Budde U, Eikenboom JC, et al:
Update on the pathophysiology and classification of von Willebrand
disease: A report of the Subcommittee on von Willebrand Factor. J
Thromb Haemost. 4:2103–2114. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nichols WL, Rick ME, Ortel TL, et al:
Clinical and laboratory diagnosis of von Willebrand disease: a
synopsis of the 2008 NHLBI/NIH guidelines. Am J Hematol.
84:366–370. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Keeney S, Mitchell M and Goodeve A; the UK
Haemophilia Center Doctors’ Organisation Harmophilia Genetics
Laboratory Network. The molecular analysis of haemophilia A: a
guideline from the UK haemophilia centre doctors’ organization
haemophilia genetics laboratory network. Haemophilia. 11:387–397.
2005.
|
|
14
|
Boekhorst J, Verbruggen B, Lavergne JM, et
al: Thirteen novel mutations in the factor VIII gene in the
Nijmegen haemophilia A patient population. Br J Haematol.
131:109–117. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fernández-López O, García-Lozano JR,
Núñez-Vázquez R, Pérez-Garrido R and Núñez-Roldán A: The spectrum
of mutations in Southern Spanish patients with hemophilia A and
identification of 28 novel mutations. Haematologica. 90:707–710.
2005.PubMed/NCBI
|
|
16
|
Shen BW, Spiegel PC, Chang CH, et al: The
tertiary structure and domain organization of coagulation factor
VIII. Blood. 111:1240–1247. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kopp J and Schwede T: The SWISS-MODEL
repository of annotated 3-dimensional protein structure homology
models. Nucleic Acids Res. 32:D230–D234. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guex N and Peitsch MC: SWISS-MODEL and the
Swiss-PdbViewer: an environment for comparative protein modeling.
Electrophoresis. 18:2714–2723. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pavlova A, Brondke H, Müsebeck J, et al:
Molecular mechanisms underlying hemophilia A phenotype in seven
females. J Thromb Haemost. 7:976–982. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Favier R, Lavergne JM, Costa JM, et al:
Unbalanced X-chromosome inactivation with a novel FVIII gene
mutation resulting in severe hemophilia A in a female. Blood.
96:4373–4375. 2000.PubMed/NCBI
|
|
21
|
Knobe KE, Sjörin E, Soller MJ, Liljebjörn
H and Ljung RC: Female haemophilia A caused by skewed X
inactivation. Haemophilia. 14:846–848. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nair PS, Shetty S and Ghosh K: A
homozygous female hemophilia A. Indian J Hum Genet. 18:134–136.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Renault NK, Dyack S, Dobson MJ, et al:
Heritable skewed X-chromosome inactivation leads to haemophilia A
expression in heterozygous females. Eur J Hum Genet. 15:628–637.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cai XH, Wang XF, Dai J, et al: Female
hemophilia A heterozygous for a de novo frameshift and a novel
missense mutation of factor VIII. J Thromb Haemost. 4:1969–1974.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Martín-Salces M, Venceslá A, Alvárez-Román
MT, et al: Clinical and genetic findings in five female patients
with haemophilia A: Identification of a novel missense mutation,
p.Phe2127Ser. Thromb Haemost. 104:718–723. 2010.PubMed/NCBI
|
|
26
|
Venceslá A, Fuentes-Prior P, Baena M, et
al: Severe haemophilia A in a female resulting from an inherited
gross deletion and a de novo codon deletion in the F8 gene.
Haemophilia. 14:1094–1098. 2008.PubMed/NCBI
|
|
27
|
Jayandharan G, Shaji RV, Baidya S, et al:
Identification of factor VIII gene mutation in 101 patients with
haemophilia A: mutation analysis by inversion screening and
multiplex PCR and CSGE and molecular modeling of 10 novel missense
substitutions. Haemophilia. 11:481–491. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lakich D, Kazazian HH Jr, Antonarakis SE
and Gitschier J: Inversions disrupting the factor VIII gene are a
common cause for severe haemophilia A. Nat Genet. 5:236–241. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jacquemin M, De Maeyer M, D’Oiron R, et
al: Molecular mechanisms of mild and moderate hemophilia A. J
Thromb Haemost. 1:456–463. 2003. View Article : Google Scholar
|
|
30
|
Pipe SW: Functional roles of the factor
VIII B domain. Haemophilia. 15:1187–1196. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Green PM, Bagnall RD, Waseem NH and
Giannelli F: Haemophilia A mutations in the UK: results of
screening one-third of the population. Br J Haematol. 143:115–128.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Repessé Y, Slaoui M, Ferrandiz D, et al:
Factor VIII (FVIII) gene mutations in 120 patients with hemophilia
A: detection of 26 novel mutations and correlation with FVIII
inhibitor development. J Thromb Haemost. 5:1469–1476.
2007.PubMed/NCBI
|