Introduction
Transitional cell carcinoma (TCC), also termed
urothelial cell carcinoma, is the most common type of bladder
cancer in the United States. It comprises >90% of all primary
bladder neoplasms. In 2013, there were 70,530 new cases of TCC
reported and 14,680 TCC-associated mortalities in the United States
(1). Although transurethral
resection of the tumor, combined with intravesical bacillus
Calmette-Guerin immunotherapy has become the standard treatment for
this type of bladder cancer, the tumor recurrence rate remains high
(60–70%). Additionally, the complications of chemotherapy are
severe, including frequent micturition, urgent micturition,
urination pain, hematuria, immune suppression, febrise and
lumbodynia (2).
It is known that patients with bladder cancer have
mutations in cell cycle checkpoint genes that are associated with
malignant growth and genetic instability. The alteration of
multiple cell cycle regulators renders patients with high-grade TCC
with a survival rate of only 8% (3). Cell cycle progression is governed by
different cyclin-dependent kinases (Cdks) in a step-wise manner,
including Cdk4/6 during the G1 phase and Cdk2 from the late G1
phase to the early S phase. Combined with D-type (D1, D2, D3)
cyclin, Cdk4/6 is activated and phosphorylates the retinoblastoma
protein (Rb), promoting the escape of E2F from phosphorylated Rb,
and therefore inducing cell transit from the G1 phase into the S
phase. Several previous studies on cell cycle regulation and
bladder cancer genetics have demonstrated that the overexpression
of cyclin D1 and Cdk4/6 and high levels of phosphorylation of Rb
contribute to the dysregulation of the cell cycle during
oncogenesis and tumor progression in bladder cancer (4–6).
It has been demonstrated that the mitogen-activated
protein kinase (MAPK) cascade (Ras/Raf/Mek/Erk), which induces
cyclin and Cdk gene expression and promotes cell cycle progression
by activating upstream transcription factors (7), is responsible for TCC oncogenesis and
tumor growth (8). Previous studies
have demonstrated that the Raf/Mek/Erk cascade is a target for
cancer treatment and may be inhibited by the cAMP/protein kinase A
(PKA) pathway (9,10). However, whether PKA activators
negatively regulate cell proliferation in different types of cancer
remains to be elucidated. One of the PKA activators, cholera toxin,
has been demonstrated to inhibit pancreatic cancer and leukemia
cell growth in vitro, which is dependent upon intracellular
cAMP accumulation (11,12). Our previous studies confirmed the
ability of PKA activators to inhibit the growth of malignant human
glioma cells (13,14). However, the anti-tumor effects of
cholera toxin on bladder carcinoma have not yet been clearly
demonstrated, therefore, the present study investigated whether
targeting PKA with cholera toxin has therapeutic effects against
bladder cancer cells.
In the present study, it was hypothesized that
cholera toxin may arrest cells in the G1 phase and inhibit cell
proliferation in two high-grade invasive cell lines of bladder TCC,
T24 and UM-UC-3. Furthermore, it was hypothesized that the
inhibition of proliferation by the cholera toxin may be due to
PKA-dependent inactivation of the Raf/Mek/Erk pathway and thus, the
expression of downstream genes, including cyclin D1, Cdk4 and Cdk6,
may be downregulated. Therefore, the present study investigated
whether cholera toxin is a potential candidate for anti-cancer
treatment with a certain activity on bladder carcinoma.
Materials and methods
Cell culture and drug treatment
T24 cells were purchased from the Wuhan Cell Bank of
China (Wuhan, Hubei, China) and maintained in RPMI-1640 culture
medium (Invitrogen Life Technologies, Grand Island, NY, USA)
supplemented with 10% fetal bovine serum (FBS; Invitrogen Life
Technologies), 100 U/ml penicillin and 100 U/ml streptomycin, in a
humidified atmosphere of 5% CO2 at 37°C. UM-UC-3 cells
were obtained from American Type Culture Collection (Manassas, VA,
USA) and maintained in minimum essential medium (Invitrogen Life
Technologies). KT5720 and UO126 cells (Calbiochem Bioscience, La
Jolla, CA, USA) were dissolved in dimethyl sulfoxide (Sigma, St.
Louis, MO, USA) and cholera toxin (Sigma-Aldrich, St. Louis, MO,
USA) was dissolved in sterile ultra-purified water. The cells were
plated and allowed to grow overnight in medium containing 10% FBS.
The medium was then replaced with serum-free medium during the
treatment.
Cell survival assay
Cell viability was evaluated using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT)
assay (Sigma). The optical density (OD) was determined at 570 nm
using an EXL800 microimmunoanalyzer (Bio-Tek Instruments,
Burlington, VT, USA). The cell viability rate was calculated using
the following formula: ODexperiment/ODcontrol
× 100%.
Cell proliferation and clone formation
assay
The cells were placed in a six-well plate at a
density of 500 cells/well and maintained with or without 10 ng/ml
cholera toxin in normal medium containing 10% FBS for 10 days.
Colonies were fixed with methanol and stained with 0.1% crystal
violet in 20% methanol for 15 min and images of the representative
colonies were captured.
Hoechst staining and lactate
dehydrogenase (LDH) release assay
For Hoechst 33342 staining, the cells were incubated
with Hoechst 33342 (5 μg/ml; Sigma) for 20 min in the dark and then
observed using a fluorescent microscope (Olympus, Melvie, NY, USA)
with a 340 nm excitation filter.
Cytotoxicity of diazepam was evaluated using the LDH
release assay. LDH release was quantified using a CytoTox
96® nonradioactive cytotoxicity assay kit (Promega
Corporation, Madison, WI, USA) in accordance with the
manufacturer’s instructions. Absorbance was measured at 490 nm
using an iMark™ Microplate Absorbance Reader (Bio-Rad, Hercules,
CA, USA). Lysis solution was used as a positive control for maximum
LDH release. To calculate the %Cytotoxicity of each
treatment group, the following formula was used, in accordance with
the manufacturer’s instructions: %Cytotoxicity =
Experimental LDH release (OD490)/Maximum LDH release (OD490). The
LDH release (%control) was calculated as the
%Cytotoxicity of the treatment group over the
%Cytotoxicity of the vehicle control (0 μM) group.
Cell cycle analysis
The cells were seeded in six-well culture plates at
a density of 8×105 cells/well and incubated overnight.
Cholera toxin (0–50 ng/ml) was added to the serum-free medium for
0–24 h. The cells were harvested by trypsinization, suspended in
100 μl medium and then fixed in 75% dehydrated alcohol overnight in
4°C. The cells were subsequently incubated with the DNA-binding dye
propidium iodide (PI; 50 mg/ml), RNase (4 Ku/ml), NaF (0.3 mg/ml)
and sodium citrate (1 mg/ml) for 1–2 h at room temperature in the
dark. Red fluorescence from 488 mm laser-excited PI in every cell
was analyzed using an EPICS Altra flow cytometer (Beckman Coulter,
Fullerton, CA, USA) using a peak fluorescence gate to discriminate
aggregates. The percentage of cells in the G1, S and G2/M phases
were determined from the DNA content histograms using MultiCycle
for windows software (Phoenix Flow Systems, San Diego, CA,
USA).
Raf kinase assay and western blot
analysis
The Raf kinase assay kit was purchased from Upstate
Biotechnology (Charlottesville, VA, USA) and used in accordance
with the manufacturer’s instructions. Western blot analysis was
performed as previously described and the blots were visualized by
peroxidase reaction using enhanced chemiluminescence (Amersham
Pharmacia Biotech, Piscataway, NJ, USA) (15). The following antibodies were used:
cyclin D1, cyclin D3, Cdk4, Cdk6, p-RbSer807/Ser811,
c-Raf, p-c-RafSer259, B-Raf, p-B-RafSer445,
p-Mek1/2Ser217/221, Mek,
p-Erk1/2Thr202/Tyr204, Erk and GAPDH (Cell Signaling
Technology, Beverly, MA, USA); cyclin E, Cdk2 and Rb (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) and
p-c-RafSer43 and p-c-RafSer621 (Abcam,
Cambridge, MA, USA); β-actin (Neomarker, Fremont, CA, USA) and
α-tubulin (Sigma). The blots are representative of three
independent experiments.
Statistical analysis
Statistical analysis was performed using PASS 11
(NCSS, LLC, Kaysville, Utah, USA) using one way analysis of
variance. P<0.05 was considered to indicate a statistically
significant difference.
Results
Cholera toxin inhibits proliferation and
arrests the cell cycle in the G1 phase of human bladder TCC
cells
The effect of cholera toxin on human bladder TCC
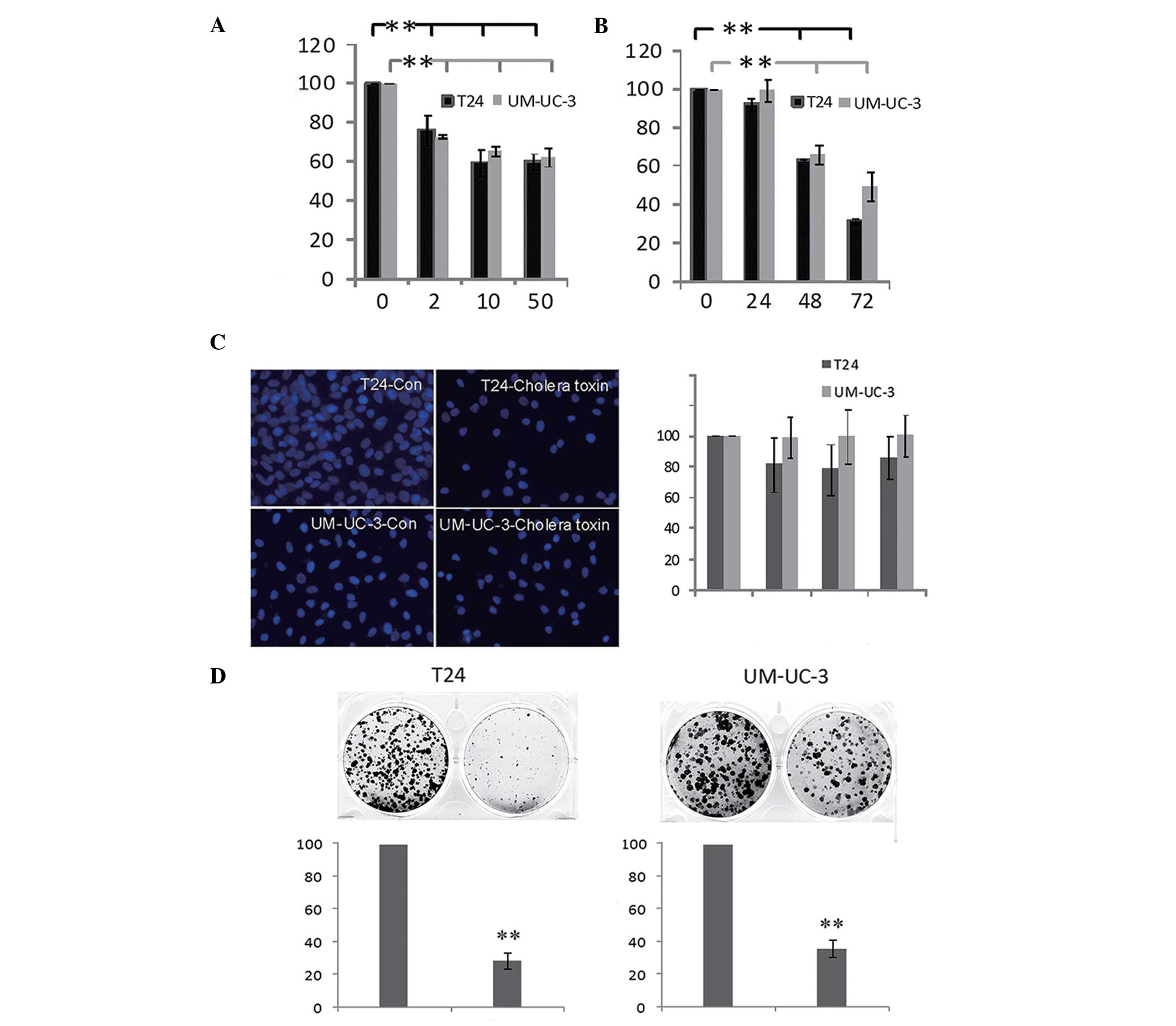
cell growth was investigated. As shown in Fig. 1A, the viability of T24 and UM-UC-3
cells decreased following incubation with the cholera toxin
(between 2 and 50 ng/ml) for 48 h. The inhibitory effect of cholera
toxin (10 ng/ml) increased time dependently between 24 and 72 h
(Fig. 1B) in the two cell lines.
However, cell apoptosis and cell necrosis were not detected at 72 h
(Fig. 1C). In combination with the
result that fewer and smaller colonies are observed when cells are
incubated with 10 ng/ml cholera toxin for 10 days (Fig. 1D), these results indicated that
cholera toxin significantly inhibited T24 and UM-UC-3 cell
proliferation in a dose- and time-dependent manner.
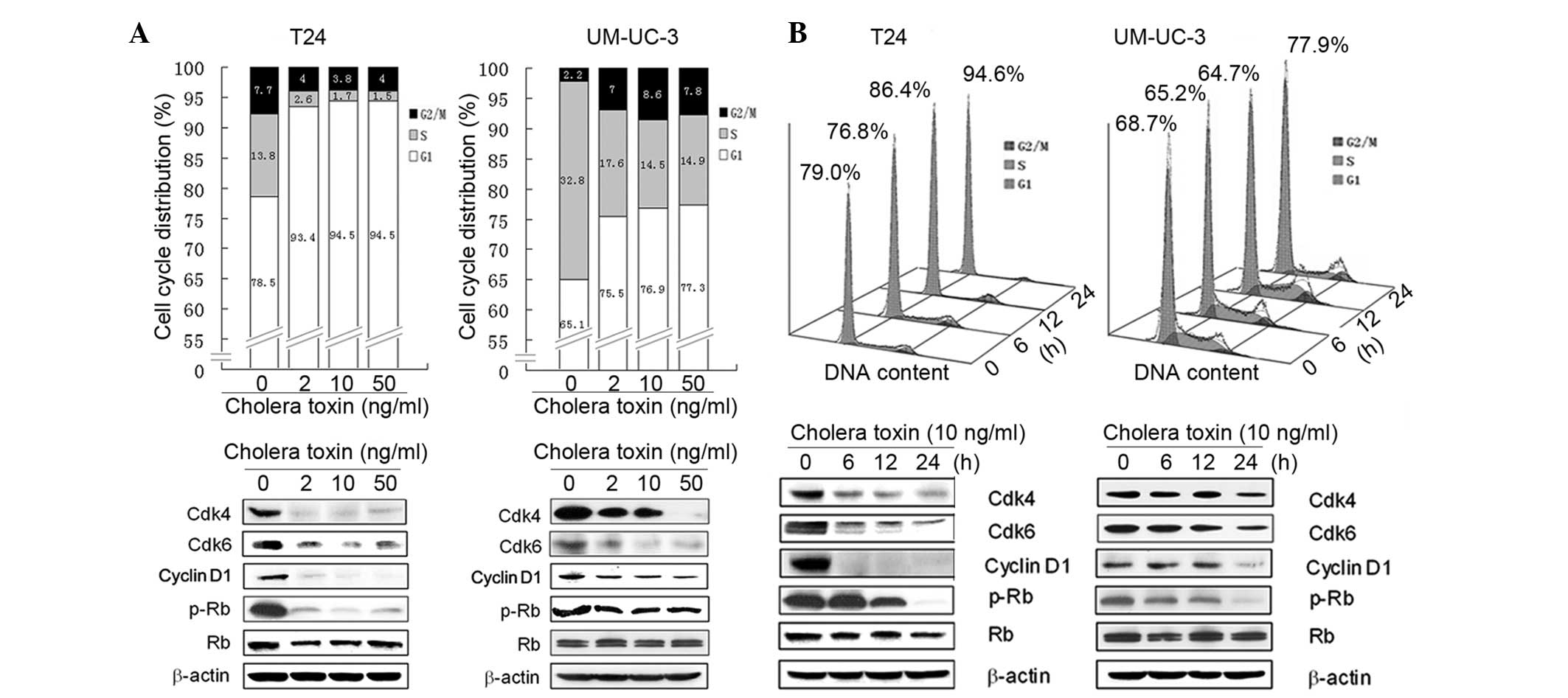
To further investigate the mechanism underlying the
anti-proliferative activity of cholera toxin, its effect on cell
cycle distribution was investigated using flow cytometry. In
cholera toxin-treated T24 cells there was an accumulation of 93.4,
94.5 and 94.5% cells, respectively, in the G1 phase following 24 h
incubation with cholera toxin at 2, 10 and 50 ng/ml, respectively,
compared with the control group comprising of 78.5% cells. This was
accompanied by a decrease in the proportion of cells in the S phase
(Fig. 2A). In the time-response
study, an increase in the percentage of cells in the G1 phase was
observed with 10 ng/ml cholera toxin treatment for 12 (P<0.05)
and 24 h (P<0.001) compared with the control cells (Fig. 2B).
Incubation with cholera toxin at 2, 10 and 50 ng/ml
for 24 h, resulted in an increase in the number of UM-UC-3 cells in
the G1 phase to 75.5, 76.9 and 77.3%, respectively, compared with
65.1% observed in the control cells (Fig. 2A). However, the effect of cholera
toxin was weaker in UM-UC-3 cells compared with T24 cells. Similar
to T24 cells, the effect of cholera toxin on G1 arrest in UM-UC-3
cells was also largely accompanied by a decrease in S phase cells
(Fig. 2B). Overall, these results
demonstrated that cholera toxin induced a significant G1 arrest of
the cell cycle in T24 and UM-UC-3 cells.
Cholera toxin downregulates the G1-S
transition promoters in human TCC cells
Based on the G1 phase arrest induced by cholera
toxin in T24 and UM-UC-3 cells, the expression of cell cycle
regulatory molecules involved in G1 phase progression was then
investigated. Cell cycle progression through the G1 phase is
primarily regulated by the sequential activation of the cyclin
D-Cdk4/6 and cyclin E-Cdk2 complexes, which induce Rb
phosphorylation and E2F release. However, Cdk1 (e.g.
p27Kip1) is capable of deactivating the cyclin D-Cdk4/6
and cyclin E-Cdk2 complexes and thus decreases the phosphorylation
of Rb (16). In the present study,
it was found that cholera toxin was able to notably reduce the
protein expression levels of cyclin D1, Cdk4 and Cdk6 in a dose-
and time-dependent manner. In addition, Rb phosphorylation markedly
decreased in the two cell lines (Fig.
2A and B). However, cholera toxin was not observed to have an
effect on other G1 regulators, including cyclin D3, cyclin E, Cdk2
and P27Kip1 (data not shown). These results suggested
that cholera toxin reduces the activation of Rb by downregulating
the expression of cyclin D1, Cdk4 and Cdk6 thus inducing G1 phase
arrest.
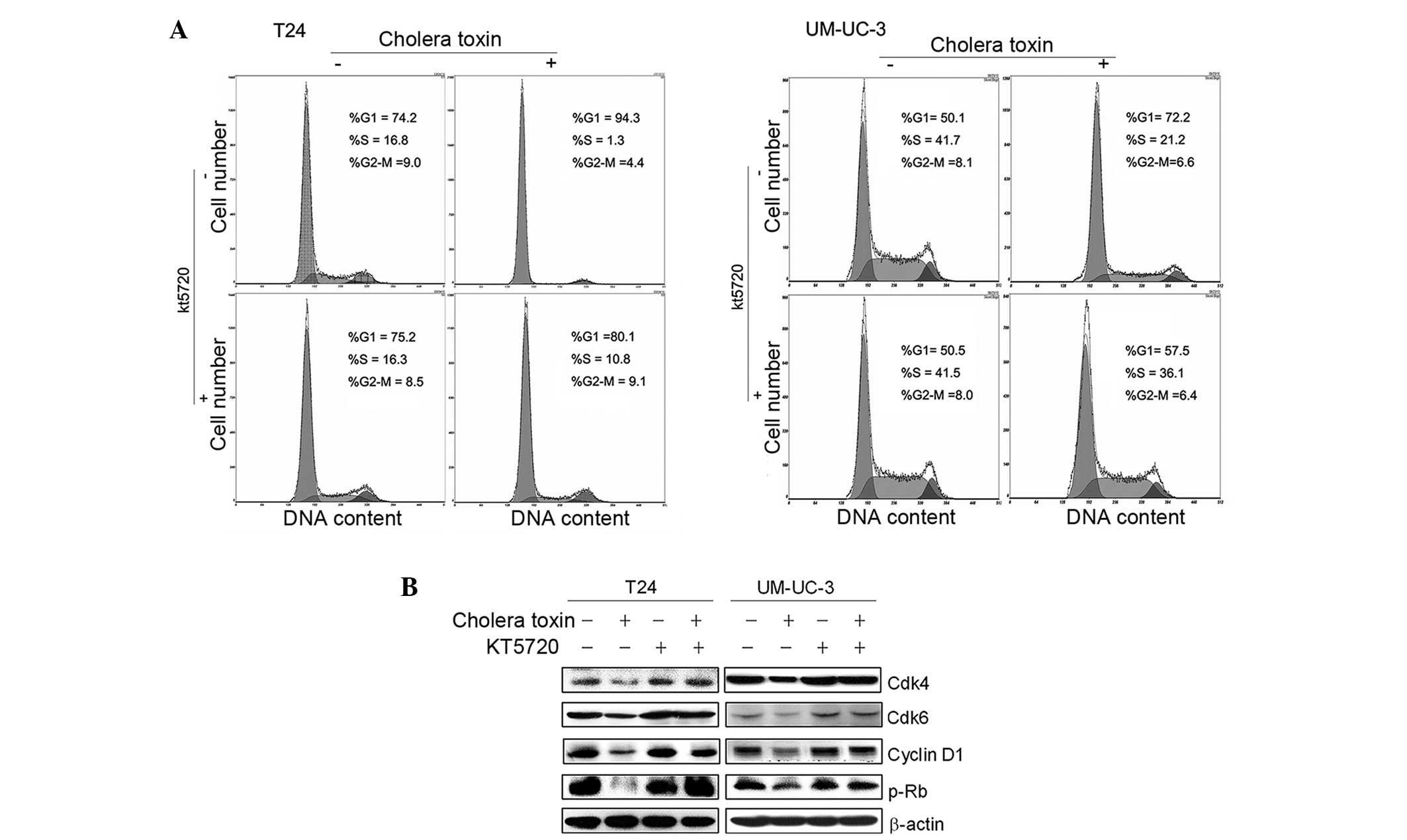
PKA activity is required for cholera
toxin-induced G1 phase arrest and downregulation of G1-S transition
promoters
Our previous studies confirmed that PKA activators,
cholera toxin or forskolin, repressed cell growth via cell cycle
arrest in the G1 phase in malignant glioma cells via PKA activation
(13,14). To examine whether PKA activation is
also involved in the G1 growth arrest of cholera toxin in T24 and
UM-UC-3 cells, a specific pharmacological inhibitor of PKA, KT5720,
was used. As shown in Fig. 3A,
pretreatment with KT5720 (1 μM) for 2 h eradicated cholera
toxin-induced G1 phase arrest in the two cell lines. Decreased
expression levels of cyclin D1, Cdk4 and Cdk6, as well as the
reduced phosphorylation of Rb due to cholera toxin, were rescued by
KT5720 pretreatment (Fig. 3B).
This indicated that in TCC cell lines T24 and UM-UC-3, PKA
stimulation was responsible for the cholera toxin-induced
downregulation of G1-S transition promoters and G1 phase
arrest.
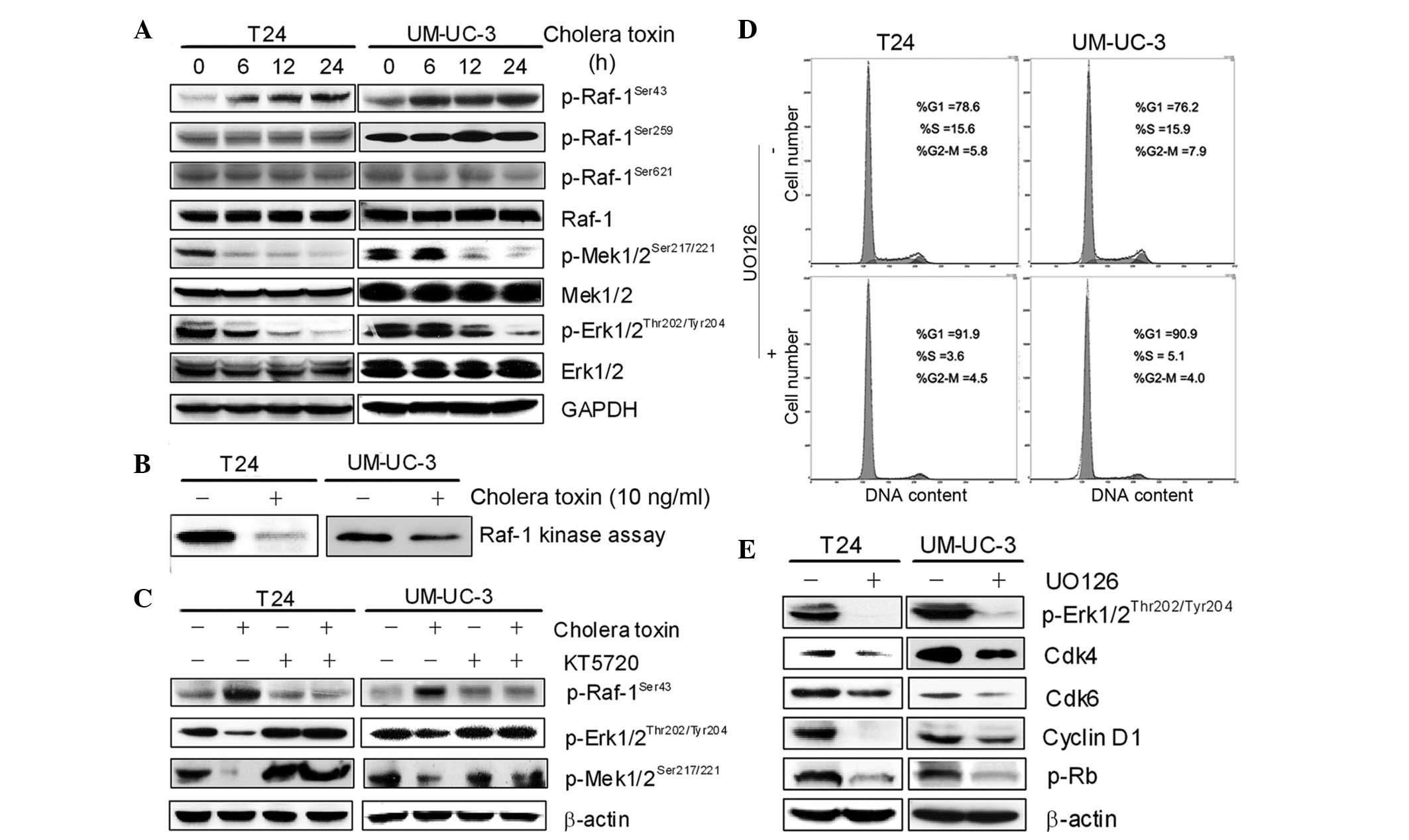
PKA-dependent inactivation of the
c-Raf/Mek/Erk pathway mediates G1 phase arrest
A number of pathways are involved in the regulation
of cell proliferation, including the c-Raf/Mek/Erk pathway, and
functions via activation of multiple transcription factors, which
switch on their target genes, for example cyclin D1 (7). To further investigate the mechanism
by which cholera toxin inhibits proliferation and arrests cells in
the G1 phase in the two TCC cell lines, the level of c-Raf
phosphorylation was assessed at serine 43, 259 and 621 following
cholera toxin treatment for 24 h. The result demonstrated that the
phosphorylation of serine 43 by c-Raf was elevated by cholera toxin
in a time-dependent manner (Fig.
4A).
The activity of the c-Raf/Mek/Erk pathway was then
detected using the Raf kinase assay and western blot analysis. As
shown in Fig. 4B, c-Raf activity
was attenuated in cholera toxin-treated T24 and UM-UC-3 cells.
Among the four major MAPK modules, ERK5/p38/JNK/Erk1/2, the one
converging on the activation of ERK and its upstream activator MAPK
and ERK kinase (MEK) is the most extensively studied and perhaps
the most relevant to cancer pathogenesis and therapy.
p-Mek1/2Ser217/221 and p-Erk1/2Thr202/Tyr204
are markers of activated Mek1/2 and Erk1/2 and were found to be
time-dependently downregulated in response to 10 ng/ml cholera
toxin. Furthermore, KT5720 significantly inhibited c-Raf
phosphorylation at serine 43 and upregulated the expression of
p-MekSer217/221 and p-ErkThr202/Tyr204 in T24
and UM-UC-3 cells (Fig. 4C). These
results demonstrated that PKA activation was required for the
inactivation of the c-Raf/Mek/Erk pathway by cholera toxin.
To further identify the role of the c-Raf/Mek/Erk
pathway in G1-S phase transition and cell proliferation, UO126, a
specific inhibitor of Mek activity, was used. Treatment with 1 μM
UO126 for 24 h arrested the cell cycle in the G1 phase, accompanied
by a decrease in cell population in the S phase in T24 and UM-UC-3
cells (Fig. 4D). In addition, the
expression of p-ErkThr202/Tyr204, cyclin D1, Cdk4, Cdk6
and p-Rb was downregulated in UO126-treated cells (Fig. 4E). The effect of UO126 on the two
cell lines is consistent with cholera toxin. This suggested that
the inactivation of the c-Raf/Mek/Erk pathway is responsible for
cholera toxin-induced decreases in cyclin D1, Cdk4 and Cdk6 and
results in G1 phase arrest.
Discussion
In the present study, the biological effect of
cholera toxin in two bladder TCC cell lines was investigated. The
results demonstrated that cholera toxin markedly induced G1 phase
arrest and inhibited cell proliferation in two human invasive
bladder TCC cell lines. However, no growth-inhibitory effect of
cholera toxin was observed in non-tumorigenic urothelium SV-HUV-1
cells (data not shown). This indicates that cholera toxin functions
as a selective inhibitor of TCC cell proliferation, however, not
normal bladder uroepithelial cell proliferation.
Tumorigenesis and the progression of bladder cancer
is considered to be closely associated with the dysregulation of
multiple cell cycle modulators (8), including Cdk4, cyclin D1 and p-Rb,
and these cell cycle regulators are regarded as prognostic
indicators of bladder cancer progression (17). The amplification of the CDK4/6 gene
occurs more frequently in higher stage/grade bladder cancer
(4). Cdk4/6 and cyclin D1 are also
significantly correlated with the recurrence of bladder cancer
(5,18). In the present study, it was
demonstrated that cholera toxin simultaneously downregulated cyclin
D1, Cdk4 and Cdk6, and markedly decreased the phosphorylation of
Rb, therefore inhibiting cell cycle progression and cell
proliferation in the bladder TCC cell lines.
Approximately 82% of grade one bladder cancers show
constitutive activation of the RAF/MEK/ERK pathways. The crosstalk
between cAMP/PKA and RAF/MEK/ERK pathways in cell death,
proliferation and differentiation is known to be important. Our
previous study demonstrated that cholera toxin inhibited
proliferation and induced cell-cycle arrest in glioma cells by
downregulating cyclin D1, and that the PKA/CREB pathway was
involved (13). In the present
study, the anti-proliferative mechanism of cholera toxin in bladder
TCC cells was consistent with previous results. It was revealed
that cholera toxin inactivated the c-Raf/Mek/Erk cascade and
downregulated the expression of the regulatory molecules (cyclin
D1, Cdk4 and Cdk6), in a PKA-dependent manner.
The RAF/MEK/ERK pathway has recently emerged as a
promising target for anticancer therapies, as excessive activation
of this pathway is often observed in various types of tumor
(19). PKA negatively regulates
the activity of c-Raf and functions as an inhibitor of the
c-Raf/Mek/Erk pathway via c-Raf. The phosphorylation of c-Raf in
several inhibitory sites by PKA, for example phosphorylation at ser
43, disrupts the Ras/c-Raf association, inactivates c-Raf, inhibits
the cRaf/Mek/Erk pathway and finally inhibits cell growth (20,21).
In the present study, an increase in c-Raf phosphorylation at ser
43 by cholera toxin, and consequently the inactivation of
c-Raf/Mek/Erk, was observed in T24 and UM-UC-3 cells. Sorafenib, an
FDA-approved agent for the treatment of human hepatocellular
carcinoma (HCC), has been demonstrated to inhibit HCC cell growth
by also inducing c-Raf phosphorylation at Ser-43 and Ser-259
(22).
Furthermore, UO126, the specific Mek inhibitor,
induced the downregulation of cyclin D1, Cdk4 and Cdk6 in T24 and
UM-CU-3 cells, which is consistent with the effect of cholera
toxin. All these data indicated that deactivation of the
c-Raf/Mek/Erk cascade by PKA was, at least partially responsible
for the downregulation of cyclin D1, Cdk4 and Cdk6 and cell cycle
arrest in the G1 phase by cholera toxin.
In summary, in the present study, it was confirmed
that cholera toxin specifically inhibited cell proliferation and
induced G1 phase arrest in human bladder TCC cells. This tumor
inhibitory effect was revealed to be due to the PKA-dependent
inactivation of the c-Raf/Mek/Erk pathway and a marked reduction of
cyclin D1, Cdk4 and Cdk6. These findings support the use of cholera
toxin in target specific therapy for the treatment of bladder
cancer.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (nos. 30830111, 81202554 and 81202555)
and the Guangdong Natural Science Foundation (S10451008901004893
and S2012010009237).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar
|
|
2
|
Shelley MD, Mason MD and Kynaston H:
Intravesical therapy for superficial bladder cancer: a systematic
review of randomised trials and meta-analyses. Cancer Treat Rev.
36:195–205. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chatterjee SJ, Datar R, Youssefzadeh D, et
al: Combined effects of p53, p21, and pRb expression in the
progression of bladder transitional cell carcinoma. J Clin Oncol.
22:1007–1013. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Simon R, Struckmann K, Schraml P, et al:
Amplification pattern of 12q13-q15 genes (MDM2, CDK4, GLI) in
urinary bladder cancer. Oncogene. 21:2476–2483. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shin KY, Kong G, Kim WS, Lee TY, Woo YN
and Lee JD: Overexpression of cyclin D1 correlates with early
recurrence in superficial bladder cancers. Br J Cancer.
75:1788–1792. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chatterjee SJ, George B, Goebell PJ, et
al: Hyperphosphorylation of pRb: a mechanism for RB tumour
suppressor pathway inactivation in bladder cancer. J Pathol.
203:762–770. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chang F, Steelman LS, Lee JT, et al:
Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from
cytokine receptors to transcription factors: potential targeting
for therapeutic intervention. Leukemia. 17:1263–1293. 2003.
View Article : Google Scholar
|
|
8
|
Mitra AP and Cote RJ: Molecular
pathogenesis and diagnostics of bladder cancer. Annu Rev Pathol.
4:251–285. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sevetson BR, Kong X and Lawrence JC Jr:
Increasing cAMP attenuates activation of mitogen-activated protein
kinase. Proc Natl Acad Sci USA. 90:10305–10309. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pessina A, Giuliani A, Croera C, et al:
Selection of a WEHI-3B leukemia cell subclone resistant to
inhibition by cholera toxin. Mol Cell Biochem. 233:19–26. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ohmura E, Wakai K, Isozaki O, et al:
Inhibition of human pancreatic cancer cell (MIA PaCa-2) growth by
cholera toxin and 8-chloro-cAMP in vitro. Br J Cancer. 67:279–283.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li Y, Yin W, Wang X, Zhu W, Huang Y and
Yan G: Cholera toxin induces malignant glioma cell differentiation
via the PKA/CREB pathway. Proc Natl Acad Sci USA. 104:13438–13443.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
He S, Zhu W, Zhou Y, et al:
Transcriptional and post-transcriptional down-regulation of cyclin
D1 contributes to C6 glioma cell differentiation induced by
forskolin. J Cell Biochem. 112:2241–2249. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kurien BT and Scofield RH: Western
blotting. Methods. 38:283–293. 2006. View Article : Google Scholar
|
|
16
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: a changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mitra AP, Lin H, Datar RH and Cote RJ:
Molecular biology of bladder cancer: prognostic and clinical
implications. Clin Genitourin Cancer. 5:67–77. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Weinberg RA: The retinoblastoma protein
and cell cycle control. Cell. 81:323–330. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
McCubrey JA, Steelman LS, Chappell WH, et
al: Roles of the Raf/MEK/ERK pathway in cell growth, malignant
transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu J, Dent P, Jelinek T, Wolfman A, Weber
MJ and Sturgill TW: Inhibition of the EGF-activated MAP kinase
signaling pathway by adenosine 3′,5′-monophosphate. Science.
262:1065–1069. 1993.PubMed/NCBI
|
|
21
|
Stork PJ and Schmitt JM: Crosstalk between
cAMP and MAP kinase signaling in the regulation of cell
proliferation. Trends Cell Biol. 12:258–266. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Carr BI, Wang Z, Wang M, Cavallini A,
D’Alessandro R and Refolo MG: c-Met-Akt pathway-mediated
enhancement of inhibitory c-Raf phosphorylation is involved in
vitamin K1 and sorafenib synergy on HCC growth inhibition. Cancer
Biol Ther. 12:531–538. 2011. View Article : Google Scholar : PubMed/NCBI
|