Introduction
Fluoride is an essential trace element for all
mammalian species; however, in excess, it is known to be toxic to
animals and cultured cells (1).
Excessive exposure to fluoride in drinking water, or in combination
with exposure to fluoride from other sources, may give rise to a
number of adverse effects, including dental fluorosis, skeletal
fluorosis and vascular calcification (2–4).
However, the cellular mechanisms underlying fluoride-induced
cytotoxicity in osteoblasts remain to be fully elucidated.
Apoptosis induced by fluoride has been demonstrated; however, to
the best of our knowledge, the occurrence of fluoride-induced
autophagy has not been studied. The objectives of the present study
were to determine the effects of fluoride treatment on the
viability, apoptosis and autophagy of the osteoblastic cell line
MC3T3-E1, the association between them.
Autophagy is an evolutionarily conserved lysosomal
degradation process by which cells recycle damaged/obsolete
macromolecules and organelles (5).
Autophagy primarily fulfils a survival role during adaptation to
unfavorable growth conditions or following cellular stress
(6). Previous studies also
demonstrated its involvement in general processes, including
differentiation, development, defense against pathogens, ageing,
apoptosis and cell death (7–10).
Specifically, the association between autophagy and apoptosis is
close and complex. Therefore, further studies to enhance the
understanding of the molecular processes contributing to autophagy
have provided insight into the association between autophagy and
apoptosis. Recently studies suggest that autophagy serves a largely
cytoprotective role under physiologically relevant conditions
(11,12). The cytoprotective function of
autophagy is mediated by the negative modulation of apoptosis
(13); however, the mechanisms
mediating the complex counter-regulation of apoptosis and autophagy
are have yet to be fully elucidated.
In the present study, it was demonstrated that
fluoride induces apoptosis as well as autophagy in MC3T3-E1 cells.
Fluoride-induced autophagy may protect MC3T3-E1 cells against
fluoride-induced apoptosis. It was also demonstrated that the
expression of phosphorylated c-Jun N-terminal kinase (p-JNK) was
upregulated by fluoride, which contributes to the induction of
autophagy. These results offer an inference that drugs targeting
autophagy may improve therapy for fluoride damage.
Materials and methods
Reagents
α-Minimal essential medium (α-MEM),
penicillin-streptomycin (5,000 U/ml penicillin; 5,000 U/ml
streptomycin), fetal bovine serum (FBS), rapamycin (RAPA) and
3-methyladenine (3-MA) were obtained from Invitrogen (Carlsbad, CA,
USA). MTT dimethylsulfoxide (DMSO) and Triton X-100 were purchased
from Sigma (St. Louis, MO, USA). Fluoride was obtained from Xinhua
Pure Chemical Industries (Shenyang, China). All reagents used were
trace element analysis grade. All water used was glass
distilled.
Cell culture
Osteoblastic MC3T3-E1 cells (CRL-2593; American Type
Culture Collection, Manassas, VA, USA) were cultured in regular
growth medium (α-MEM with 10% FBS, and 1% penicillin/streptomycin)
at 37°C in a 5% CO2 incubator. They were subcultured
every three days using 0.2% trypsin plus 0.02% EDTA. For
experiments, cells were cultured for 24 h to obtain monolayers
containing 3 ml α-MEM with 10% FBS. Following rinsing of the cells
with phosphate-buffered saline (PBS), the medium was exchanged with
medium containing either sodium fluoride (NaF) or other agents and
the cells were further cultured.
MTT assay
Cell proliferation by treatment with various
concentrations of NaF during 24–72 h was detected by MTT assay.
Briefly, MC3T3-E1 cells were seeded into 96-well plates
(6×103 cells/well) and maintained in growth media for 24
h under 5% CO2 at 37°C. At 60% confluence, the cells
were treated with (0–10 mmol/l) NaF for 24, 48 and 72 h,
respectively. Thereafter, 10 μl of MTT solution (5 mg/ml) was added
to each well and the cells were incubated for another 4 h at 37°C.
Following the formation of formazan crystals, MTT medium was then
replaced with 150 μl of DMSO for dissolving the formazan crystals
and the plates were agitated for 5 min. The absorbance of each well
was recorded with a microplate spectrophotometer at 570 nm.
Relative cellular growth was determined from the ratio of the
average absorbance in treated cells versus the average absorbance
in control cells. The cell viability was calculated as the ratio of
optical densities.
Analysis of apoptosis
MC3T3-E1 cells were cultured at 4×106
cells/ml and seeded in six-well plates. Cells were harvested by
trypsinization, then washed twice with cold PBS and centrifuged at
110 × g. Cells (1×105–1×106) were then
resuspended in 300 μl of 1X binding buffer, centrifuged again at
110 × g for 5 min and then the supernatant was removed. Cells were
resuspended in 300 μl of 1X binding buffer and transferred to a
sterile flow cytometry glass tube. 10 μl of Annexin V-fluorescein
isothiocyanate (FITC) was added and cells were incubated in the
dark for 30 min at room temperature. Then cells were incubated in
the dark with 5 μl propidium iodide (PI) and analyzed using a flow
cytometer (FACSCalibur, Becton-Dickinson, Franklin Lakes, NJ, USA).
Cellular apoptosis was determined using the Annexin V-FITC
Apoptosis Detection kit I (Clontech Laboratories Inc, Mountain
View, CA, USA).
Terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) method
Following incubation with 1% paraformaldehyde for 5
min, cells were fixed in 2:1 v/v ethanol - acetic acid for 10 min
at room temperature. Following three washes with PBS, cells were
incubated with 100 U/ml terminal deoxyuridine transferase, 0.5
μg/ml biotinylated uridine in 1 M potassium cacodylate and 125 mM
Tris-HC1, 2.5 mM cobalt chloride (Boeringher, Mannheim, Germany),
at pH 6.6 for 1 h at 37°C in a humidified chamber. Following
washes, a 1:40 solution of fluoresceinated streptavidin
(Boeringher) was incubated for 30 min at room temperature. Slides
were counter-stained with 0.3 μg/ml of PI (Sigma) in PBS for 1 min
at room temperature. An epifluorescent microscope (Ernst Leitz,
Inc., Rockleigh, NJ, USA) was used to detect apoptotic cells, which
were quantified by counting the number of fluorescein-positive
cells relative to the total number of cells in at least 10
microscopic fields. For each experiment 200 cells were counted.
Preparation of proteins in the
mitochondrial and cytosolic fractions and western blot
analysis
The methods for preparation of proteins in the
mitochondrial and cytosolic fractions and western blot analysis
have been described previously (14). The cells were washed twice in
ice-cold PBS and resuspended in five volumes of ice-cold extraction
buffer (20 mM western blotting Hepes-KOH, 1.5 mM MgCl2,
1 mM EDTA, 1 mM ethylene glycol tetraacetic acid, 1 mM
dithiothreitol and 0.1 mM phenylmethanesulfonyl fluoride, pH 7.5.
The resuspended cells were homogenized with ten strokes of a Teflon
homogenizer. The homogenates were centrifuged twice at 750 × g for
10 min at 4°C. The supernatants were centrifuged at 10,000 × g for
15 min at 4°C to obtain the mitochondrial pellets. Cytosolic
fractions were obtained following further centrifugation at 100,000
× g for 1 h at 4°C. The protein concentrations of the resulting
supernatants and mitochondrial fractions were measured. The samples
(10 μg protein) were separated by sodium dodecyl sulfate
polyacrylamide gel electrophoresis with 10 and 8% polyacrylamide
gel. The following primary antibodies were used: anti-B-cell
lymphoma 2-associated x (Bax), anti-apoptosis-inducing factor
(AIF), anti-cytochrome-C, anti-β-actin (Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA), anti-caspase 3,
anti-microtubule-associated protein 1 light chain 3α (LC3) and
anti-Beclin 1 (Cell Signaling Technology, Inc., Danvers, MA, USA).
These separated proteins in SDS-PAGE were electrotransferred onto a
Hybond-polyvinylidene fluoride (PVDF) membrane. The individual SDS
gels were distinguished by placing the protein molecular weight
markers (Invitrogen Life Technologies, Carlsbad, CA, USA) in a
different but consistent position. The PVDF membrane was then
soaked in a blocking solution (5% nonfat milk in TBST buffer; 20 mM
Tris-HCl, pH 7.5, 0.5 M NaCl, 0.1% Tween 20) for 2 h at room
temperature. The soaked PVDF membrane was then incubated in
Tris-buffered saline/Tween 20 (TBST) containing primary antibodies
overnight at 4°C, washed with TBST buffer three times for 5 min
each and incubated at room temperature for 2 h in TBST containing
horseradish peroxidase (HRP)-conjugated goat anti-mouse and goat
anti-rabbit immunoglobulin G (IgG) (Santa Cruz Biotechnology,
Inc.). The membrane was washed with TBST buffer three times for 10
min each. The membranes were incubated with enhanced
chemoluminescence reagent (Pierce Biotechnology, Inc., Rockford,
IL, USA) for HRP (30 sec) and exposed to autoradiography films for
visualization of the bands. The relative amounts of various
proteins were analyzed. The results were quantified by Quantity One
Software.
Fluorescence microscopy
MC3T3-E1 cells were seeded into six-well plates and
incubated for 24 h. Then cells were washed once with ice-cold PBS
and fixed with 4% paraformaldehyde for 30 min at 4°C. Following
washing with PBS three times, the cells were incubated with 1%
Triton X-100 for 10 min. The cells were blocked at nonspecific
antibody binding sites by incubating with 10% goat serum in PBS
containing 0.3% Triton X-100 and 0.5% bovine serum albumin for 30
min at room temperature, followed by incubation with a mouse
monoclonal antibody against Beclin 1 (1:400 in PBS, Cell Signaling
Technology) overnight. Cells were incubated with FITC-conjugated
goat anti-mouse IgG (1:100 in PBS) for 0.5 h at room temperature.
Heochst 33342 was added to the cells for 15 min. Following washing
three times with PBS, cells were visualized using fluorescence
microscopy.
Statistical analysis
Statistical analysis was performed using SPSS
(Version 17.0; SPSS, Inc., Chicago, IL, USA). The experiments were
repeated at least three times. Data are expressed as the mean ±
standard deviation. Differences in the results for the two groups
were evaluated by the Student’s t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Fluoride induces MC3T3-E1 cell apoptosis
via a mitochondrial-mediated pathway
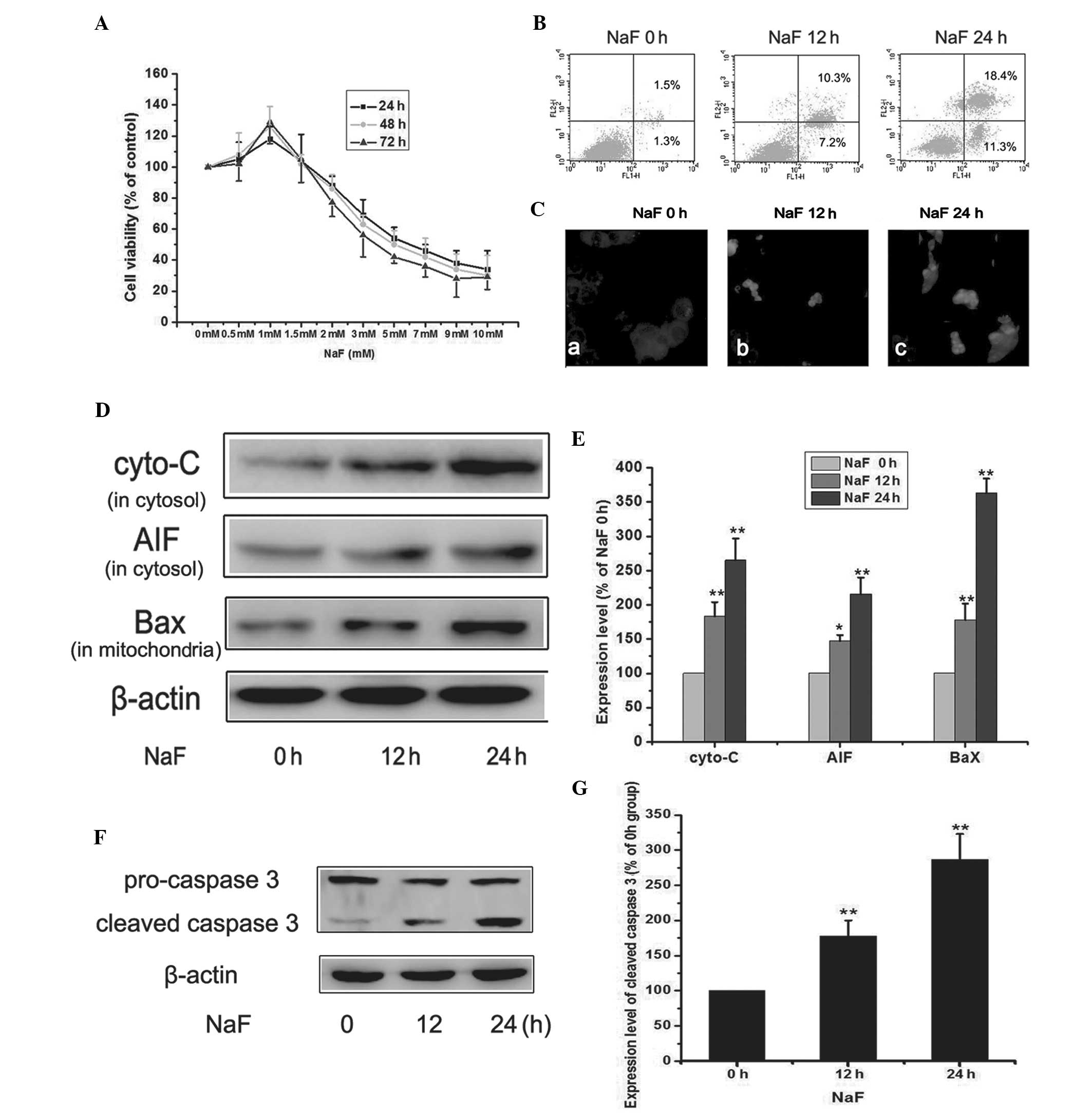
The effects of fluoride on osteoblastic MC3T3-E1
cells and the mechanism(s) by which fluoride affected MC3T3-E1 cell
apoptosis were assessed at different concentrations. Osteoblastic
MC3T3-E1 cells were treated with fluoride at concentrations ranging
from 0 to 10 mM for 24, 48 or 72 h, respectively. Cell viability
assays demonstrated that fluoride inhibited cell growth in a dose-
and time-dependent manner (Fig.
1A). The IC50-values of fluoride at 24, 48 and 72 h
were 3.51, 5.02 and 6.36 mM. Cells were treated with 5.0 mM
fluoride for 0, 12 and 24 h and apoptosis was determined by flow
cytometry followed by Annexin V/PI double staining. Flow cytometry
assays demonstrated a substantial increase in the apoptotic
population among cells treated with fluoride at 12 and 24 h
(Fig. 1B). From the results of the
TUNEL assay, it was demonstrated that apoptosis was induced
following incubation of MC3T3-E1 cells with fluoride. To further
confirm that apoptosis was induced by fluoride, western blot
analysis was performed to detect the expression of AIF (in
cytosol), cytochrome-C (in cytosol) and Bax (in the mitochondria)
as well as caspase 3. As displayed in Fig. 1D, the expression of Bax in
mitochondria was significantly increased in the fluoride groups
compared with the control group (P<0.01), suggesting that the
translocation of Bax into the mitochondria was involved in cell
death induced by fluoride. Mitochondria-mediated apoptosis
comprises caspase-dependent and caspase-independent processes,
while AIF is involved in the caspase-independent response. As
displayed in Fig. 1D, the
expression of AIF and cytochrome-C in the cytosol were
significantly increased in the fluoride groups compared with the
control group (P<0.01) while the expression of Bax (in the
mitochondria) was upregulated in the fluoride group. These results
demonstrate that the release of AIF from the mitochondria into the
cytosol was involved in cell death. Similarly, caspase-3 was
examined by western blot analysis. Levels of cleaved caspase 3 in
fluoride groups were upregulated compared with the control group
(Fig. 1F). These data indicated
that fluoride-induced apoptosis in MC3T3-E1 cells proceeded via a
mitochondrial pathway involving the caspase-dependent (caspase 3)
and the caspase-independent (AIF) pathways.
Fluoride induces MC3T3-E1 cell
autophagy
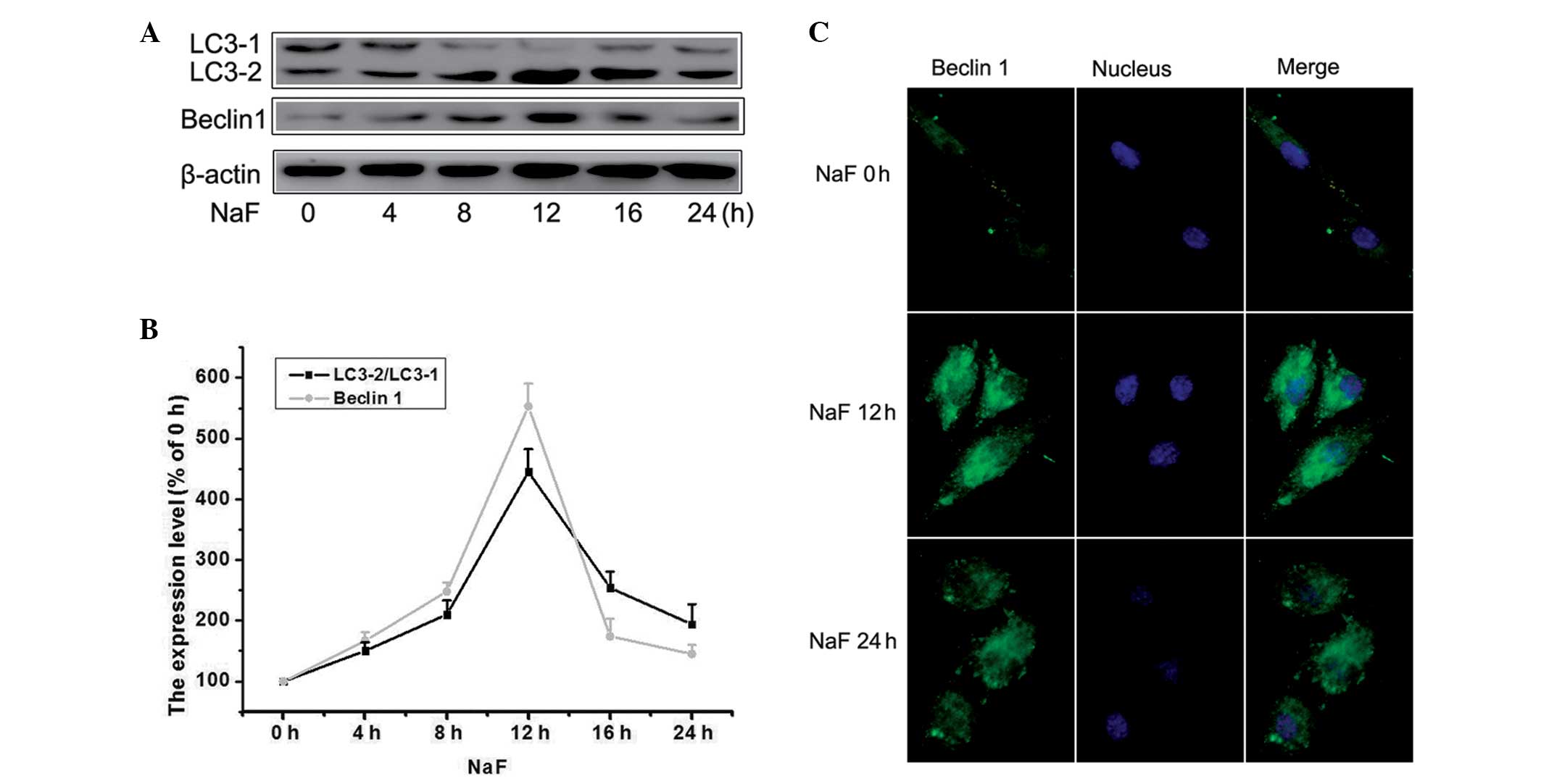
To determine whether mitochondrial damage was a
cause or consequence of autophagy, cells were treated with 5.0 mM
of fluoride for 0, 4, 8, 12, 16 and 24 h and the expression of
Beclin 1 and Lc3-2/1 were detected by western blot analysis. The
occurrence of autophagy following exposure of cells to fluoride was
assessed. As shown in Fig. 2A and
B, the levels of Lc3-2/1 began to increase after 8 h, peaked at
12 h, remained at this level until 16 h and decreased after 16 h of
incubation. Similar changes were observed in regard to the
expression of Beclin 1, suggesting that fluoride not only induced
apoptosis but also induced autophagy in MC3T3-E1 cells. Cultured
with fluoride, autophagy was quickly activated along with
apoptosis. From the results of fluorescence microscopy, fluoride
led to enhanced levels of Beclin 1, with induction being sustained
upon fluoride stimulation compared with the control (Fig. 2C). The fluorescence intensity in
the 12 h group was greater than that in the 24 h group. These
findings indicated that fluoride did not block autophagic flux, but
induced autophagic activity.
Fluoride treatment activates JNK to
induce autophagy
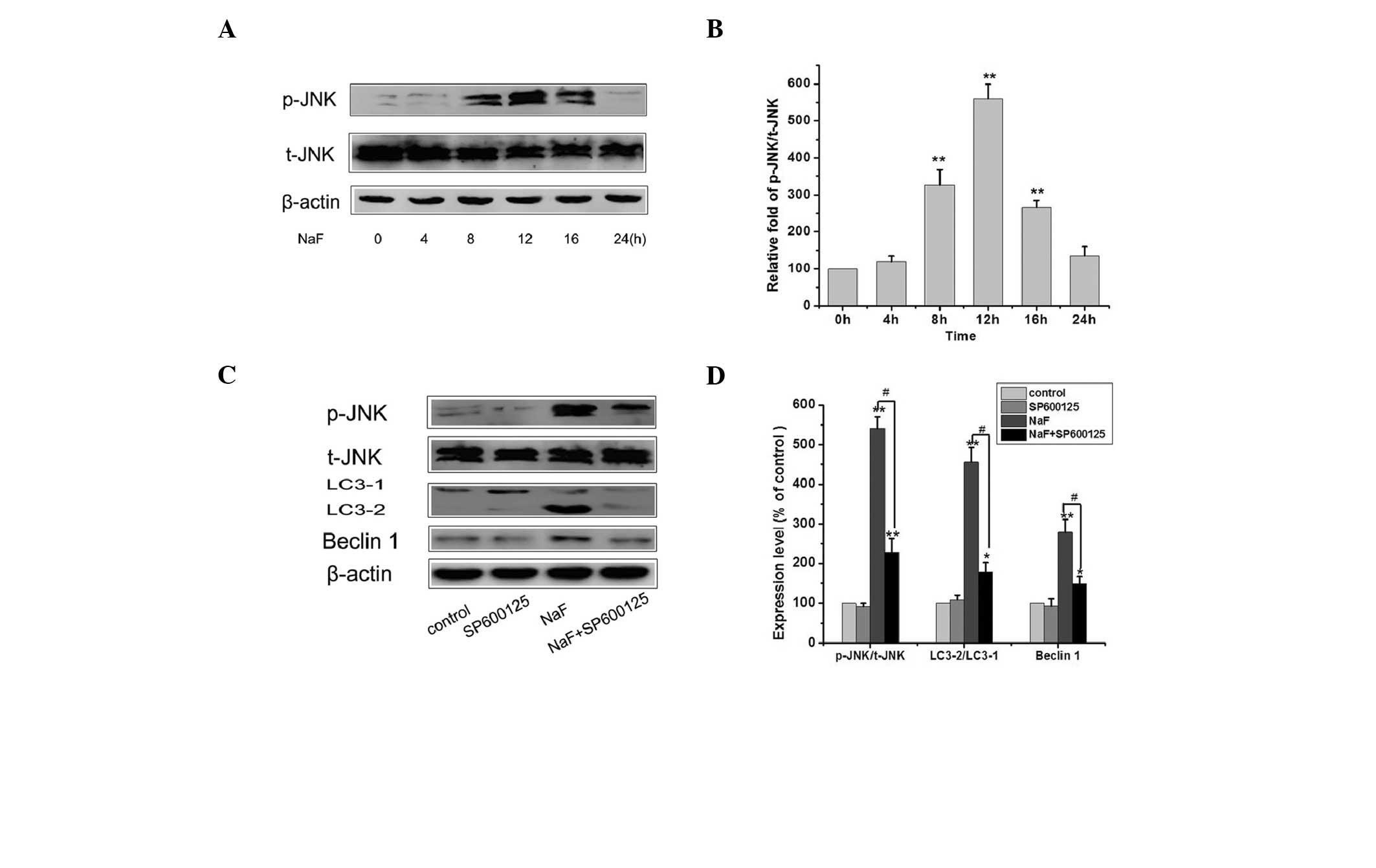
Studies have shown that JNK activation promotes
autophagy in different cell lines (15,16).
To investigate the role of JNK in fluoride-induced autophagy,
MC3T3-E1 cells were treated with 5.0 mM fluoride for 0–24 h and the
JNK level was detected using western blotting. As displayed in
Fig. 3A, incubation with fluoride
increased the phosphorylation of JNK from 8 to 16 h. However, the
co-treatment with 1 μM SP600125 (an inhibitor of the MAPK/JNK
pathway; concentration from reference 17), effectively inhibited JNK
phosphorylation and suppressed fluoride-induced LC3-2 protein
(Fig. 3C). These results suggest
that the MAPK/JNK pathway is involved in to the autophagy of
MC3T3-E1 cells induced by fluoride. Fluoride-induced autophagy is
mediated by JNK activation.
Autophagy induced by fluoride protects
MC3T3-E1 cells from undergoing apoptosis
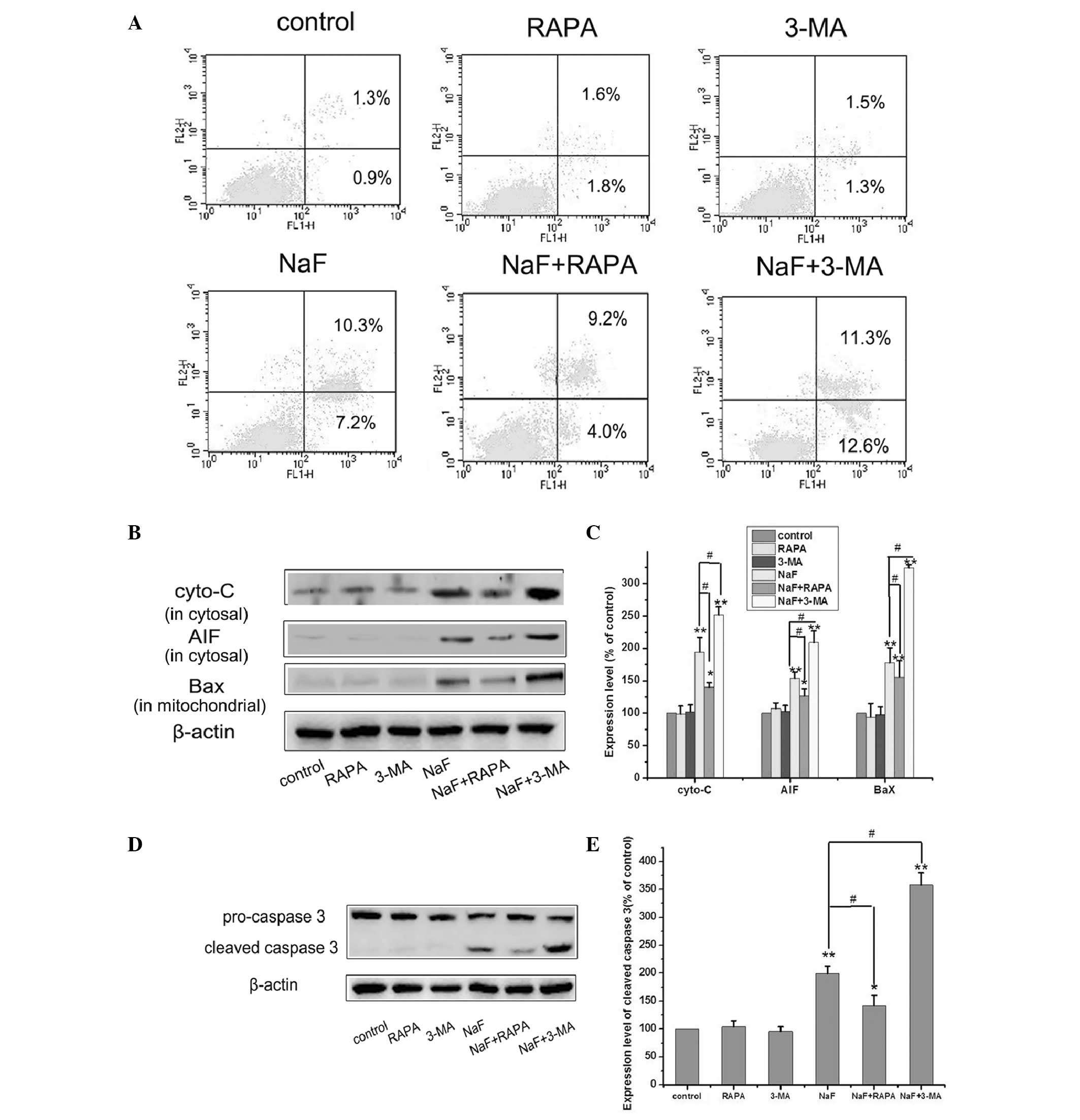
To investigate the effect of autophagy on apoptosis,
3-methyladenine (3-MA), a potent pharmacological inhibitor of
autophagy, was used to suppress fluoride-induced autophagy. It was
demonstrated that pretreatment with 5 mM 3-MA (the concentration of
3-MA from reference 18 was used)
was able to block autophagy in MC3T3-E1 cells without significant
cytotoxicity. 3-MA itself scarcely induced apoptosis and cell
death; however it significantly increased apoptosis at 12 h
following fluoride exposure (fluoride group: 17.5%; fluoride plus
3-MA: 23.8%) (Fig. 4A). These
results suggested that suppression of autophagy by 3-MA was able to
increase fluoride-induced injury in MC3T3-E1 cells. By contrast,
fluoride-induced apoptosis decreased following co-treatment with 2
μM of the autophagy-inducer RAPA (the concentration of RAPA from
reference 19 was used) relative
to cells treated with fluoride alone (Fig. 4A) (fluoride group: 17.5%; fluoride
plus RAPA: 13.2%). Treatment of normal MC3T3-E1 cells with RAPA or
3-MA alone did not affect cell viability (Fig. 4A). The western blot analysis
offered further evidence. The inhibitory effect of 3-MA the
inductive effect of RAPA on autophagy in fluoride-induced apoptosis
were also evaluated by western blot analysis. The upregulation of
the expression of the apoptotic protein AIF (in the cytosol),
cytochrome-C (in the cytosol), Bax (in the mitochondria) and
cleaved caspase 3 were more apparent when cells were co-treated
with 3-MA but were attenuated by co-treatment with RAPA, as shown
in Fig. 4B–E. These results
indicate that autophagy induced by fluoride served a protective
function and blockage of autophagy enhanced the apoptotic effect of
fluoride in MC3T3-E1 cells.
Discussion
Fluoride is an essential trace element, which has
been demonstrated to increase bone formation and promote cell
proliferation in low concentrations for all mammalian species
(20). Previous studies
demonstrated that fluoride leads to the proliferation of
osteoblasts, promotes their differentiation and modulates the
activity of growth factors, including insulin-like growth factor
(20). Fluoride also increases
alkaline phosphatase-specific activity and promotes bone formation
in rats in vivo (21).
However, excess fluoride may give rise to a number of adverse
effects. Studies have demonstrated that excess fluoride is able to
downregulate the expression of the collagen I gene and induce
apoptosis in newborn rat osteoblasts (22). The expression of insulin-like
growth factor-I was also decreased when cultured with high
concentrations of fluoride in mouse osteoblasts (23). Fluoride was able to induce
osteoblast cell apoptosis; however, the mechanism of apoptosis was
not clear. The expression of Bax is closely associated with
mitochondrial apoptosis, which is important in the regulation of
cell apoptosis (24). The present
study demonstrated that fluoride increased the expression of Bax.
Thus, this type of apoptosis is a mitochondria-mediated apoptosis.
Mitochondria-mediated apoptosis comprises caspase-dependent and
caspase-independent processes. Cytochrome-C is linked to
caspase-dependent apoptotic signaling, whereas AIF is involved in
the caspase-independent response (25). The present study demonstrated that
the levels of the two proteins significantly increased in MC3T3-E1
cells induced by fluoride. Therefore, it is hypothesized that
apoptosis caused by fluoride appears to proceed via mitochondrial
dysfunction linked to caspase-dependent/-independent pathways.
The link between apoptosis and autophagy is complex
and difficult to elucidate. The process of apoptosis is often
accompanied by the occurrence of autophagy (26). The present study demonstrated that
fluoride not only induced apoptosis but also induced autophagy in
MC3T3-E1 cells. As displayed in Fig.
2, the autophagy proteins, including Beclin 1 and LC3, were
upregulated by fluoride. Furthermore, in accordance with previous
studies, the present study revealed that fluoride activated
autophagy by initiation of the JNK signaling pathway (27). Autophagy is generally considered to
be a survival mechanism of cells (28). It was demonstrated that the
suppression of autophagy by 3-MA was able to increase
fluoride-induced injury in MC3T3-E1 cells. By contrast,
fluoride-induced apoptosis was decreased following co-treatment
with the autophagy-inducer RAPA. The results suggest that autophagy
is a pro-survival mechanism for fluoride-treated MC3T3-E1 cells and
reduces the incidence of apoptosis, facilitating the survival of
the cells. Several mechanisms for autophagy-limiting apoptosis have
been discussed. For example, the release of B-cell lymphoma 2 and
FLICE-like inhibitory protein (FLIP: A member of the tumour
necrosis factor signaling pathway and a regulator of apoptosis)
from autophagocytosis-associated protein Atg3 protein complexes may
inhibit the pathways of apoptosis (29). Hypoxia-inducible factor and
autophagy are closely correlated and may be important in
anti-apoptotic signaling (30).
However, the precise mechanism for autophagy-mediated
anti-apoptosis remains to be elucidated and is under investigation
in our laboratory.
Abbreviations:
|
α-MEM
|
α-Minimal essential medium
|
|
MAPK
|
mitogen-activated protein kinase
|
|
3-MA
|
3-methyladenine
|
|
JNK
|
c-Jun N-terminal kinase
|
|
RAPA
|
autophagy-inducer rapamycin
|
References
|
1
|
Alonso Á and Camargo JA: A video-based
tracking analysis to assess the chronic toxic effects of fluoride
ion on the aquatic snail Potamopyrgus antipodarum (Hydrobiidae,
Mollusca). Ecotoxicol Environ Saf. 81:70–75. 2012.PubMed/NCBI
|
|
2
|
Mascarenhas AK: Risk factors for dental
fluorosis: a review of the recent literature. Pediatr Dent.
22:269–277. 2000.PubMed/NCBI
|
|
3
|
Rawlani S and Rawlani S and Rawlani S:
Assessment of Skeletal and Non-skeletal Fluorosis in Endemic
Fluoridated Areas of Vidharbha Region, India: A Survey. Indian J
Community Med. 35:298–301. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li Y, Berenji GR, Shaba WF, Tafti B, et
al: Association of vascular fluoride uptake with vascular
calcification and coronary artery disease. Nucl Med Commun.
33:14–20. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stroikin Y, Dalen H, Lööf S, et al:
Inhibition of autophagy with 3-methyladenine results in impaired
turnover of lysosomes and accumulation of lipofuscin-like material.
Eur J Cell Biol. 83:583–590. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dutta D, Xu J, Kim JS, et al: Upregulated
autophagy protects cardiomyocytes from oxidative stress-induced
toxicity. Autophagy. 9:328–344. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jacquel A, Obba S, Boyer L, et al:
Autophagy is required for CSF-1-induced macrophagic differentiation
and acquisition of phagocytic functions. Blood. 119:4527–4531.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Graziotto JJ, Cao K, Collins FS, et al:
Rapamycin activates autophagy in Hutchinson-Gilford progeria
syndrome: implications for normal aging and age-dependent
neurodegenerative disorders. Autophagy. 8:147–151. 2012. View Article : Google Scholar
|
|
9
|
Joubert PE, Werneke SW, de la Calle C, et
al: Chikungunya virus-induced autophagy delays caspase-dependent
cell death. J Exp Med. 209:1029–1047. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou F, Yang Y and Xing D: Bcl-2 and
Bcl-xL play important roles in the crosstalk between autophagy and
apoptosis. FEBS J. 278:403–413. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bhogal RH, Weston CJ, Curbishley SM, et
al: Autophagy: a cyto-protective mechanism which prevents primary
human hepatocyte apoptosis during oxidative stress. Autophagy.
8:545–558. 2012. View Article : Google Scholar
|
|
12
|
Hui B, Shi YH, Ding ZB, et al: Proteasome
inhibitor interacts synergistically with autophagy inhibitor to
suppress proliferation and induce apoptosis in hepatocellular
carcinoma. Cancer. 118:5560–5571. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ponnusamy M, Liu N, Sellamuthu R, et al:
Autophagy protects against necrotic renal epithelial cell-induced
death of renal interstitial fibroblasts. Am J Physiol Renal
Physiol. 303:F83–F91. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wessel D and Flügge UI: A method for the
quantitative recovery of protein in dilute solution in the presence
of detergents and lipids. Anal Biochem. 138:141–143. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu P, Das M, Reilly J, et al: JNK
regulates FoxO-dependent autophagy in neurons. Genes Dev.
25:310–322. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Komiya K, Uchida T, Ueno T, et al: Free
fatty acids stimulate autophagy in pancreatic β-cells via JNK
pathway. Biochem Biophys Res Commun. 401:561–567. 2010.PubMed/NCBI
|
|
17
|
Yang CM, Chien CS, Yao CC, et al:
Mechanical strain induces collagenase-3 (MMP-13) expression in
MC3T3-E1 osteoblastic cells. J Biol Chem. 279:22158–22165. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zeng Y, Yang X, Wang J, et al:
Aristolochic acid I induced autophagy extenuates cell apoptosis via
ERK 1/2 pathway in renal tubular epithelial cells. PLoS One.
7:e303122012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Amarnath S, Flomerfelt FA, Costanzo CM, et
al: Rapamycin generates anti-apoptotic human Th1/Tc1 cells via
autophagy for induction of xenogeneic GVHD. Autophagy. 6:523–541.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ren G, Wang K, Chang R, et al:
Simultaneous administration of fluoride and selenite regulates
proliferation and apoptosis in murine osteoblast-like MC3T3-E1
cells by altering osteoprotegerin. Biol Trace Elem Res.
144:1437–1448. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mousny M, Omelon S, Wise L, et al:
Fluoride effects on bone formation and mineralization are
influenced by genetics. Bone. 43:1067–1074. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yan X, Yan X, Morrison A, et al: Fluoride
induces apoptosis and alters collagen I expression in rat
osteoblasts. Toxicol Lett. 200:133–138. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Z, Yang X, Yang S, et al: Sodium
fluoride suppress proliferation and induce apoptosis through
decreased insulin-like growth factor-I expression and oxidative
stress in primary cultured mouse osteoblasts. Arch Toxicol.
85:1407–1417. 2011. View Article : Google Scholar
|
|
24
|
Chipuk JE, McStay GP, Bharti A, et al:
Sphingolipid metabolism cooperates with BAK and BAX to promote the
mitochondrial pathway of apoptosis. Cell. 148:988–1000. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gu B, Zhang GF, Li LY, et al: Human
herpesvirus 6A induces apoptosis of primary human fetal astrocytes
via both caspase-dependent and -independent pathways. Virol J.
8:5302011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang T, Li Y, Park KA, et al:
Cucurbitacin induces autophagy through mitochondrial ROS production
which counteracts to limit caspase-dependent apoptosis. Autophagy.
8:559–576. 2012. View Article : Google Scholar
|
|
27
|
Rodríguez-Blanco J, Martín V,
García-Santos G, et al: Cooperative action of JNK and AKT/mTOR in
1-methyl-4-phenylpyridinium-induced autophagy of neuronal PC12
cells. J Neurosci Res. 90:1850–1860. 2012.PubMed/NCBI
|
|
28
|
Li S, Zhou Y, Fan J, et al: Heat shock
protein 72 enhances autophagy as a protective mechanism in
lipopolysaccharide-induced peritonitis in rats. Am J Pathol.
179:2822–2834. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Marquez RT and Xu L: Bcl-2: Beclin 1
complex: multiple, mechanisms regulating autophagy/apoptosis toggle
switch. Am J Cancer Res. 2:214–221. 2012.PubMed/NCBI
|
|
30
|
Yeh CH, Hsu SP, Yang CC, et al: Hypoxic
preconditioning reinforces HIF-alpha-dependent HSP70 signaling to
reduce ischemic renal failure-induced renal tubular apoptosis and
autophagy. Life Sci. 86:115–123. 2010. View Article : Google Scholar
|