Introduction
The serine/threonine kinase LKB1 is causally linked
to the Peutz-Jeghers syndrome (PJS), which in turn increases the
risk of developing cancer; LKB1 is one of the most commonly mutated
genes associated with multiple sporadic cancer types (1,2).
LKB1 is ubiquitously expressed in healthy adult and embryonic
tissues (3,4). Studies in the past 14 years have
established LKB1 as a critical tumor suppressor protein, regulating
cell proliferation and survival (5,6).
Currently, LKB1 is unanimously considered a central player of an
important signaling network affecting numerous cell processes,
whose deregulation contributes to pathologies such as PJS and
carcinomas, although the underlying mechanisms remain elusive
(7,8).
Among the various molecular mechanisms altered in
human neoplasias, those involving control of the cell division
cycle are believed to be fundamental for carcinogenesis (9). Modulation of the expression and the
function of cell cycle regulatory proteins greatly contributes to
growth inhibition (10). A number
of studies have provided compelling evidence that reintroduction of
LKB1 into human cancer cells with impaired LKB1 activity results in
reduced growth by inhibiting the G1/S transition
(11–13). However, it is noteworthy that most
of these experiments were performed in cancer cells with an
LKB1-null or -deficient genetic background, and the regulation and
activity of endogenous LKB1 were rarely investigated, considerably
limiting our understanding of LKB1 function.
A previous study by our group demonstrated that LKB1
knockdown accelerates the G1/S transition through the
p53 and p16 pathways in two healthy cell lines, suggesting that
abolishment of LKB1 expression in healthy cells contributes to the
formation of malignancies (14).
It is crucial to determine whether the enhanced expression of LKB1
can inhibit proliferation of cells with endogenous LKB1 expression.
In the present study, we examined the effects of enhanced LKB1
expression on the cell cycle profile via transfection of cells with
exogenous LKB1 and investigated the underlying mechanisms in two
healthy cell lines, the human embryonic kidney 293T (HEK-293T) and
the human umbilical vein endothelial cells (HUVECs), and in one
human hepatocellular carcinoma cell line, HepG2. Our results reveal
that an enhanced expression of LKB1 activates LKB1/AMPK signaling
and confers an observable G1 arrest phenotype in cells
with endogenous LKB1 activity.
Materials and methods
Materials
Cell culture media, supplements and the
Lipofectamine 2000 reagent were purchased from Invitrogen Life
Technologies (Carlsbad, CA, USA). Primary antibodies targeting
AMP-activated protein kinase (AMPK), phospho-AMPK (Thr172),
acetyl-coenzyme A carboxylase (ACC), phospho-ACC (Ser79),
retinoblastoma protein (Rb), phospho-Rb (Ser807/811),
cyclin-dependent kinase (CDK) 4, CDK6, cyclin D1, cyclin D3, cyclin
E, p21, p27 and p15 were purchased from Cell Signaling Technology
(Beverly, MA, USA). Primary antibodies targeting LKB1, p53, p19,
p16 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were
obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Horseradish peroxidase (HRP)-conjugated secondary antibodies were
obtained from Jackson ImmunoResearch Laboratories, Inc. (West
Grove, PA, USA). Unless otherwise indicated, chemicals and organic
solvents were purchased from Sigma-Aldrich (St. Louis, MO, USA) and
were of the highest grade.
Cell culture and transfection
Culturing of HEK-293T cells and HUVECs was performed
as previously described (14).
HepG2 cells were grown in Dulbecco’s modified Eagle’s medium
(DMEM). The cells were maintained in the presence of 10% fetal
bovine serum, without antibiotics, at 37°C in a humidified
atmosphere containing 5% CO2. Cultured cells from
passages 3–10 were used. Transfection was performed with indicated
plasmids using the Lipofectamine 2000 reagent, following the
manufacturer’s protocols.
Vector constructs
A plasmid for the eukaryotic expression of enhanced
green fluorescent protein (EGFP) bearing a wild-type copy of the
LKB1 gene (LKB1-WT) and an EGFP empty plasmid were kindly
provided by Dr Junying Yuan (Harvard Medical School). An LKB1
kinase-deficient EGFP plasmid (LKB1-K78M) was generated from
LKB1-WT by converting the lysine residue at codon 78 into
methionine using the QuikChange Site-Directed Mutagenesis Kit
(Stratagene, La Jolla, CA, USA) using the primers: forward,
5′-CATAAGCTTGCCACCATGGAG GTGGTGGACCCGCA-3′ and reverse,
5′-AATTCTAGA TCACTGCTGCTTGCAGGCCGAC-3′. Constructs were confirmed
by sequencing in both directions.
Flow cytometric analysis
Exponentially growing cells were transfected with
indicated plasmids for 48 h, detached and fixed with 80% ethanol
overnight. Propidium iodide/RNase A (Sigma-Aldrich) was used to
stain the nuclei. Cell cycle distribution was determined by flow
cytometry using a FACSVantage SE cell sorter, and the percentage of
cells at different phases of the cell cycle was analyzed using the
CellQuest™ Pro software (both from BD Biosciences, Franklin Lakes,
NJ, USA).
Western blot analysis
Cells were harvested and lysed as previously
described (14). The protein
concentration was determined by the BCA method. Equal amounts of
protein lysates were separated by SDS-PAGE electrophoresis on 8,
10, 12 or 15% gels and transferred onto polyvinylidene difluoride
membranes (Immobilon-P; Millipore, Billerica, MA, USA). Protein
loading was normalized with the housekeeping gene GAPDH.
Membranes were blocked with 5% non-fat milk and incubated with
specific primary antibodies targeting LKB1, AMPK, phospho-AMPK,
ACC, phospho-ACC, Rb, phospho-Rb, CDK4, CDK6, cyclin D1, cyclin D3,
cyclin E, p15, p16, p19, p21, p27, p53 and GAPDH. Detection of HRP
on immunoblots was performed using the SuperSignal West Pico
Chemiluminescent Substrate kit (Pierce Biotechnology, Rockford, IL,
USA). The protein signals were quantified using the Quantity One
software (Bio-Rad, Hercules, CA, USA).
Results
Enhanced expression of LKB1 activates
LKB1/AMPK signaling in a kinase-dependent manner
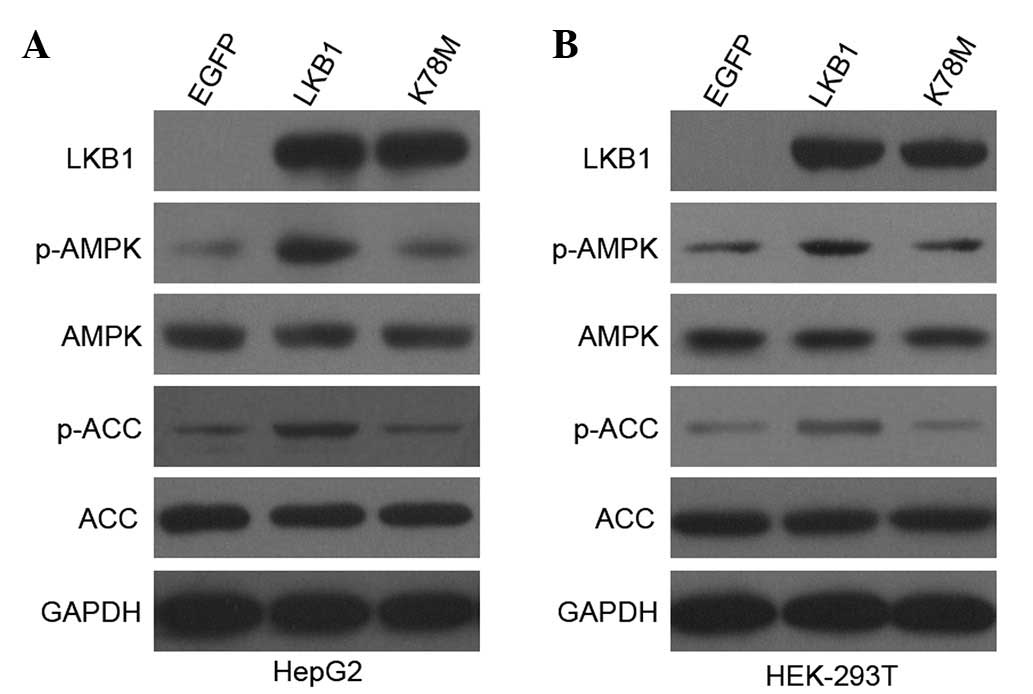
Plasmids encoding EGFP-tagged wild-type LKB1
(LKB1-WT) were transfected into HepG2 and HEK-293T cells, with the
LKB1-empty vector (EGFP) used as a negative control. An invariant
nucleotide binding site mutant plasmid (LKB1-K78M), which confers
deficiency in the kinase activity of LKB1 (15), was also included to test for kinase
dependency. Consistent with previous reports (14,16),
endogenous LKB1 was expressed in both HepG2 and HEK-293T cell lines
(data not shown). As shown in Fig.
1, EGFP-tagged recombinant LKB1 was expressed at comparable
levels in cells transfected with LKB1-WT and LKB1-K78M constructs,
but not in cells transfected with the control EGFP vector. Thus,
the enhanced expression of LKB1 protein was successfully achieved
in HepG2 and HEK-293T cells.
A well-known substrate of LKB1 is AMPK, which is
likely to mediate most, if not all, of the tumor suppressor effects
of LKB1 (6,17). As expected (Fig. 1), transfection with wild-type but
not kinase-inactive LKB1 into HepG2 and HEK-293T cells
substantially increased phosphorylation of AMPKα at Thr172, and
phosphorylation of ACC at Ser79, which is a useful indicator of
increased AMPK activity in vivo. Total AMPK and ACC protein
levels were not altered in transfected cells. These results
indicate that the enhanced expression of LKB1 (via expression of
exogenous LKB1 in addition to the endogenous one) enhanced
LKB1/AMPK activation in HepG2 and HEK-293T cells in a LKB1
kinase-dependent manner.
Exogenous activation of LKB1/AMPK
signaling arrests the cell cycle at the G1 phase
Cell cycle control is primarily achieved at the
‘restriction point’, the transition from the G1 to the S
phase, beyond which mitogenic signaling is no longer required and
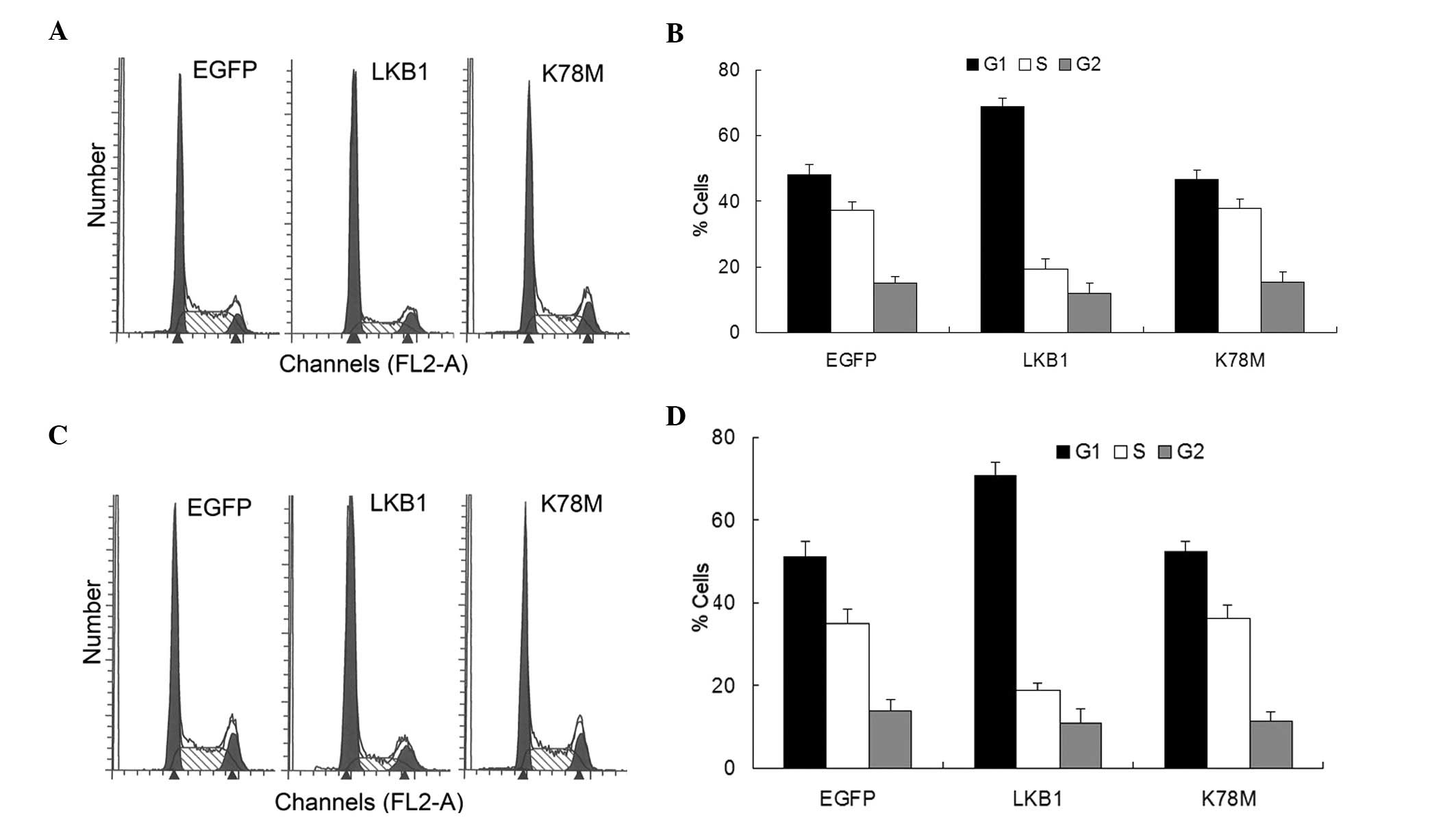
the cell is committed to complete the replication cycle (18). Transfection with LKB1-WT into HepG2
and HEK-293T cells led to the accumulation of cells at
G1 and a reduction in the proportion of cells at the S
phase (Fig. 2). Limited or no
effect was noted in the LKB1-K78M-transfected cells (Fig. 2). These results agree with earlier
reports that ectopic LKB1 expression in LKB1-deficient cancer cells
blocks the G1/S transition (11–13),
and indicate that exogenous activation of LKB1/AMPK signaling is
sufficient to induce G1 arrest, even in cells with
endogenous expression of LKB1.
Exogenous activation of LKB1/AMPK
signaling reduces phosphorylation of Rb
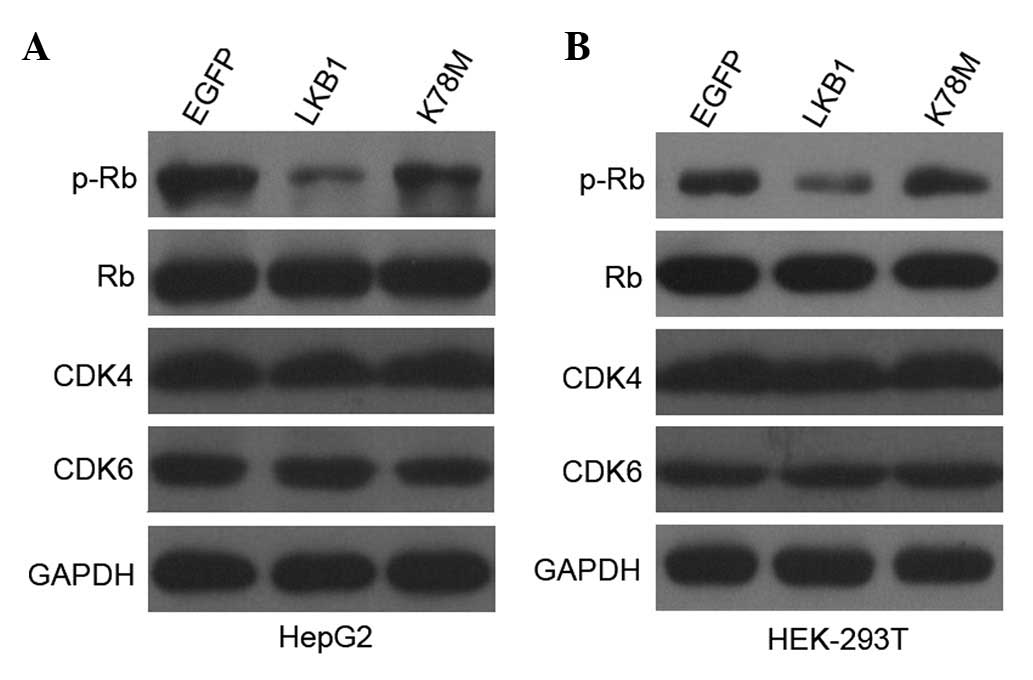
Progression through the G1 phase is
characterized by the hyperphosphorylation of Rb, which releases E2F
transcription factors to induce synthesis of proteins necessary for
DNA synthesis (19). Compatible
with the increase of cell populations at the G1 phase,
transfection with LKB1-WT, but not with LKB1-K78M, considerably
reduced phosphorylation of Rb in HepG2 and HEK-293T cells, with no
differences observed in the total Rb protein level (Fig. 3). LKB1 and
Ca2+/CaM-dependent protein kinase kinase (CaMKK) are
essential activators of the AMPK signaling pathway (20). To examine the potential role of
CaMKK activation in controlling the G1/S transition, we
pretreated HepG2 cells and HEK-293T cells with STO-609, a selective
CaMKK inhibitor. No effect was observed on cell cycle distribution
and Rb phosphorylation (data not shown). These results indicate
that activation of LKB1/AMPK signaling, but not of CaMKK/AMPK
signaling, leads to hypophosphorylation of Rb, which in turn
contributes to G1 arrest in HepG2 and HEK-293T
cells.
Exogenous activation of LKB1/AMPK
signaling does not affect CDK4 and CDK6 protein levels
The cell cycle machinery comprises a variety of
proteins that ensure the proper progression through distinct
phases, with different CDKs proteins playing central roles, by
phosphorylating Rb and leading to cell cycle entry into the S phase
(21). To investigate whether the
LKB1-induced G1 arrest is due to changes in major
G1-phase CDKs, we examined the protein levels of CDK4
and CDK6. Transfection with LKB1-WT and LKB1-K78M into HepG2 and
HEK-293T cells did not affect the expression of CDK4 or CDK6
(Fig. 3), indicating that neither
CDK4 nor CDK6 are involved in the regulation of the G1/S
arrest mediated by LKB1, and excluding the possibility that
hypophosphorylation of Rb results from the decreased expression of
CDK4 or CDK6 in HepG2 and HEK-293T cells.
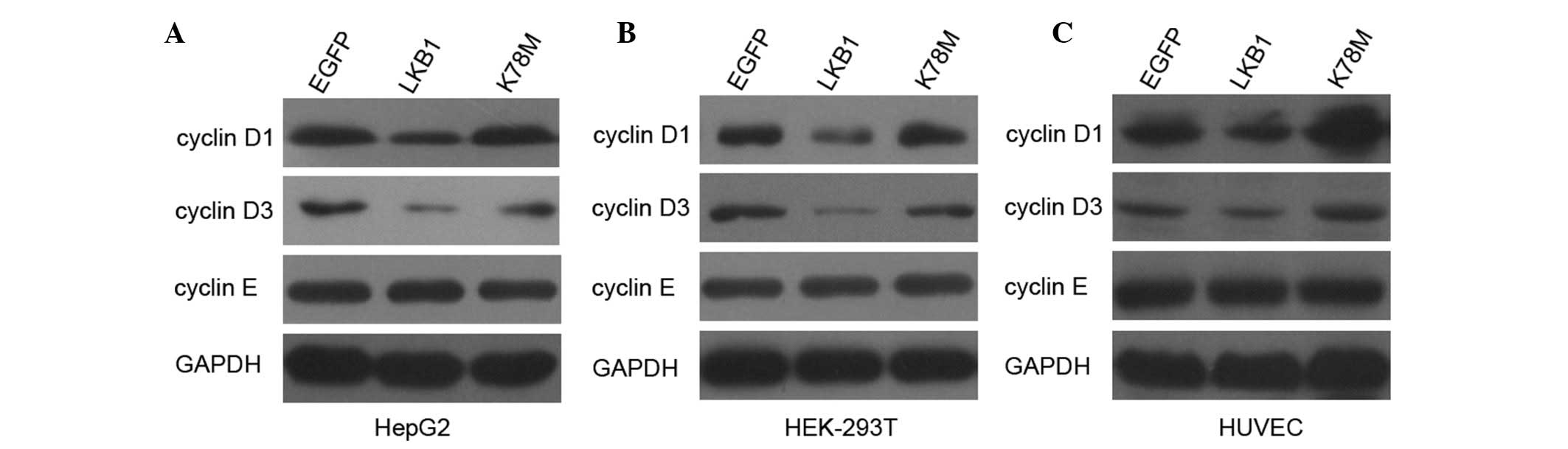
Exogenous activation of LKB1/AMPK
signaling decreases the expression of cyclin D1 and cyclin D3, but
not cyclin E
The enzymatic activity of CDKs is dependent on
physical interactions with cyclins, which are the regulatory
subunits of CDKs (22). We
assessed whether enhanced LKB1 expression affects major
G1 regulatory cyclins in HepG2 and HEK-293T cells. As
shown in Fig. 4, protein levels of
cyclin D1 and cyclin D3 were markedly decreased following
transfection with LKB1-WT, but not with LKB1-K78M. To confirm this
finding, we extended the analysis to HUVECs. Transfection of HUVECs
with LKB1 WT also decreased the expression of cyclin D1 and cyclin
D3 (Fig. 4). These results
indicate that activation of LKB1/AMPK signaling inhibits the
expression of cyclin D1 and cyclin D3, which may reduce the
activity of CDKs and lead to hypophosphorylation of Rb in HepG2 and
HEK-293T cells. Protein profiles of cyclin E were also
investigated, however, no change was detected in the level of this
cyclin, indicating that cyclin E is not involved in the regulation
of the G1/S transition, affected by LKB1.
Exogenous activation of LKB1/AMPK
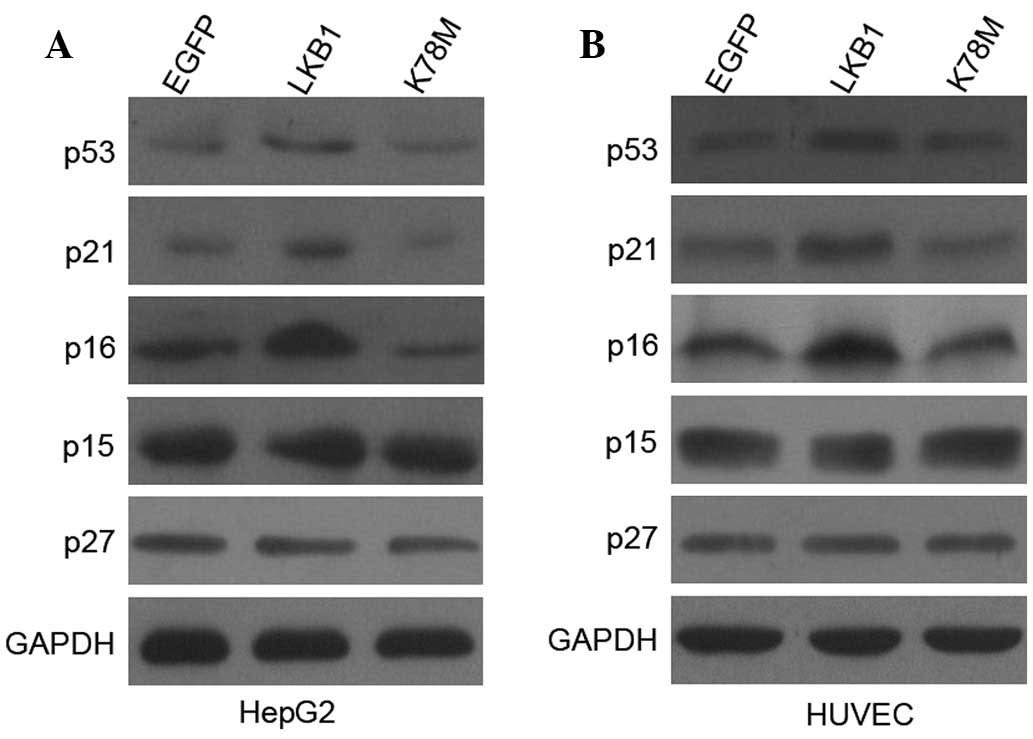
signaling increases the expression of p53, p21 and p16, but not p15
or p27
Induction of CDK inhibitors (CKIs) is a
well-characterized mechanism to inhibit G1 CDK activity
(23). p21 (WAF1/Cip1) is a CKI
with a broad spectrum of activities, known to inhibit the activity
of most CDKs, and generally considered a key downstream effector of
the p53 tumor suppressor protein, since its expression is
transcriptionally induced by p53 (24,25).
p16 (INK4a) is also a critical CKI involved in G1/S
control, which binds directly to CDK4 and CDK6 and prevents the
association with D-type cyclins (26,27).
In the present study, the observation that the ectopic expression
of LKB1 can considerably increase the protein levels of p53, p21,
and p16 in HepG2 cells (Fig. 5) is
in agreement with our previous study, which showed, to the best of
our knowledge, for the first time that both p53 and p16 are the
targets of LKB1, regulating G1/S transition (14). Notably, transfection with LKB1-WT
had no effect on p53, p21, and p16 expression in HEK-293T cells
(data not shown). However in HUVECs, the ectopic expression of LKB1
elicited similar increases in the expression of p53, p21, and p16
compared with the ones observed in HUVECs (Fig. 5). These results indicate that in
both cancer and healthy cells, the exogenous activation of
LKB1/AMPK signaling induces the expression of p53, p21 and p16,
which in turn contributes to the inactivation of CDKs, and further
leads to hypophosphorylation of Rb.
p15 (INK4b), p19 (INK4d) and p27 (Kip1) are
important CKIs participating in cell cycle regulation (22,28).
As shown in Fig. 5, no observable
differences were detected in p15 and p27 expression following
transfection with LKB1 constructs into HepG2 cells and HUVECs,
indicating that neither p15 or p27 is involved in LKB1-mediated
G1 arrest. The protein level of p19 was not detectable
in our study.
Discussion
The present study shows, to the best of our
knowledge, for the first time that ectopic LKB1 expression
activates the LKB1/AMPK signaling pathway and leads to
G1 arrest in healthy and cancer cells with endogenous
LKB1 expression in a kinase-dependent manner, which is in agreement
with our previous study demonstrating that the suppression of
endogenous LKB1 activity in two healthy cell lines accelerates
G1/S transition (14).
Our results indicate that G1/S cell cycle arrest induced
by exogenous LKB1 may not be limited to tumor cells with
inactivated LKB1, and are in disagreement with results by Tiainen
et al, who found that cells with endogenous LKB1 are
resistant to the inhibitory effects of exogenous LKB1 on cell
growth (12). This discrepancy may
be explained, in part, by cell type-specific differences in
signaling mediated by LKB1. Alternatively, the discrepancy could be
due to differences in the delivery of LKB1. It is notable that the
ectopic expression of the phosphatase and tensin homolog (PTEN)
tumor suppressor protein strongly inhibited cell proliferation in
both cancer and healthy cell lines maintaining endogenous wild-type
PTEN expression, and this effect was also dependent on the kinase
activity of PTEN (29). Germline
mutations in LKB1 and PTEN lead to closely related diseases,
including PJS and Cowden disease, suggesting that tumor suppressors
LKB1 and PTEN exert negative effects on the growth of a wide
variety of cell lines, and that these proteins may be involved in
the same or related pathways that inhibit growth (30,31).
We found that the overexpression of LKB1 inhibited
the synthesis of cyclin D1 and cyclin D3, but stimulated the
synthesis of p53, p21 and p16. These effects were specific, as the
expression of other components of the cell cycle machinery,
including CDK4, CDK6, cyclin E, p15 and p27, was not affected.
These results indicate a cell model whereby growth inhibition
mediated by LKB1 depends on the combined action of cyclins and
CKIs. The reduced expression of cyclin D1 and cyclin D3, and an
increased expression of p53, p21 and p16 jointly inhibit the
activity of CDKs, resulting in hypophosphorylation of Rb and
consequently, cell cycle arrest at the G1 phase.
Notably, HEK-293T cells are an exception to this model, in that the
expression of p53, p21 and p16 remained unaltered in response to
LKB1 overexpression, indicating that the p53 and p16 pathways may
not be involved in G1 phase arrest in these cells.
Supporting evidence for this hypothesis comes from the observation
that inhibition of ubiquitin carboxy-terminal hydrolase (UCH) L1
expression arrested both HEK-293T and KR4 cell lines at the
G1 phase, while p53 upregulation was only observed in
KR4 cells, but not in HEK-293T cells (32). One plausible explanation is that
HEK-293T cells constitutively express the simian virus 40 (SV40)
large T antigen, which inactivates the endogenous p53 protein and
restricts its activity to the baseline level (33,34).
The basal p53 level may be further downregulated, as shown in our
previous study (14). However, due
to the inhibition of SV40 large T antigen, p53 pathways were not
activated. Since p21 is the major transcriptional target of p53
(24) and p16 and p53 may have
coordinated expression (35,36),
the detection of unaltered p21 and p16 protein levels in HEK-293T
cells is expected. Additional studies are nevertheless required to
validate the above-described model.
The finding that in HepG2 cells and HUVECs, the
ectopic LKB1 expression leads to an increase in the expression of
p53, p21 and p16 is inconsistent with early studies on
LKB1-deficient cancer cells, which showed that either p53 or p16 is
sufficient for the function of LKB1 in the cell cycle control
(11–13), however, it is in agreement with our
previous study reporting, to the best of our knowledge, for the
first time that LKB1 targets both p53 and p16 pathways in healthy
cells (15). Our results further
suggest that in cells with endogenous LKB1 expression, there might
be a cross-talk between p53 and p16 pathways, and that these
pathways act as important components of LKB1 signaling, which is
required for regulating the G1/S transition.
Nevertheless, which and how many molecules and cellular events
mediate the link between LKB1 activity and the p53 and p16 pathways
remains to be determined.
The results indicate that combined alterations in
the levels of cyclins and CKI regulatory proteins may have
synergistic effects, resulting in decelerated cell cycle
progression and thereby, inhibiting cell proliferation in response
to exogenous activation of LKB1 signaling. This may explain, at
least in part, the association between concurrent aberrations in
the activities of p53, p21, p16 and cyclin D proteins, as well as
the aggressive clinical behavior observed in PJS patients, since
increased neoplastic cell proliferation is a negative prognostic
factor in related tumors (37–40).
The extent to which these molecules individually contribute to the
observed growth-arrest phenotype is still a subject of debate.
In summary, the results described in the present
study extend earlier observations in LKB1-deficient neoplastic
cells, by demonstrating that LKB1/AMPK activation via ectopic LKB1
expression can cause cell cycle arrest at the G1 phase,
even in cells with endogenous LKB1 expression. The results further
suggest that, unlike LKB1-mediated growth inhibition in LKB1-null
or -deficient genetic backgrounds, the antiproliferative activity
of LKB1 on cells expressing endogenous LKB1 is related to a
multifaceted regulation of multiple target molecules that are
critically involved in cell cycle progression. Overall, this study
has provided novel insights into the growth-inhibitory effect of
the tumor suppressor protein LKB1.
Acknowledgements
We thank Dr Junying Yuan for kindly providing the
pEGFP-LKB1-WT and pEGFP plasmids. This study was supported by a
grant from the National Natural Science Foundation of China (no.
81201544).
Abbreviations:
|
PJS
|
Peutz-Jeghers syndrome
|
|
HEK-293T cells
|
human embryonic kidney 293T cells
|
|
HUVECs
|
human umbilical vein endothelial
cells
|
|
CDK
|
cyclin-dependent kinase
|
|
CKI
|
cyclin-dependent kinase inhibitor
|
|
AMPK
|
AMP-activated protein kinase
|
|
CaMKK
|
Ca2+/CaM-dependent protein
kinase kinase
|
References
|
1
|
McGarrity TJ and Amos C: Peutz-Jeghers
syndrome: clinicopathology and molecular alterations. Cell Mol Life
Sci. 63:2135–2144. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rustgi AK: The genetics of hereditary
colon cancer. Genes Dev. 21:2525–2538. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hemminki A, Markie D, Tomlinson I, et al:
A serine/threonine kinase gene defective in Peutz-Jeghers syndrome.
Nature. 391:184–187. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jenne DE, Reimann H, Nezu J, et al:
Peutz-Jeghers syndrome is caused by mutations in a novel serine
threonine kinase. Nat Genet. 18:38–43. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Williams T and Brenman JE: LKB1 and AMPK
in cell polarity and division. Trends Cell Biol. 18:193–198. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shackelford DB and Shaw RJ: The LKB1-AMPK
pathway: metabolism and growth control in tumour suppression. Nat
Rev Cancer. 9:563–575. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hearle N, Schumacher V, Menko FH, et al:
Frequency and spectrum of cancers in the Peutz-Jeghers syndrome.
Clin Cancer Res. 12:3209–3215. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Partanen JI, Tervonen TA, Myllynen M, et
al: Tumor suppressor function of Liver kinase B1 (Lkb1) is linked
to regulation of epithelial integrity. Proc Natl Acad Sci USA.
109:E388–E397. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Manning AL and Dyson NJ: pRB, a tumor
suppressor with a stabilizing presence. Trends Cell Biol.
21:433–441. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sclafani RA and Holzen TM: Cell cycle
regulation of DNA replication. Annu Rev Genet. 41:237–280. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tiainen M, Vaahtomeri K, Ylikorkala A and
Mäkelä TP: Growth arrest by the LKB1 tumor suppressor: induction of
p21(WAF1/CIP1). Hum Mol Genet. 11:1497–1504. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tiainen M, Ylikorkala A and Mäkelä TP:
Growth suppression by Lkb1 is mediated by a G1 cell
cycle arrest. Proc Natl Acad Sci USA. 96:9248–9251. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gurumurthy S, Hezel AF, Sahin E, Berger
JH, Bosenberg MW and Bardeesy N: LKB1 deficiency sensitizes mice to
carcinogen-induced tumorigenesis. Cancer Res. 68:55–63. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liang X, Wang P, Gao Q, Xiang T and Tao X:
Endogenous LKB1 knockdown accelerates G1/S transition
through p53 and p16 pathways. Cancer Biol Ther. 9:156–160. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Karuman P, Gozani O, Odze RD, et al: The
Peutz-Jegher gene product LKB1 is a mediator of p53-dependent cell
death. Mol Cell. 7:1307–1319. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hou X, Xu S, Maitland-Toolan KA, et al:
SIRT1 regulates hepatocyte lipid metabolism through activating
AMP-activated protein kinase. J Biol Chem. 283:20015–20026. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lizcano JM, Göransson O, Toth R, et al:
LKB1 is a master kinase that activates 13 kinases of the AMPK
subfamily, including MARK/PAR-1. EMBO J. 23:833–843. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Drury LS and Diffley JF: Factors affecting
the diversity of DNA replication licensing control in eukaryotes.
Curr Biol. 19:530–535. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Giacinti C and Giordano A: RB and cell
cycle progression. Oncogene. 25:5220–5227. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Green MF, Anderson KA and Means AR:
Characterization of the CaMKKβ-AMPK signaling complex. Cell Signal.
23:2005–2012. 2011.
|
|
21
|
Senderowicz AM: Inhibitors of
cyclin-dependent kinase modulators for cancer therapy. Prog Drug
Res. 63:183–206. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shapiro GI: Cyclin-dependent kinase
pathways as targets for cancer treatment. J Clin Oncol.
24:1770–1783. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cánepa ET, Scassa ME, Ceruti JM, et al:
INK4 proteins, a family of mammalian CDK inhibitors with novel
biological functions. IUBMB Life. 59:419–426. 2007.PubMed/NCBI
|
|
24
|
Abbas T and Dutta A: p21 in cancer:
intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Garner E and Raj K: Protective mechanisms
of p53-p21-pRb proteins against DNA damage-induced cell death. Cell
Cycle. 7:277–282. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pei XH and Xiong Y: Biochemical and
cellular mechanisms of mammalian CDK inhibitors: a few unresolved
issues. Oncogene. 24:2787–2795. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Satyanarayana A and Rudolph KL: p16 and
ARF: activation of teenage proteins in old age. J Clin Invest.
114:1237–1240. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Maddika S, Ande SR, Panigrahi S, et al:
Cell survival, cell death and cell cycle pathways are
interconnected: implications for cancer therapy. Drug Resist Updat.
10:13–29. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li J, Simpson L, Takahashi M, et al: The
PTEN/MMAC1 tumor suppressor induces cell death that is rescued by
the AKT/protein kinase B oncogene. Cancer Res. 58:5667–5672.
1998.PubMed/NCBI
|
|
30
|
Winship IM and Dudding TE: Lessons from
the skin - cutaneous features of familial cancer. Lancet Oncol.
9:462–472. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jass JR: Colorectal polyposes: from
phenotype to diagnosis. Pathol Res Pract. 20:431–447. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bheda A, Shackelford J and Pagano JS:
Expression and functional studies of ubiquitin C-terminal hydrolase
L1 regulated genes. PLoS One. 4:e67642009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Aulestia FJ, Redondo PC, Rodríguez-García
A, et al: Two distinct calcium pools in the endoplasmic reticulum
of HEK-293T cells. Biochem J. 435:227–235. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Santi SA and Lee H: The Akt isoforms are
present at distinct subcellular locations. Am J Physiol Cell
Physiol. 298:C580–C591. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gallagher SJ, Thompson JF, Indsto J, et
al: p16INK4a expression and absence of activated B-RAF are
independent predictors of chemosensitivity in melanoma tumors.
Neoplasia. 10:1231–1239. 2008.PubMed/NCBI
|
|
36
|
Zhang Y, Xiong Y and Yarbrough WG: ARF
promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus
deletion impairs both the Rb and p53 tumor suppression pathways.
Cell. 92:725–734. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Morton JP, Jamieson NB, Karim SA, et al:
LKB1 haploinsufficiency cooperates with Kras to promote pancreatic
cancer through suppression of p21-dependent growth arrest.
Gastroenterology. 139:586–597. 5972010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
De Leng WW, Westerman AM, Weterman MA, et
al: Cyclooxygenase 2 expression and molecular alterations in
Peutz-Jeghers hamartomas and carcinomas. Clin Cancer Res.
9:3065–3072. 2003.PubMed/NCBI
|
|
39
|
Scott KD, Nath-Sain S, Agnew MD and
Marignani PA: LKB1 catalytically deficient mutants enhance cyclin
D1 expression. Cancer Res. 67:5622–5627. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ji H, Ramsey MR, Hayes DN, et al: LKB1
modulates lung cancer differentiation and metastasis. Nature.
448:807–810. 2007. View Article : Google Scholar : PubMed/NCBI
|