Introduction
Vascular smooth muscle cells (VSMCs) are one of the
major types of cell in the blood vessel, and their proliferation is
associated with cardiovascular diseases and atherosclerosis (AS)
(1). Numerous clinical and
experimental studies have investigated the pathogenic mechanism
underlying VSMC proliferation (2–5) in
order to identify potential therapeutic targets for VSMC
proliferation in cardiovascular diseases. However, at present, such
targets are yet to be elucidated.
Evidence has revealed a marked stimulatory effect of
homocysteine (Hcy) on VSMC proliferation (6). Hcy has been reported to be an
independent risk factor for AS (7), and mild to moderate increases in Hcy
serum levels, which may be due to nutritional or genetic factors,
are frequently observed in patients with AS; therefore, the
stimulatory effect of Hcy on VSMC proliferation may be highly
involved in AS pathogenesis. The identification of the molecular
mechanisms underlying Hcy-induced VSMC proliferation may be
particularly beneficial.
Hcy is a non-protein, sulfur-containing amino acid,
which is formed exclusively upon demethylation of
S-adenosylmethionine (SAM), the active form of methionine. SAM
serves as the primary methyl donor for >100 transmethylation
reactions, including DNA methylation modification (8). Following the transfer of its methyl
group, SAM is converted to S-adenosylhomocysteine (SAH), which is a
potent inhibitor of SAM-dependent methyltransferases (9). SAH then undergoes hydrolysis to form
Hcy. In the methionine cycle, Hcy is remethylated to generate
methionine and further activated to produce SAM. Hcy may also
undergo hydrolysis. Increased Hcy levels may interfere with the
methionine cycle and, consequently, DNA methylation
modification.
Modification of DNA methylation is the primary
mechanism of epigenetic gene regulation (10). Generally, hypermethylation
inhibits, while hypomethylation promotes, gene expression (11). The involvement of aberrant DNA
methylation has been confirmed in the pathogenesis of various
diseases, including cancer and AS (12). Our previous study showed that
Hcy-induced aberrant DNA methylation patterns in AS affect numerous
genes, including peroxisome proliferator-activated receptor α and
apolipoprotein E (13). However,
the methylation pattern of the platelet-derived growth factor
(PDGF) gene, which is a potent mitogen for VSMC proliferation, is
yet to be elucidated in Hcy-treated VSMCs.
PDGF has significant roles in developmental and
physiological processes, and has also been implicated in various
proliferative disorders. PDGF has been shown to be a potent
stimulator of VSMC growth (2).
Furthermore, PDGF has been identified as an important mediator of
VSMC proliferation, with potential mechanisms including activation
of the Ras pathway, phosphoinositol 3′-kinase, promotion of VSMC
extracellular Ca2+ influx and release of intracellular
Ca2+ (14–17). The impact of Hcy on the epigenetic
regulation of PDGF, which may influence PDGF expression and be
involved in VSMC proliferation, may provide novel insight into the
pathogenesis of AS.
In the present study, Hcy-induced VSMC
proliferation, PDGF expression and the methylation pattern of the
PDGF gene were investigated. Furthermore, their causative
correlation and the potential mechanism were also investigated, in
order to identify a useful target for the prevention and treatment
of AS induced by Hcy.
Materials and methods
Cell culture
The study was approved by the ethics committee of
Ningxia Medical University (Yinchuan, China). Primary cultured
VSMCs were obtained from human umbilical vein media (Department of
Obstetrics, General Hospital of Ningxia Medical University,
Yinchuan, China). Cells were cultured in Dulbecco’s Modified
Eagle’s medium-Ham’s F12 media (Gibco-BRL, Gaithersburg, MD, USA)
supplemented with 20% fetal calf serum (FCS; Gibco-BRL), 100 U/ml
penicillin and streptomycin (Sigma-Aldrich Trading Co., Ltd,
Shanghai, China) at 37°C in an incubator with 5% CO2.
The purity of the primary VSMC culture was confirmed by the
characteristic ‘hill-and-valley’ growth pattern and
immunocytochemistry of α-actin. VSMCs were used at between three
and five passages for the experiment. Cells were seeded onto
six-well plates and grown to 80% confluence. Cells were then
serum-deprived for 24 h to reach synchrony, followed by the
addition of 5% FCS for a further 24 h and the administration of Hcy
(Sigma-Aldrich Trading Co., Ltd). Hcy was administered at the
following concentrations: 0 (control), 50, 100, 200 and 500 μM and
500 μM plus folate (Sigma-Aldrich Trading Co., Ltd), respectively.
Hcy was replenished every 8 h in compensation for its short
half-life (total three times).
Cell viability assay
To assess cell proliferation, VSMCs were seeded in
96-well plates at 103–104 cells in 100
μl/well. The aforementioned concentrations of Hcy were then added
and the cells were incubated for 72 h. Hcy was replenished every 8
h. A total of 20 μl MTT (5 mg/ml; Sigma-Aldrich Trading Co., Ltd)
was added to each well and incubated at 37°C for 4 h. The
supernatant was removed using a pipette, and 150 μl dimethyl
sulfoxide was added to each well. After 10 min of incubation at
room temperature, plates were read on a micro-enzyme-linked
immunosorbent assay reader (Bio-Tek Instruments, Inc., Winooski,
VT, USA) at 490 nm. Values were normalized using the control
value.
Flow cytometric analysis
Following treatment with various concentrations of
Hcy, cells were analyzed using a FACStar™ Plus flow cytometer
(Becton, Dickinson and Company, Franklin Lakes, NJ, USA). In brief,
cells were washed once with phosphate-buffered saline (PBS; 0.01
mol/l, pH 7.2) and fixed at −10°C for 5 min in
paraformaldehyde-lysine-periodate fixation solution. Cells were
then treated with 0.1% Triton X-100 and 0.5% RNase A, followed by
the addition of propidium iodide. All samples were passed through
70-mm mesh prior to flow cytometric analysis; 30,000 nuclei were
examined in each analysis. The percentage of cells was estimated
using the Cell FIT analysis version 2.0 software (Becton, Dickinson
and Company). Three separate experiments were performed with three
different populations of cells.
Quantitative polymerase chain reaction
(qPCR)
Total RNA was isolated using TRIzol®
reagent (Invitrogen Life Technologies, Grand Island, NY, USA) and
reverse transcribed using the RevertAid First Strand cDNA Synthesis
kit (MBI Fermentas, Vilnius, Lithuania). GAPDH was used as an
endogenous control, and the primer sequences were as follows: PDGF,
5′-TCTGCTGCTACCTGCGTCTGG-3′ (forward) and
5′-CACTGCACGTTGCGGTTGTT-3′ (reverse) and GAPDH,
5′-AGAAGGCTGGGGCTCATTTG-3′ (forward) and 5′-AGGGGCCATCCACAGTCTTC-3′
(reverse). qPCR analysis was performed using an FTC-3000 Real-Time
PCR detection system (Funglyn Biotech Corp. Ltd., Toronto, ON,
Canada). The RNA levels of each gene were calculated using the
cycle threshold (Ct) value of the sample relative to that of GAPDH
using the following formula:
Ct=Ct(GAPDH)−Ct(sample). Final results,
expressed as n-fold differences in target gene expression relative
to the calibrator, termed Ntarget, were calculated using
the following formula:
Ntarget=2Ct(sample)−Ct(calibrator), where Ct
values of the calibrator and sample were determined by subtracting
the Ct value of the target gene from the Ct value.
Western blot analysis
Proteins were extracted from the cells using cell
lysis buffer and separated using 12% SDS-PAGE. The proteins and the
prestained marker (MBI Fermentas, Amherst, NY, USA) were then
transferred onto a polyvinylidene fluoride Immobilon®-P
Transfer Membrane (Millipore, Billerica, MA, USA) with a pore size
of 0.45 mm using a Trans-Blot® Semi-Dry Transfer Cell
model 755 (Bio-Rad, Hercules, CA, USA) for 90 min. The membrane was
incubated at room temperature in PBS-Tween 20 (PBS-T) buffer
containing 5% non-fat milk for 4 h. Membranes were then cut as
required and placed in a hybridization bag with 1 ml anti-β-actin
or -PDGF primary antibodies (Sigma-Aldrich, St. Louis, MO, USA) and
incubated at 4°C overnight. The membranes were subsequently washed
three times using PBS-T buffer and the secondary antibody was added
for 2 h at room temperature. The membranes were washed a further
three times with Tris buffered saline with Tween 20 and incubated
with horseradish peroxidase substrate (BeyoECL Plus A/B; Beyotime
Institute of Biotechnology, Shanghai, China) for 1 min. Blots were
developed using X-film (Kodak, Tokyo, Japan).
Detection of PDGF gene methylation using
nested methylation-specific (nMS)-PCR
Genomic DNA was isolated from VSMCs using the
Wizard® Genomic DNA purification kit (Promega Corp.,
Madison, WI, USA) and nMS-PCR was performed for the detection of
PDGF gene methylation. Following standard sodium bisulfite DNA
modification using the EZ DNA Methylation-Gold™ kit (Zymo Research,
Irvine, CA, USA), two-step PCR amplifications were performed.
nMS-PCR initially uses an outer primer pair that does not contain
any CpG sites, followed by second step PCR using conventional PCR
primers. CpG islands were identified in the PDGF promoter region
using a CpG Island Search engine (http://www.uscnorris.com/cpgislands2/cpg.aspx;
http://www.cbs.dtu.dk/services/promoter/). The primers
used in the nMS-PCR assay were as follows: PDGF-outer primer,
5′-TTTTTTTGTTTTGAAATTTTGGTTAA-3′ (forward) and
5′-CAAAATCCCAACAAAAAAAATCTCC-3′ (reverse); PDGF-methylated primer,
5′-TTTGGAAATTAATGATAAGTTAGGC-3′ (forward) and
5′-AAGCATCATAAAAAACAAACGCATC-3′ (reverse); PDGF-unmethylated
primer, 5′-TTGGAAATTAATGATAAGTTAGGTGA-3′ (forward) and
5′-AAACATCATAAAAAACAAACACATCA-3′ (reverse). To reduce mispriming
and to increase efficiency, touchdown PCR was used for the
amplification. The PCR products were separated using
electrophoresis on a 2% agarose gel containing ethidium bromide.
DNA bands were visualized using ultraviolet light and methylation
was calculated using the following formula: Methylation % =
methylation/(methylation + unmethylation) × 100.
SAM and SAH concentrations examined by
high-performance liquid chromatography (HPLC)
The concentrations of SAM and SAH were determined
using HPLC. Cells were centrifuged, washed twice with cold PBS and
maintained on ice. The cell pellets were subsequently homogenized
in four volumes of 0.4 μM HClO4. A 200 μl aliquot of the
acid extract was loaded into a C18 column (Shimadzu, Kyoto, Japan),
run by a Hitachi L2000 HPLC system (Hitachi, Tokyo, Japan). The
absorption of the eluted compounds was monitored at an excitation
wavelength of 254 nm. Elution of SAM and SAH was achieved at a flow
rate of 1.0 ml/min using mobile-phase ammonium formate solution.
Chromatograms were recorded using a D-2000 Elite integrator. SAM
and SAH standards were used to identify the elution peaks, and the
SAM and SAH tissue values were calculated using the standard
curve.
Endogenous C-5 DNA methyltransferase (C-5
MT-ase) activity assay
A modification of the assay developed by Hattori
et al (18) was used to
determine the activities of DNA methyltransferases. VSMCs
(1×106) were homogenized using a glass pestle containing
500 μl lysis buffer (100 mmol/l NaCl, 10 mmol/l Tris-HCl, pH 8.0,
25 mmol/l EDTA, 0.5% SDS and proteinase K 0.2 g/l). The suspension
was freeze-thaw cycled between −70 and 37°C three times. VSMC
protein exacts were stored at −70°C prior to analysis. The reaction
contained cell homogenates (5 μg protein), 0.25 μg
poly(deoxyinosinic-deoxycytidylic) acid and 11.1×1010%
Bq [methyl-3H] SAM in a total volume of 20 μl, and was
incubated at 37°C for 2 h. RNA was removed by adding 20 μl RNase A
(2 g/l) and incubating at room temperature for 5 min. DNA was
purified using the E.Z.N.A.® Cycle-Pure kit (Omega
Bio-Tek, Inc., Norcross, GA, USA), and the purified genomic DNA was
spotted onto a Whatman GF/C filter disc and dried at 80°C for 5
min, prior to counting in a Packard 1600 TR Liquid Scintillation
Counter (Packard Instrument Co., Meriden, CT, USA) for
determination of C-5 MT-ase activity. Each reaction was performed
in triplicate and the assay was repeated three times to blind the
source of the samples. In order to exclude background protein, all
samples were initially assayed with the control containing the
whole cell lysate, but without poly(deoxyinosinic-deoxycytidylic)
acid. C-5 MT-ase activity was expressed as the quantity of
incorporated [methyl-3H] groups into the
poly(deoxyinosinic-deoxycytidylic) acid (cpm/1 μg protein).
Statistical analysis
Each experiment was repeated three times. Results
are expressed as the mean ± standard deviation. Statistical
comparisons of single parameters between two groups were performed
using the paired Student’s t-test. Kruskal-Wallis one-way analysis
of variance was used to compare the means of multiple groups,
followed by the Dunn test. A value of P≤0.05 was considered to
indicate a statistically significant difference.
Results
Hcy increases VSMC viability and
stimulates the cell cycle
The primary culture of confluent VSMCs exhibited a
typical elongated ribbon or spindle-shaped appearance with a
characteristic ‘hill and valley’ pattern (Fig. 1A), and immunocytochemistry revealed
98% positive α-actin staining (Fig.
1B). MTT assay showed that VSMC viability significantly
increased by 1.2-, 2.2-, 3.6- and 4.3-fold subsequent to treatment
with 50, 100, 200 and 500 μM Hcy, respectively, compared with the
control group (P<0.01). Furthermore, folate treatment had an
antagonistic effect against Hcy-induced VSMC proliferation, with a
63% decrease in VSMC viability observed in the folate plus 500 μM
Hcy group compared with the 500 μM Hcy group (P<0.01) (Fig. 1C).
Flow cytometry revealed that, as Hcy concentration
increased, the proportion of the cell population in S phase
increased, and that in G0/G1 phase decreased.
Folate showed an inhibitory effect against Hcy on the VSMC cell
cycle (Fig. 2).
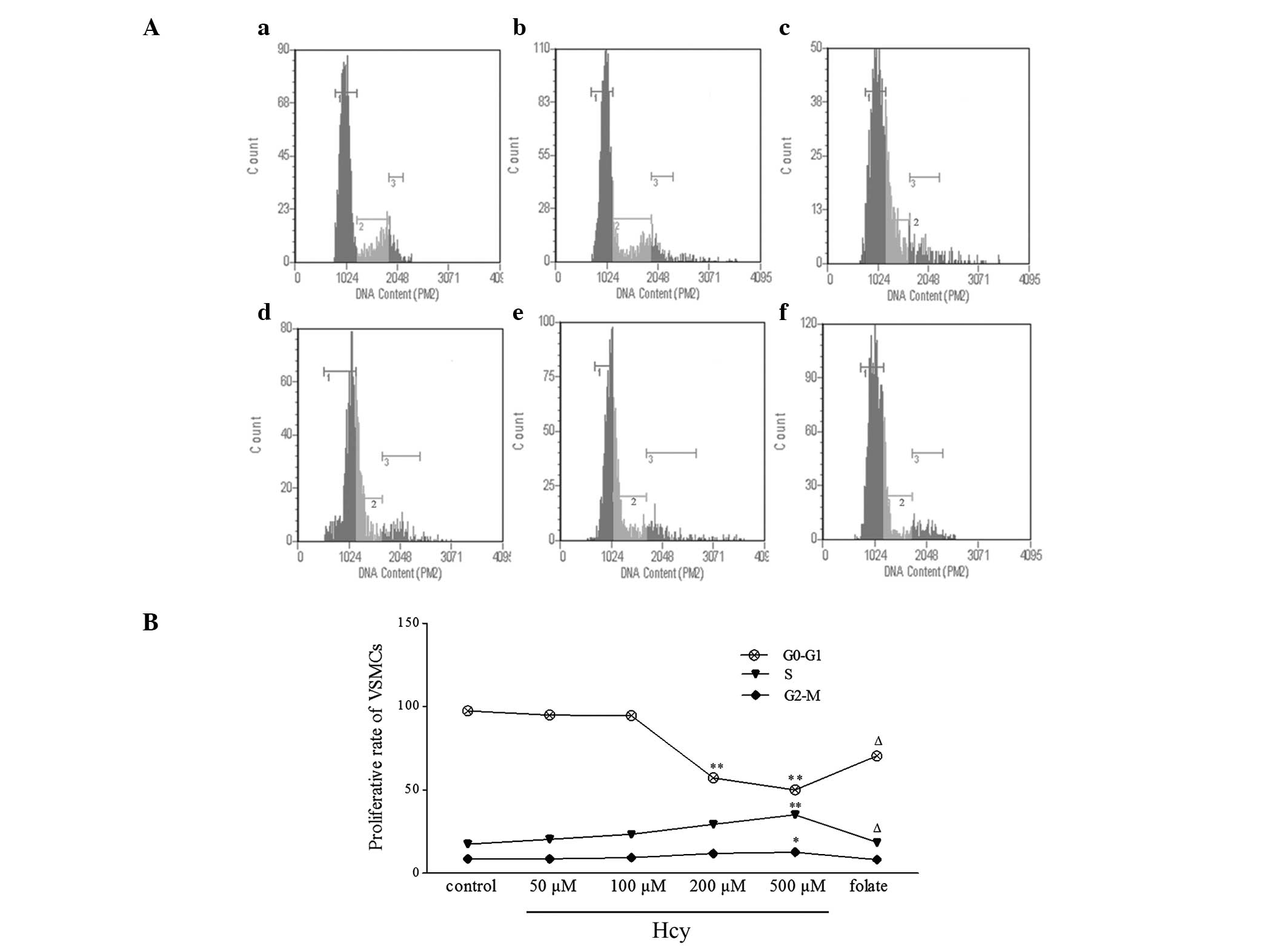 | Figure 2VSMC cell cycle phases following Hcy
treatment, detected using flow cytometry. (A) Representative flow
cytometric profiles demonstrating VSMC cell cycle distribution. (a)
0, (b) 50, (c) 100, (d) 200 and (e) 500 μM Hcy; 1,
G0/G1 phase; 2, S phase; 3, G2/M
phase. (B) Summary of cell cycle analysis, revealing an increased S
fraction and a decreased G0/G1 fraction in
Hcy-treated VSMCs. The values for each experiment were converted to
percentages relative to the corresponding control group (100%).
Values are presented as the mean ± standard deviation.
*P<0.05 and **P<0.01 vs. control group;
ΔP<0.05 vs. 500 μM Hcy group. VSMC, vascular smooth
muscle cell; Hcy, homocysteine. |
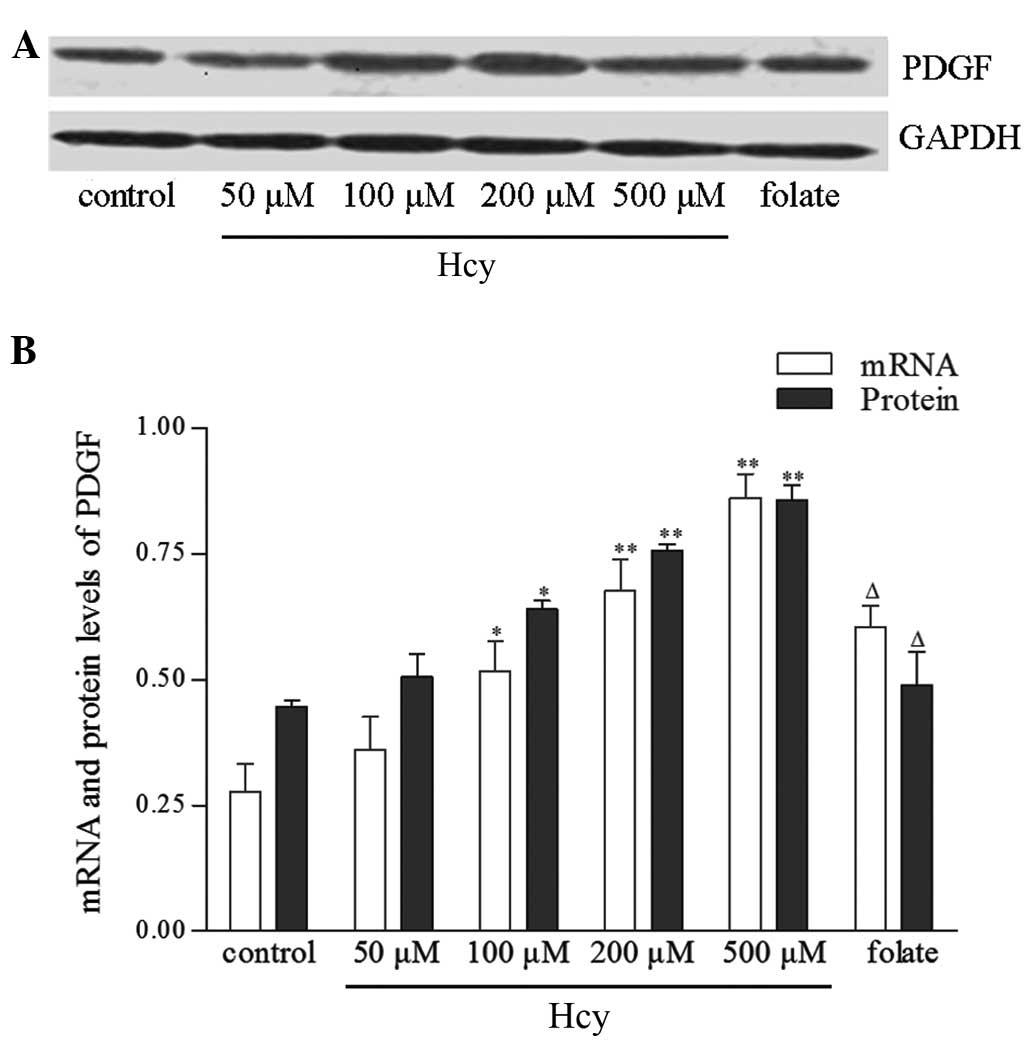
Hcy increases PDGF mRNA and protein
expression
qPCR and western blot analyses revealed a
dose-dependent increase in PDGF mRNA and protein expression with
Hcy treatment, in parallel with the increase in VSMC proliferation
observed under Hcy treatment. PDGF has a key role in VSMC
proliferation; therefore, this parallel alteration in VSMC PDGF
expression and proliferation suggests a causative role for PDGF in
Hcy-induced VSMC proliferation (Fig.
3).
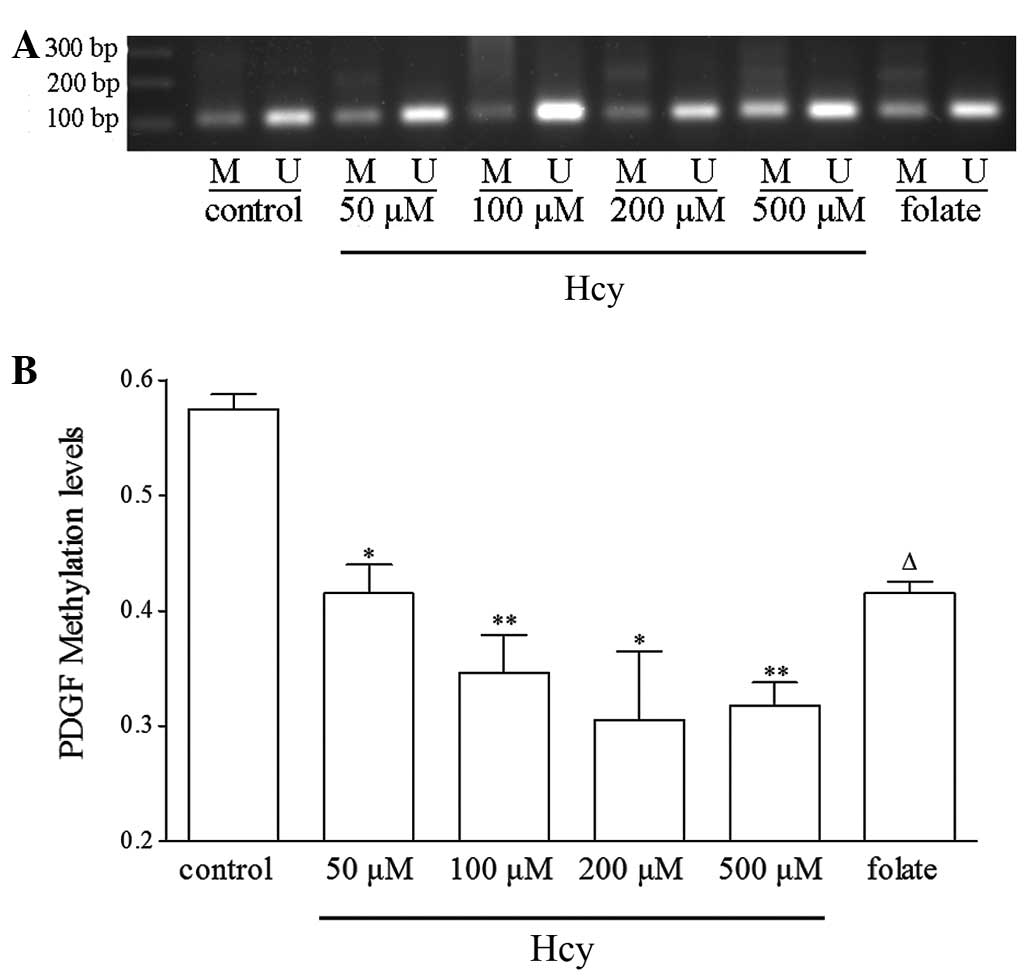
Hcy induces hypomethylation of the PDGF
gene
DNA methylation occurs almost exclusively at CpG
dinucleotides, and CpG methylation is often associated with gene
silencing and vice versa. The DNA methylation level of the CpG
islands in the PDGF gene promoter was analyzed using nMS-PCR. The
PDGF gene promoter was found to be significantly hypomethylated in
a dose-dependent manner upon Hcy treatment (Fig. 4). These data indicate that Hcy
treatment inhibits the methylation of the PDGF gene, and this
demethylation effect may be involved in the upregulation of PDGF
expression and VSMC proliferation in the pathogenesis of AS.
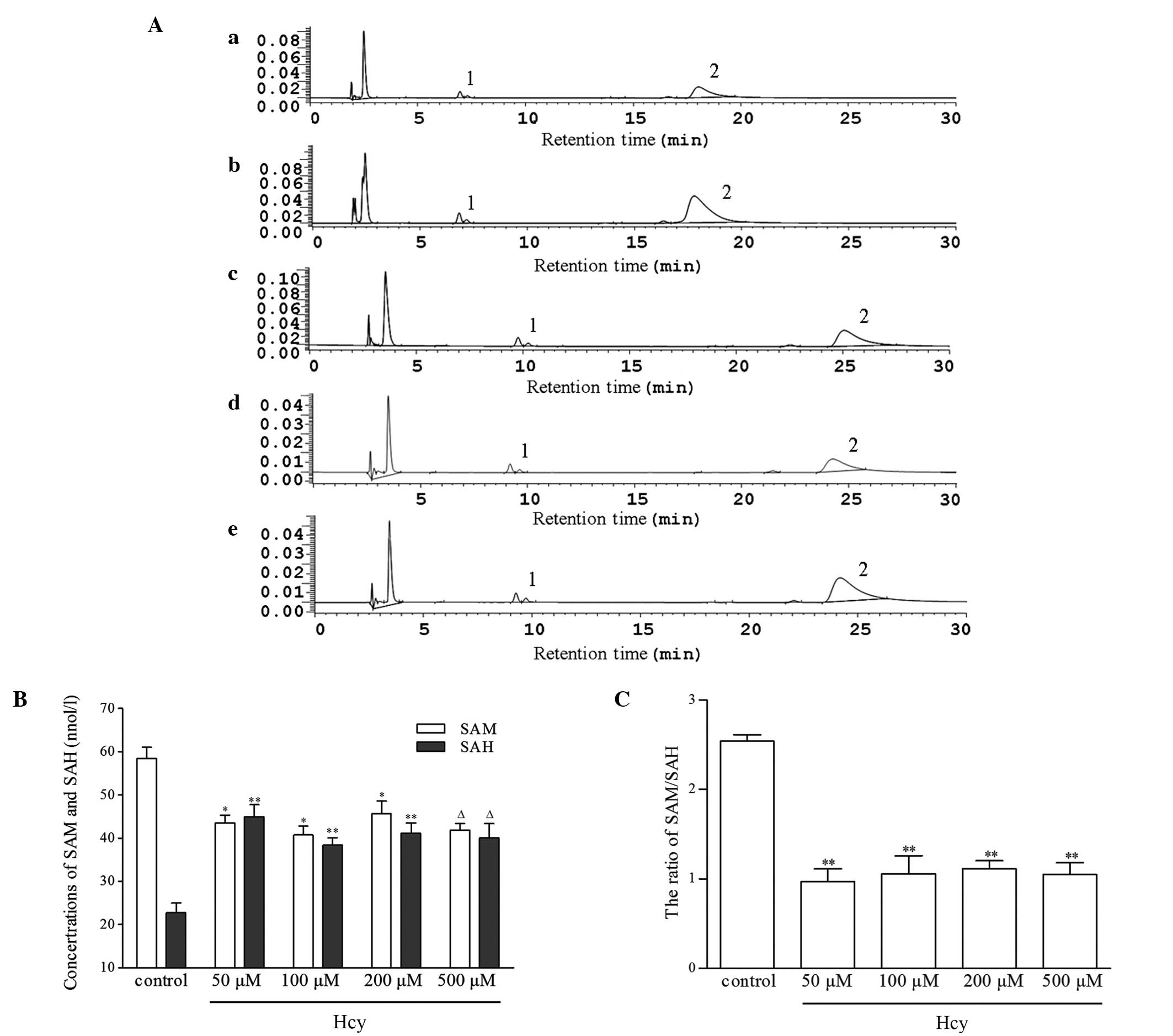
Hcy affects SAM and SAH levels and C-5
MT-ase activity
SAM and SAH concentrations are important factors in
the transmethylation process. In the present study, SAM and SAH
levels were assessed using HPLC. In the Hcy-treated groups, the
intracellular levels of SAH were significantly higher than those in
the control group, showing up to a three-fold increase. However,
upon Hcy treatment the concentration of SAM was observed to
decrease, leading to a significant decrease in the ratio of
SAM/SAH. SAM is a primary methyl-donor while SAH is a potent
inhibitor of methyltransferase activity; therefore, the SAM/SAH
ratio may be critical in the modification of DNA methylation.
However, the changes in SAM and SAH levels did not exhibit the
dose-effect trend with Hcy concentration (Fig. 5).
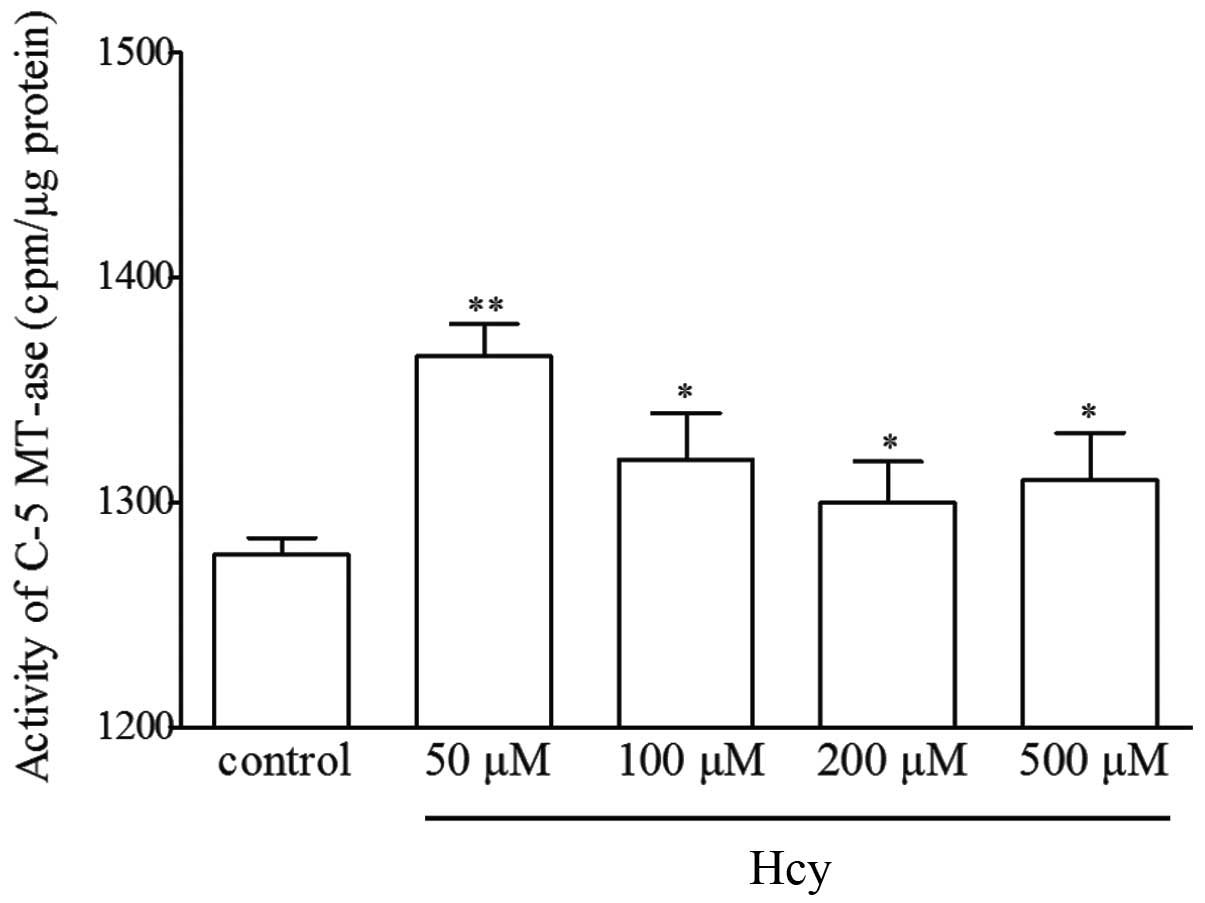
C-5 MT-ase activity is another key regulator of the
transmethylation reaction. Hcy was found to significantly
upregulate the activity of C-5 MT-ase compared with the control
cells (Fig. 6); however, no
significant dose-dependent increase was observed.
Discussion
This study has indicated that a significant
association exists among Hcy concentration, PDGF promoter
hypomethylation, upregulated PDGF expression and cell proliferation
in VSMCs. Despite the lack of direct evidence, the data of the
present study suggest a causative interrelation among these
processes. To the best of our knowledge, this is the first study to
suggest that Hcy-induced VSMC proliferation may be mediated by
epigenetic regulation of PDGF by Hcy.
VSMC proliferation is considered to be one of the
important pathological factors in AS (19). AS begins with eccentric thickening
of the intima, leading to complex lesions over several decades
(20). The thickening neointima is
predominantly composed of VSMCs, mesenchymal intimal cells and
inflammatory cells (21). The
pathogenesis of AS primarily involves changes in the expression and
function of genes, rather than gene mutations, and the
proliferation of VSMCs requires significant alterations in gene
expression, particularly in mitogenic genes (22). Numerous studies, including our
previous study (23), have
demonstrated that Hcy is capable of stimulating VSMC proliferation
(24). In the present study, PDGF
mRNA and protein levels showed a dose-dependent increase with Hcy
treatment, and cell cycle analysis further revealed an increased
proportion of cells advancing into S phase from
G0/G1-phase, suggesting that VSMCs
proliferated under Hcy treatment.
It is well established that PDGF is a potent
mitogenic factor, and has a critical role in normal embryonic
development, cellular differentiation and proliferation (25). Increased PDGF expression subsequent
to arterial injury has been associated with neointimal cellular
proliferation in studies of rabbits, birds, rats and humans
(26–29), demonstrating an association between
VSMC proliferation and PDGF. Furthermore, the pathological
processes that occur in proliferative diseases, such as AS, have
been successfully suppressed by inhibitors of PDGF signaling
(30). Sirois et al
(31) demonstrated that inhibition
of PDGF suppressed intimal thickening in the rat carotid artery
following balloon injury. Additionally, the administration of
anti-PDGF antibodies has been found to induce intimal atrophy in a
baboon graft model (32). These
observations support a pathogenic role for PDGF in proliferative
vascular diseases (33). In the
present study, Hcy was observed to induce a parallel alteration in
PDGF expression and VSMC proliferation, suggesting that Hcy may
stimulate VSMC proliferation through the PDGF signaling
pathway.
In the present study, PDGF promoter methylation was
detected in order to investigate the mechanism underlying
Hcy-induced PDGF overexpression. A positive, dose-dependent
correlation was identified between Hcy concentration, decreased
methylation of the PDGF promoter and upregulation of PDGF
expression. DNA methylation is a key form of the epigenetic genomic
modification that regulates gene expression, and hypomethylation of
the CpG islands in gene promoter regions is often associated with
increased gene expression. Therefore, Hcy-induced PDGF
hypomethylation may be responsible for the increased PDGF
expression.
DNA methylation status is significantly correlated
with the ratio of SAM/SAH (34).
In the present study, a decrease in the SAM/SAH ratio was observed
upon Hcy treatment, which may partially explain the demethylation
effect of Hcy on the PDGF gene. In the methionine cycle, which
connects Hcy metabolism and the transmethylation reaction of DNA,
SAH is hydrolyzed to form Hcy; therefore, increased levels of Hcy
inhibit the decomposition of SAH, resulting in SAH accumulation.
SAH is a potent inhibitor of SAM-dependent methyltransferases
(9); therefore, increased levels
of SAH may promote genomic demethylation, while decreased levels of
SAM, which is the sole methyl-donor for DNA methylation, may limit
its availability and coordinate the demethylation effect of SAH.
Hiltunen et al (35) showed
that only a few rounds of replication are required to develop
significant hypomethylation of the smooth muscle cell genome; the
decrease in the SAM/SAH ratio may thus be an important mechanism
for PDGF hypomethylation in VSMCs. The increase in C-5 MT-ase
activity observed in the study may be a compensatory response
against the inhibition of SAH in this in vitro
experiment.
In the present study, folate supply exhibited an
antagonistic effect against the Hcy-induced aberrant PDGF
methylation, increase in PDGF expression and increase in VSMC
proliferation. In the methionine cycle, Hcy is converted to
methionine by acquiring a methyl group from
N-5-methyltetrahydrofolate, a reaction catalyzed by methionine
synthase (36). Methionine is then
activated to SAM. Folate increases the production of
N-5-methyltetrahydrofolate, promotes the transformation of Hcy to
SAM and decreases levels of Hcy and SAH. Therefore, the
antagonistic effect of folate against Hcy is closely associated
with the methionine cycle. This is consistent with the effect of
folate on the methylation status of the PDGF gene and VSMC
proliferation observed in the present study. Hcy was found to
interfere with the epigenetic regulation of PDGF gene expression;
since the epigenetic regulation of PDGF gene expression may be
involved in VSMC proliferation, these findings may be beneficial
for patients with cardiovascular disorders and AS.
In conclusion, the present study has demonstrated
that Hcy-induced VSMC proliferation may be mediated through PDGF
signaling by interference with the epigenetic regulation of PDGF.
Hcy induces hypomethylation of the promoter region of the PDGF gene
and upregulates PGDF mRNA and protein expression, which eventually
causes VSMC proliferation. These data may provide evidence for a
useful target for the prevention and treatment of AS caused by
Hcy.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (nos. 81260105 and 81200118),
the Ningxia Education Department Scientific and Technological
Project (nos. NGY2012056 and NGY2013082) and a grant from the
Ningxia Science and Technique Project (no. NZ1195).
References
|
1
|
Zhang J, Guo C, Wang R, Huang L, Liang W,
Liu R and Sun B: An Egr-1-specific DNAzyme regulates Egr-1 and
proliferating cell nuclear antigen expression in rat vascular
smooth muscle cells. Exp Ther Med. 5:1371–1374. 2013.PubMed/NCBI
|
|
2
|
Little PJ, Rostam MA, Piva TJ, et al:
Suramin inhibits PDGF-stimulated receptor phosphorylation,
proteoglycan synthesis and glycosaminoglycan hyperelongation in
human vascular smooth muscle cells. J Pharm Pharmacol.
65:1055–1063. 2013. View Article : Google Scholar
|
|
3
|
Jia G, Cheng G, Gangahar DM and Agrawal
DK: Involvement of connexin 43 in angiotensin II-induced migration
and proliferation of saphenous vein smooth muscle cells via the
MAPK-AP-1 signaling pathway. J Mol Cell Cardiol. 44:882–890. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Adhikari N, Basi DL, Townsend D, Rusch M,
Mariash A, Mullegama S, Watson A, Larson J, Tan S, Lerman B, Esko
JD, Selleck SB and Hall JL: Heparan sulfate Ndst1 regulates
vascular smooth muscle cell proliferation, vessel size and vascular
remodeling. J Mol Cell Cardiol. 49:287–293. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Luo X, Xiao Y, Song F, Yang Y, Xia M and
Ling W: Increased plasma S-adenosyl-homocysteine levels induce the
proliferation and migration of VSMCs through an oxidative
stress-ERK1/2 pathway in apoE(−/−) mice. Cardiovasc Res.
95:241–250. 2012.PubMed/NCBI
|
|
6
|
Zhang D, Chen Y, Xie X, Liu J, Wang Q,
Kong W and Zhu Y: Homocysteine activates vascular smooth muscle
cells by DNA demethylation of platelet-derived growth factor in
endothelial cells. J Mol Cell Cardiol. 53:487–496. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Janda K, Aksamit D, Drozdz M, et al:
Influence of elevated homocystein level and selected lipid
parameters in kidney transplant patients on the progression of
atherosclerotic changes assessed by intima-media thickness index
(CCA-IMT). Przegl Lek. 69:670–674. 2012.(In Polish).
|
|
8
|
Huidobro C, Fernandez AF and Fraga MF: The
role of genetics in the establishment and maintenance of the
epigenome. Cell Mol Life Sci. 70:1543–1573. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tisdale MJ: Potentiation of the growth
inhibitory effects of adenosine 3′,5′-monophosphate analogues by
homocysteine. Biochem Pharmacol. 31:979–982. 1982.
|
|
10
|
Zhang CY, Wang NN, Zhang YH, Feng QZ, Yang
CW and Liu B: DNA methylation involved in proline accumulation in
response to osmotic stress in rice (Oryza sativa). Genet Mol
Res. 12:1269–1277. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
He Y, Cui Y, Wang W, Gu J, Guo S, Ma K and
Luo X: Hypomethylation of the hsa-miR-191 locus causes high
expression of hsa-mir-191 and promotes the
epithelial-to-mesenchymal transition in hepatocellular carcinoma.
Neoplasia. 13:841–853. 2011.PubMed/NCBI
|
|
12
|
Amatruda JF, Ross JA, Christensen B, et
al: DNA methylation analysis reveals distinct methylation
signatures in pediatric germ cell tumors. BMC Cancer. 13:3132013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yideng J, Zhihong L, Jiantuan X, Jun C,
Guizhong L and Shuren W: Homocysteine-mediated PPARalpha, gamma DNA
methylation and its potential pathogenic mechanism in monocytes.
DNA Cell Biol. 27:143–150. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sahni A, Wang N and Alexis J: UAP56 is an
important mediator of angiotensin II/platelet derived growth factor
induced vascular smooth muscle cell DNA synthesis and
proliferation. Biochem Biophys Res Commun. 431:636–640. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thevathasan JV, Tan E, Zheng H, Lin YC, Li
Y, Inoue T and Fivaz M: The small GTPase HRas shapes local PI3K
signals through positive feedback and regulates persistent membrane
extension in migrating fibroblasts. Mol Biol Cell. 24:2228–2237.
2013. View Article : Google Scholar
|
|
16
|
Chandra A and Angle N: VEGF inhibits
PDGF-stimulated calcium signaling independent of phospholipase C
and protein kinase C. J Surg Res. 131:302–309. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kojima N, Hori M, Murata T, Morizane Y and
Ozaki H: Different profiles of Ca2+ responses to
endothelin-1 and PDGF in liver myofibroblasts during the process of
cell differentiation. Br J Pharmacol. 151:816–827. 2007.
|
|
18
|
Hattori N, Abe T, Hattori N, et al:
Preference of DNA methyltransferases for CpG islands in mouse
embryonic stem cells. Genome Res. 14:1733–1740. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dai Y, Mercanti F, Dai D, Wang X, Ding Z,
Pothineni NV and Mehta JL: LOX-1, a bridge between GLP-1R and
mitochondrial ROS generation in human vascular smooth muscle cells.
Biochem Biophys Res Commun. 437:62–66. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Whitman SC: A practical approach to using
mice in atherosclerosis research. Clin Biochem Rev. 25:81–93.
2004.PubMed/NCBI
|
|
21
|
Shi ZD and Tarbell JM: Fluid flow
mechanotransduction in vascular smooth muscle cells and
fibroblasts. Ann Biomed Eng. 39:1608–1619. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xie C, Ritchie RP, Huang H, Zhang J and
Chen YE: Smooth muscle cell differentiation in vitro: models and
underlying molecular mechanisms. Arterioscler Thromb Vasc Biol.
31:1485–1494. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yideng J, Jianzhong Z, Ying H, Juan S,
Jinge Z, Shenglan W, Xiaoqun H and Shuren W: Homocysteine-mediated
expression of SAHH, DNMTs, MBD2, and DNA hypomethylation potential
pathogenic mechanism in VSMCs. DNA Cell Biol. 26:603–611. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu X, Shen J, Zhan R, et al: Proteomic
analysis of homocysteine induced proliferation of cultured neonatal
rat vascular smooth muscle cells. Biochim Biophys Acta.
1794:177–184. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nazarenko I, Hede SM, He X, Hedrén A,
Thompson J, Lindström MS and Nistér M: PDGF and PDGF receptors in
glioma. Ups J Med Sci. 117:99–112. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sakata Y, Xiang F, Chen Z, Kiriyama Y,
Kamei CN, Simon DI and Chin MT: Transcription factor CHF1/Hey2
regulates neointimal formation in vivo and vascular smooth muscle
proliferation and migration in vitro. Arterioscler Thromb Vasc
Biol. 24:2069–2074. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang H, Jia X, Han F, Zhao J, Zhao Y, Fan
Y and Yuan X: Dual-delivery of VEGF and PDGF by double-layered
electrospun membranes for blood vessel regeneration. Biomaterials.
34:2202–2212. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li JC, Pan JQ, Huang GQ, Tan X, Sun WD,
Liu YJ and Wang XL: Expression of PDGF-beta receptor in broilers
with pulmonary hypertension induced by cold temperature and its
association with pulmonary vascular remodeling. Res Vet Sci.
88:116–121. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Takimoto T, Suzuki K, Arisaka H, Murata T,
Ozaki H and Koyama N: Effect of N-(p-coumaroyl)serotonin and
N-feruloylserotonin, major anti-atherogenic polyphenols in
safflower seed, on vasodilation, proliferation and migration of
vascular smooth muscle cells. Mol Nutr Food Res. 55:1561–1571.
2011. View Article : Google Scholar
|
|
30
|
Choi BK, Cha BY, Yagyu T, Woo JT and Ojika
M: Sponge-derived acetylenic alcohols, petrosiols, inhibit
proliferation and migration of platelet-derived growth factor
(PDGF)-induced vascular smooth muscle cells. Bioorg Med Chem.
21:1804–1810. 2013. View Article : Google Scholar
|
|
31
|
Sirois MG, Simons M and Edelman ER:
Antisense oligonucleotide inhibition of PDGFR-beta receptor subunit
expression directs suppression of intimal thickening. Circulation.
95:669–676. 1997. View Article : Google Scholar
|
|
32
|
Englesbe MJ, Hawkins SM, Hsieh PC, Daum G,
Kenagy RD and Clowes AW: Concomitant blockade of platelet-derived
growth factor receptors alpha and beta induces intimal atrophy in
baboon PTFE grafts. J Vasc Surg. 39:440–446. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kwon HJ, Kim GE, Lee YT, Jeong MS, Kang I,
Yang D and Yeo EJ: Inhibition of platelet-derived growth factor
receptor tyrosine kinase and downstream signaling pathways by
Compound C. Cell Signal. 25:883–897. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen NC, Yang F, Capecci LM, et al:
Regulation of homocysteine metabolism and methylation in human and
mouse tissues. FASEB J. 24:2804–2817. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hiltunen MO, Turunen MP, Häkkinen TP, et
al: DNA hypomethylation and methyltransferase expression in
atherosclerotic lesions. Vasc Med. 7:5–11. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Duncan TM, Reed MC and Nijhout HF: The
relationship between intracellular and plasma levels of folate and
metabolites in the methionine cycle: a model. Mol Nutr Food Res.
57:628–636. 2013. View Article : Google Scholar : PubMed/NCBI
|