Introduction
Over recent decades, increasing evidence has
suggested that infection may be an important risk factor for the
development of atherosclerosis and coronary artery disease
(1,2). Epidemiological and clinical studies
have demonstrated that infection with Chlamydia pneumoniae,
an intracellular organism, is an independent risk factor for
cardiovascular diseases (3–5).
Endothelial cells, vascular smooth muscle cells and
monocytes/macrophages are involved in the atherogenic process, and
infected peripheral blood monocytes can transmit the C.
pneumoniae infection to smooth muscle and endothelial cells
(6).
The endothelial layer provides a nonthrombogenic and
nonadherent surface for circulating leukocytes, and is required for
the maintenance of normal vessel function (7). Furthermore, vascular endothelial
cells regulate complex processes, including hemostasis,
fibrinolysis, inflammation, vessel tone, blood pressure,
lipoprotein metabolism and angiogenesis (8). The accumulation of oxidized
low-density lipoprotein (ox-LDL) in the arterial wall can activate
the immune system and initiate a pro-inflammatory cascade of
events, including the endogenous production of reactive oxygen
species (ROS), endothelial dysfunction, the recruitment and
trans-endothelial migration of monocytes with consequent
transformation into foam cells and the proliferation of vascular
smooth muscle cells. These events ultimately lead to
atherosclerosis (9–11). Studies have shown that C.
pneumoniae infection stimulates LDL oxidization and uptake and
foam cell formation in macrophages (12,13).
Furthermore, a previous study by our group indicated that C.
pneumoniae induced macrophage-derived foam cell formation
through upregulation of the expression of scavenger receptor (SR)
A1, cluster of differentiation (CD) 36 and acyl-coenzyme A:
cholesterol acyltransferase 1 (ACAT1), and through downregulation
of the expression of ATP binding cassette transporter (ABC) A1 and
ABCGl (14). These genes regulate
intracellular cholesterol homeostasis in vascular endothelial
cells, including cholesterol synthesis, influx and efflux (15–17).
However, few studies have investigated the effect of C.
pneumoniae infection on endothelial lipid metabolism.
Materials and methods
Ethics statement
The present study involving fresh plasma was
approved by normolipidemic volunteers and the Wuhan Blood Centre
(Wuhan, China; authorization: 2010–8) and conformed to the
guidelines outlined in the Declaration of Helsinki.
Reagents and antibodies
HUVECs and epithelial HEp-2 cells were obtained from
Tongji Medical College (Wuhan, China). C. pneumoniae strain
AR-39 was purchased from the American Type Culture Collection
(Rockville, MA, USA). Dulbecco’s Modified Eagle’s Medium (DMEM) and
fetal bovine serum (FBS) were purchased from Hyclone (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). A cell lipid peroxide assay
kit was purchased from Genmed Scientifics, Inc. (Shanghai, China).
A quantitative polymerase chain reaction (qPCR) kit was purchased
from Takara Bio Inc. (Otsu, Shiga, Japan). Fluorescein
isothiocyanate (FITC)-conjugated specific anti-chlamydial
monoclonal antibodies were obtained from Dako Ltd. (Copenhagen,
Denmark), and mouse polyclonal anti-SR-A1 antibodies were obtained
from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Rat
monoclonal anti-CD36 antibodies were obtained from R&D Systems,
Inc. (Minneapolis, MN, USA). Rabbit polyclonal anti-ACAT1
antibodies were obtained from Cayman Chemical Co. (Ann Arbor, MI,
USA). Rabbit monoclonal anti-ABCG1 antibodies were obtained from
Epitomics (Burlingame, CA, USA). Mouse monoclonal anti-ABCA1
antibodies were obtained from Abcam PLC (Cambridge, UK). Mouse
polyclonal anti-β-actin antibodies and horseradish peroxidase
(HRP)-conjugated secondary antibodies were obtained from AntGene
Biotech Co., Ltd. (Wuhan, China).
Propagation of C. pneumoniae
C. pneumoniae strain AR-39 was propagated in
HEp-2 cells in the presence of 2 μg/ml cycloheximide using
centrifugation-driven infection as described previously (18). Infected HEp-2 cells were harvested
subsequent to incubation for 72 h at 37°C and 5% CO2,
and disrupted by freezing, thawing and ultrasonication. Following
centrifugation at 500 × g at 4°C for 30 min to remove the cell
debris, the supernatant containing elementary bodies was collected
and suspended in sucrose-phosphate-glutamic acid buffer and stored
at −70°C in aliquots until use. Chlamydial inclusion forming units
(IFU) were assessed by counting the chlamydial inclusions formed in
the HEp-2 cells using FITC-conjugated anti-chlamydial
antibodies.
Infection and culture of HUVECs
HUVECs were seeded onto six-well plates at a density
of 1×106 cells per well in DMEM containing 10% FBS at
37°C in 5% CO2. In the presence of 50 mg/l LDL, HUVECs
were cultured alone or with C. pneumoniae (1×106
IFU) for 48 h. In certain experiments, HUVECs were incubated in
six-well plates with 50 mg/l ox-LDL for 48 h.
LDL isolation and ox-LDL preparation
Human LDL (density, 1.006–1.063 g/ml) was isolated
from the blood of healthy donors using density-gradient
ultracentrifugation as described previously (19). The isolated LDL was dialyzed in
phosphate-buffered saline for 24 h and was condensed using polyoxyl
20,000. The concentration of LDL was measured using the Bradford
assay, and the LDL underwent sterile filtration (0.22 mm) prior to
storage in the dark at 4°C for no longer than 2 weeks before use.
LDL was analyzed using 4–20% SDS-PAGE gradient gel and Sudan black
B staining to reveal a single protein. The preparations of ox-LDL
from human plasma were performed as described previously (20). Briefly, LDL was diluted to a
concentration of 200 mg/dl in medium M199 and incubated with 5
μmol/l CuSO4, CuCl2 or copper acetate at 37°C
for 24 h, as indicated. The reactions were terminated using the
addition of 100 μmol/l EDTA. The oxidation state of LDL was
measured using the PeroXOquant Quantitative Peroxide Assay kit
(Pierce Biotechnology, Inc., Rockford, IL, USA). Peroxidation
products contained in the LDL and ox-LDL were <5 and 200–300
μmol/l H2O2, respectively. Contamination of
LDL and the modified LDL preparations by the lipopolysaccharide
(LPS) endotoxin was assessed using a Limulus Amebocyte Lysate kit
(BioWhittaker®; Lonza, Walkersville, MD, USA). All LDL
preparations had an LPS content of <50 pg/mg protein.
Measurement of thiobarbituric acid
reactive substances (TBARS)
The determination of LDL oxidation was performed as
described previously (21). The
determination of TBARS involves the measurement of malondialdehyde
(MDA) derived from the hydroperoxidation of unsaturated fatty acids
containing three or more double bonds (22). In brief, 2.5 ml 10% trichloroacetic
acid was added to a tube with 0.5 ml HUVEC supernatant or cell
lysates with or without C. pneumoniae infection, in order to
precipitate the protein. Following centrifugation, 100 μl
supernatant was mixed with 50 μl 0.67% TBA. Subsequent to vortexing
and boiling for 15 min, samples were cooled under running water.
Absorbance was measured at 532 nm in a Perkin Elmer Lambda 2
spectrophotometer (Perkin Elmer, Überlingen, Germany) against a
blank control with distilled water. The concentration of oxidation
products was determined as MDA (nmol/ml) according to a standard
curve of MDA reacting with TBA using the following formula: MDA
(nmol/ml) = absorbance × 53.4188. Experiments were repeated three
times.
Intracellular cholesterol
measurement
Cells were harvested and disrupted using
ultrasonication. Intracellular TC and free cholesterol (FC) were
detected using zymochemistry as described previously (23). TC and FC content was determined
using a standard curve (312.5, 625, 1,250 and 2,500 μmol/l standard
cholesterol) and expressed as μmol/g protein. CE content was
calculated by subtracting the content of FC from that of TC.
Experiments were repeated three times.
qPCR analysis
Total RNA was isolated using TRIzol®
reagent (Takara Bio, Inc.). cDNA was synthesized using a
PrimeScript™ RT Reagent kit (Takara Bio, Inc.) according to the
manufacturer’s instructions, and qPCR was performed using a Taq
polymerase (Takara Bio, Inc.). The oligonucleotide primers for
SR-A1, CD36, ACAT1, ABCA1, ABCG1 and β-actin were as follows:
SR-A1, 5′-GCAGTTCTCATC CCTCTCAT-3′ (forward) and 5′-GGTATTCTCTTGGAT
TTTGCC-3′ (reverse); CD36, 5′-TGCCTCTCCTAG TTGAAAAC-3′ (forward)
and 5′-GCAACAAACGATCAC CACAC-3′ (reverse); ACAT1,
5′-TCCCAGGAATCCCAC TGTAA-3′ (forward) and 5′-ACGAAGAGCACGGGA
TAGAA-3′ (reverse); ABCA1, 5′-TATGAGGGCCAGATC ACCTC-3′ (forward)
and 5′-GCTGGCTTGTTTTGCTT TTC-3′ (reverse); ABCG1,
5′-CAGGAAGATTAGACACTG TGG-3′ (forward) and
5′-GAAAGGGGAATGGAGAGAAG-3′ (reverse); β-actin,
5′-GTCCACCTTCCAGCAGATGT-3′ (forward) and 5′-CACCTTCACCGTTCCAGTTT-3′
(reverse).
Western blot analysis
Total protein was extracted from the cells and the
protein content was measured in triplicate using the Bicinchoninic
Acid Protein Assay kit (Pierce Biotechnology, Inc.). Absorption was
measured at 562 nm. Protein samples (60 μg) were separated using
10% SDS-PAGE and transferred to polyvinylidene fluoride membranes
(Invitrogen Life Technologies, Grand Island, NY, USA). Membranes
were incubated at 4°C overnight with the following antibodies:
Mouse polyclonal anti-SR-A1 (dilution, 1:200), rat monoclonal
anti-CD36 (dilution, 1:500), rabbit polyclonal anti-ACAT1
(dilution, 1:200), mouse monoclonal anti-ABCA1 (dilution, 1:1,000),
rabbit monoclonal anti-ABCG1 (dilution, 1:2,000) and mouse
polyclonal anti-β-actin (dilution, 1:1,000). The membranes were
then incubated with HRP-conjugated secondary anti-rabbit, -rat and
-mouse antibodies, each at a dilution of 1:2,000, at room
temperature for 2 h. Proteins were detected using the enhanced
chemiluminescence method. For quantification, SR-A1, CD36, ACAT1,
ABCA1 and ABCG1 protein levels were normalized to those of
β-actin.
Statistical analysis
Quantitative data are expressed as the mean ±
standard derivation. Direct comparisons between two groups were
performed using the Student’s t-test. Data from more than two
groups were compared using analysis of variance with repeated
measures, followed by the Student-Newman-Keuls multiple comparison
test. A value of P<0.05 was considered to indicate a
statistically significant difference.
Results
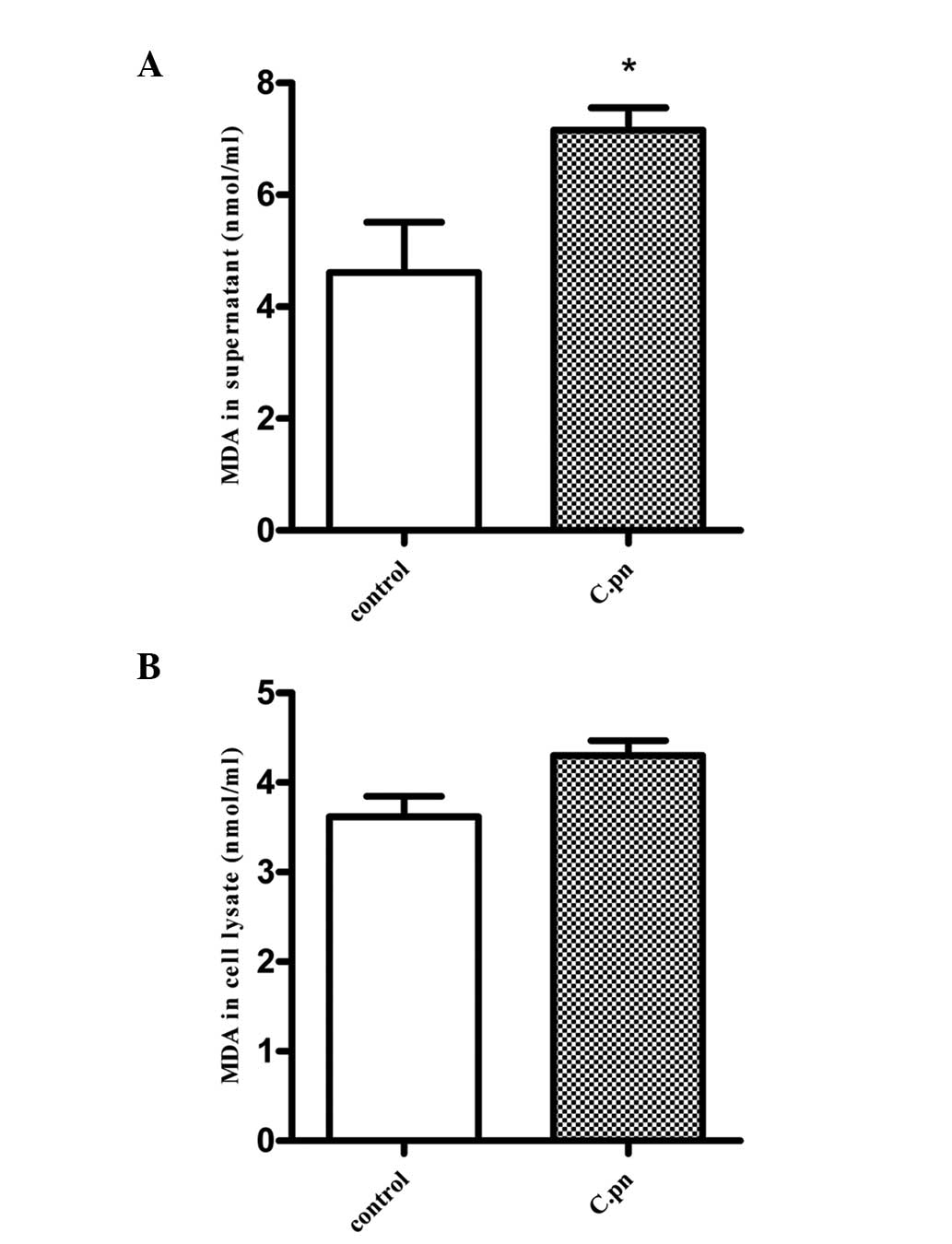
Increased lipid peroxidation products in
the supernatants, but not cell lysates, of C. pneumoniae-infected
HUVECs
Lipid peroxidation was quantified using the
measurement of TBARS. As shown in Fig.
1A, lipid peroxidation products were increased in the
supernatant of C. pneumoniae-infected HUVECs
(1×106 IFU). MDA production was observed to increase
from 4.61±0.90 nmol/ml in the control HUVEC supernatant to
7.16±0.40 nmol/ml in the infected HUVEC supernatant (P<0.05). In
contrast to the supernatant, no significant difference in LDL
oxidation was observed in the infected HUVEC lysates (Fig. 1B; P>0.05).
Effect of C. pneumoniae on intracellular
cholesterol metabolism
Cholesterol is accumulated either as CE in
cytoplasmic vesicles or as FC in the membrane (24). To determine whether intracellular
cholesterol metabolism was affected by C. pneumoniae in
HUVECs, the levels of intracellular TC and CE were assessed using
lipid mass quantification. LDL-treated HUVECs were used as a
negative control, ox-LDL-treated HUVECs were used as a positive
control and LDL-treated HUVECs infected with C. pneumoniae
were used as the infection group. As shown in Table I, C. pneumoniae infection
was found to increase the levels of TC and CE compared with the
negative control (P<0.05). In addition, levels of intracellular
TC and CE were increased in the ox-LDL-treated HUVECs compared with
those in the negative control group (P<0.05). C.
pneumoniae infection was not observed to significantly increase
TC and CE levels compared with the positive controls
(P>0.05).
 | Table IEffect of Chlamydia pneumoniae
on TC and CE levels in human umbilical vein endothelial cells. |
Table I
Effect of Chlamydia pneumoniae
on TC and CE levels in human umbilical vein endothelial cells.
| Groups | TC (μmol/g) | FC (μmol/g) | CE (μmol/g) | CE/TC (%) |
|---|
| LDL | 35.7±0.8 | 24.4±0.6 | 11.3±1.1 | 31.7 |
| LDL+C.pn | 74.5±3.1a | 41.3±1.7 | 33.2±2.1a | 44.6 |
| ox-LDL | 76.5±2.6a | 39.5±1.2 | 36.7±0.9a | 48.2 |
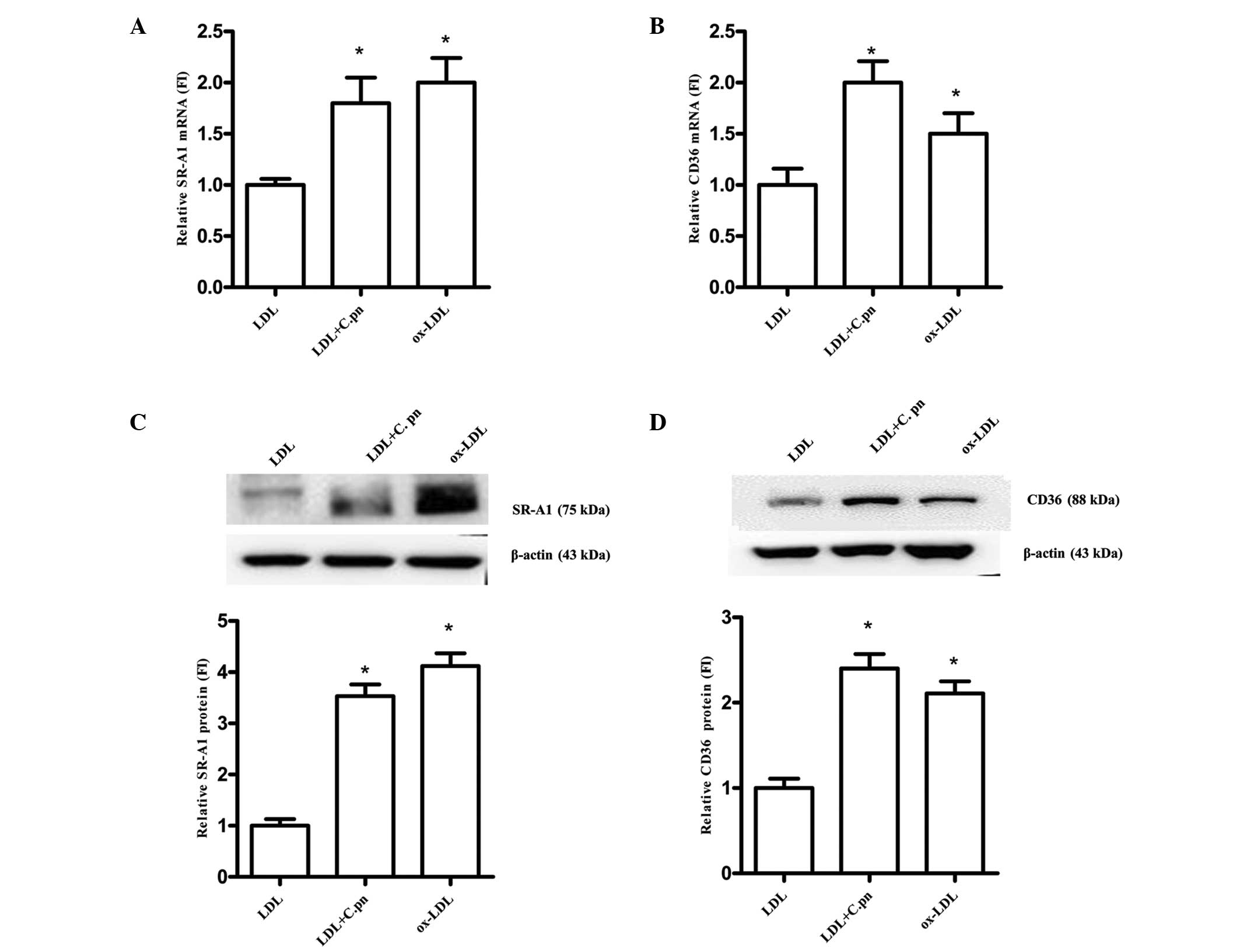
SR-A1 and CD36 expression is upregulated
in LDL-treated HUVECs with C. pneumoniae infection
The mRNA and protein expression of lipoprotein
receptors was assessed using qPCR and western blot analyses,
respectively. SR-A1 and CD36 have been demonstrated to participate
in lipoprotein uptake (25). As
shown in Fig. 2A and B, SR-A1 and
CD36 mRNA expression was upregulated by up to 1.8- and 2.0-fold in
the C. pneumoniae-infected HUVECs compared with that in the
negative controls (P<0.05). In addition, ox-LDL was found to
upregulate the mRNA expression of SR-A1 and CD36 by up to 2.0- and
1.5-fold, respectively (P<0.05). However, no significant
difference was observed in the mRNA expression of SR-A1 and CD36
between the C. pneumoniae-infected HUVECs and the positive
controls (P>0.05). Furthermore, C. pneumoniae infection
was found to increase the protein expression of SR-A1 and CD36 by
3.53- and 2.40-fold, respectively, relative to the negative control
group (P<0.05). SR-A1 and CD36 protein expression was also
increased by up to 4.12- and 2.11-fold, respectively, in the
positive control group relative to that in the negative controls
(P<0.05) (Fig. 2C and D). No
significant difference was observed in the protein expression of
SR-A1 and CD36 between the C. pneumoniae-infected HUVECs and
the positive controls (P>0.05).
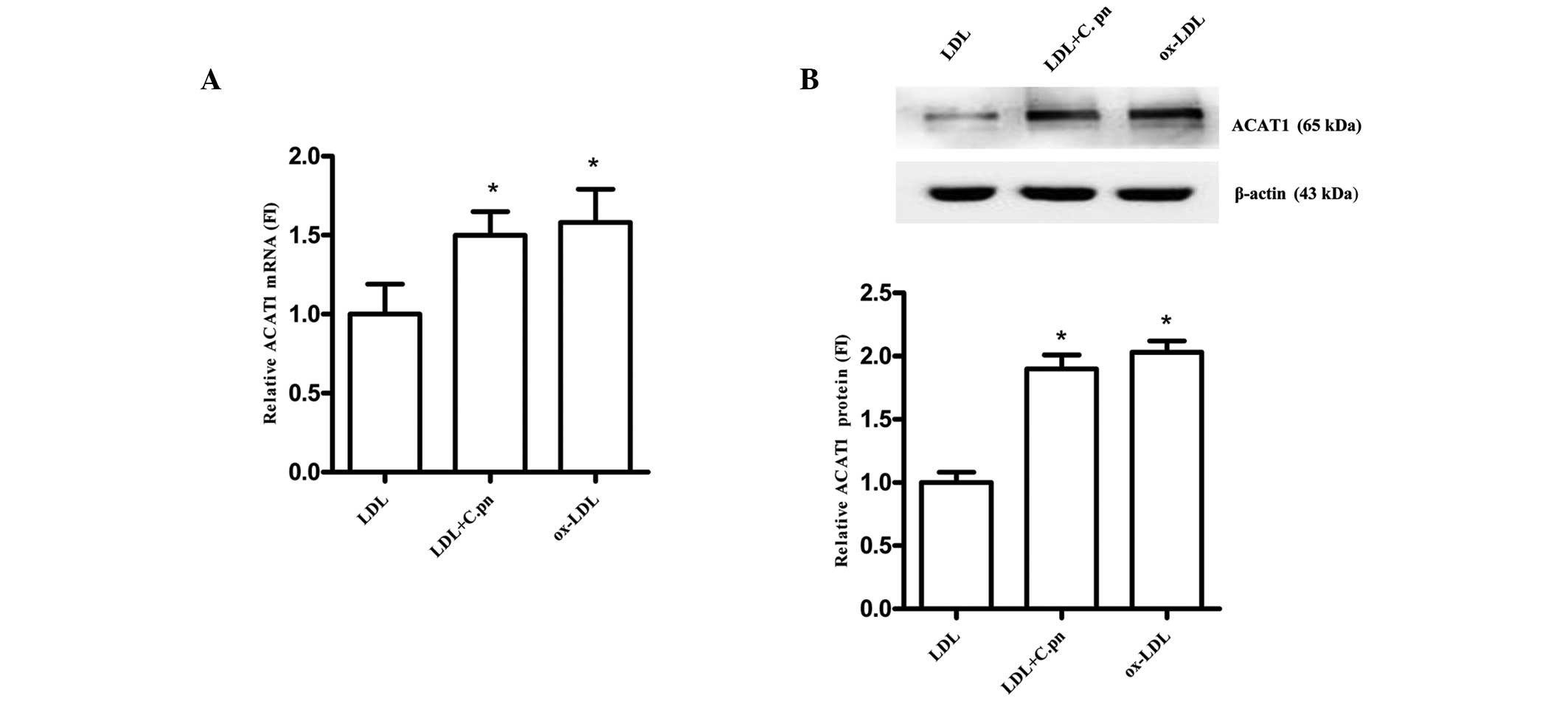
ACAT1 expression is upregulated in
LDL-treated HUVECs with C. pneumoniae infection
The conversion of cholesterol to CE is catalyzed by
the enzyme ACAT. In order to determine the role of ACAT1 in the
C. pneumoniae-induced perturbation of intracellular
cholesterol homeostasis in HUVECs, the expression of ACAT1 was
assessed using qPCR and western blot analyses. As shown in Fig. 3A and B, C. pneumoniae
infection (1×106 IFU) was found to significantly
increase the mRNA and protein expression of ACAT1 by up to 1.5- and
1.9-fold, respectively, compared with the LDL-treated HUVECs
(P<0.05). In addition, ox-LDL upregulated ACAT1 mRNA expression
by up to 1.58-fold and ACAT1 protein expression by up to 2.03-fold
relative to the negative control. No significant difference was
observed in ACAT1 mRNA and protein expression between the C.
pneumoniae-infected HUVECs and the positive controls
(P>0.05). These data show that ACAT1 may be involved in the
C. pneumoniae-induced increases in CE levels in HUVECs.
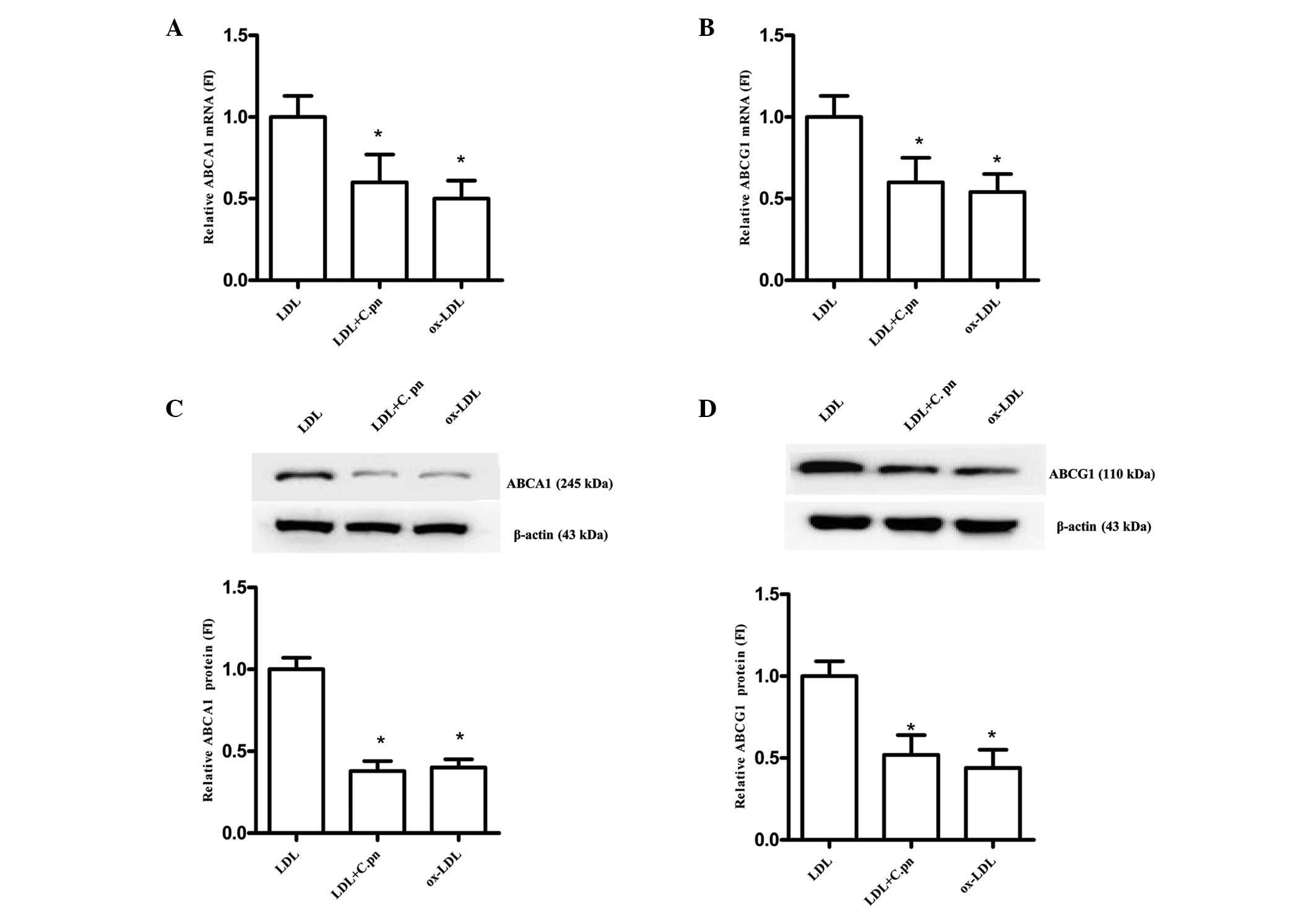
ABCA1 and ABCG1 expression is
downregulated in LDL-treated HUVECs with C. pneumoniae
infection
ABCA1 and ABCG1 are critical cell surface proteins
that mediate endothelial cholesterol efflux (15). In order to investigate the
expression and regulation of ABCA1 and ABCG1 in C.
pneumoniae-infected HUVECs, the mRNA and protein levels of
ABCA1 and ABCG1 were detected. As shown in Fig. 4A and B, the mRNA expression of both
ABCA1 and ABCG1 was reduced by 40% in the C.
pneumoniae-infected HUVECs compared with that in the
LDL-treated HUVECs (P<0.05). Furthermore, in the ox-LDL-treated
HUVECs, the mRNA expression of ABCA1 and ABCG1 was reduced by 50%
(P<0.05). In addition, ABCA1 and ABCG1 protein expression was
reduced by 62 and 48% in the C. pneumoniae-infected HUVECs
(P<0.05; Fig. 4C and D).
However, no significant difference was observed between the C.
pneumoniae-infected group and the positive control group
(P>0.05).
Discussion
Atherosclerosis is an inflammatory disease in its
genesis and progression, and it is initiated by the retention of
lipids. The present study revealed that the supernatant of C.
pneumoniae-infected HUVECs exhibited significant LDL oxidation,
and that C. pneumoniae increased the intracellular levels of
TC and CE through the regulation of target genes affecting lipid
metabolism in endothelial cells. The rate of lipid peroxidation was
determined using the TBARS method, which revealed that the
supernatant, but not the cell lysate, of C.
pneumoniae-infected HUVECs exhibited significant LDL oxidation.
Furthermore, C. pneumoniae infection was found to
significantly increase the levels of TC and CE in the LDL-treated
HUVECs; this finding was similar to the results for the
ox-LDL-treated HUVECs. The present study also demonstrated that
in vitro infection with C. pneumoniae in LDL-treated
HUVECs significantly increased the expression of SR-A1, CD36 and
ACAT1, which participate in intracellular lipid influx and CE
synthesis. In addition, C. pneumoniae was observed to
decrease the expression of ABCA1/G1, which mediate intracellular
lipid efflux. Overall, the present study has indicated that C.
pneumoniae disrupts lipid metabolism in HUVECs.
The importance of ox-LDL in terms of atherogenesis
is well established. ox-LDL inhibits the expression of the
constitutive endothelial nitric oxide synthetase enzyme and leads
to oxidative stress-induced vascular endothelial cell apoptosis
(26,27). The release of ROS induces the
expression of adhesion molecules on endothelial cells (28). Several studies have reported that
infected monocytes/macrophages are capable of transmitting C.
pneumoniae infection to endothelial cells and that C.
pneumoniae infection of macrophages enhances their adhesion to
the endothelium (29,30). In addition, ox-LDL has been
reported to enhance the expression of vascular adhesion molecules
in C. pneumoniae-infected endothelial cells (31); the increased expression of vascular
adhesion molecules consequently promotes the attraction and
adherence of monocytes and T lymphocytes to the endothelium, which
is a key process in the progression of arteriosclerosis (32). In the present study, the
supernatant of C. pneumoniae-infected HUVECs was found to
exhibit significant LDL oxidation. Thus, C. pneumoniae may
induce LDL oxidation and initiate endothelial dysfunction
chronically and continuously, eventually leading to
atherogenesis.
Vascular endothelial cells express receptors for
oxidized lipoproteins and exhibit biochemical pathways for sterol
synthesis and receptor-mediated endocytosis of lipoproteins
(33). A variety of SRs that bind
to ox-LDL and promote atherogenesis have been identified. SR-A1 and
CD36 promote inflammation and cell adhesion, and lectin-type
oxidized LDL receptor 1 (LOX-1) mediates and promotes oxidative
stress and inflammation (34). A
previous study showed that infection of HUVECs with C.
pneumoniae enhanced the uptake of ox-LDL and the expression of
LOX-1, which may directly contribute to atherogenesis (35). In the present study, C.
pneumoniae infection was found to cause a significant increase
in SR-A1 and CD36 expression in HUVECs. These findings suggest that
these receptors mediate ox-LDL uptake and lead to the accumulation
of intracellular TC.
ABCA1 and ABCG1 are considered to be the most
important genes involved in reverse cholesterol transport, through
the promotion of high-density lipoprotein (HDL) subfraction 3
and/or apolipoprotein A-I-mediated cellular cholesterol efflux
(33). Furthermore, HDL is
considered to have an important effect on endothelial health
(36). A previous study showed
that ox-LDL downregulated ABCA1 in human endothelial cells
(37). The present study found
that C. pneumoniae infection downregulated the expression of
ABCA1 and ABCG1 in HUVECs, which may have mediated a reduction in
cholesterol efflux. Therefore, C. pneumoniae infection may
impair the efflux of excessive intracellular lipids and lead to the
accumulation of intracellular TC.
The accumulation of intracellular TC has cytotoxic
effects. Oxidized lipids have been shown to induce oxidative
stress-induced endothelial cell apoptosis and reduce mitochondrial
function (38). Excess cellular
cholesterol is stored as CEs. In the majority of cell types, CEs
are present only at low levels and predominantly as cytoplasmic
lipid droplets. The appearance of CE-enriched lipid droplets in
tissues is a consequence of impaired metabolism or overnutrition
(39). ACAT1 is a key and
exclusive microsomal enzyme that esterifies FC with fatty acids to
form CE. ACAT1 is expressed in numerous tissue and cell types,
including hepatocytes and Kupffer cells in the liver, adrenal
glands, neurons, macrophages, smooth muscle cells and vascular
endothelial cells; this expression accounts for >80% of the
total ACAT enzyme activity measured in vitro (40). The present study revealed that
C. pneumoniae increased ACAT1 expression and induced the
accumulation of intracellular CE and TC. The increase in
intracellular TC induced by C. pneumoniae in HUVECs may be a
result of increased influx and reduced efflux of cholesterol. The
increase in intracellular CE may be mediated by ACAT1 and the
accumulation of intracellular CE and TC may lead to endothelial
dysfunction in atherogenesis.
In conclusion, the present study has revealed that
C. pneumoniae significantly induces LDL oxidation and the
accumulation of intracellular TC by increasing lipid influx and
reducing lipid efflux in HUVECs. In addition, C. pneumoniae
increases intracellular CE levels. The abnormalities in lipid
metabolism induced by C. pneumoniae may result in
endothelial injury and atherogenesis. These findings may provide an
enhanced understanding of the association between infection and
atherogenesis.
Acknowledgements
This study was supported by a grant from the
National Natural Science Foundation of China (no. 30900599).
References
|
1
|
Kuvin JT and Kimmelstiel CD: Infectious
causes of atherosclerosis. Am Heart J. 137:216–226. 1999.
View Article : Google Scholar
|
|
2
|
de Boer OJ, van der Wal AC and Becker AE:
Atherosclerosis, inflammation and infection. J Pathol. 190:237–243.
2000.
|
|
3
|
Kuo CC, Gown AM, Benditt EP and Grayston
JT: Detection of Chlamydia pneumoniae in aortic lesions of
atherosclerosis by immunocytochemical stain. Arterioscler Thromb.
13:1501–1504. 1993.PubMed/NCBI
|
|
4
|
Saikku P, Leinonen M, Tenkanen L, et al:
Chronic Chlamydia pneumoniae infection as a risk factor for
coronary heart disease in the Helsinki Heart Study. Ann Intern Med.
116:273–278. 1992.
|
|
5
|
Moazed TC, Kuo C, Grayston JT and Campbell
LA: Murine model of Chlamydia pneumoniae infection and
atherosclerosis. J Infect Dis. 175:883–890. 1997.PubMed/NCBI
|
|
6
|
Gieffers J, van Zandbergen G, Rupp J, et
al: Phagocytes transmit Chlamydia pneumoniae from the lungs
to the vasculature. Eur Respir J. 23:506–510. 2004.
|
|
7
|
Summersgill JT, Molestina RE, Miller RD
and Ramirez JA: Interactions of Chlamydia pneumoniae with
human endothelial cells. J Infect Dis. 181(Suppl 3): S479–S482.
2000.
|
|
8
|
Assmann G and Nofer JR: Atheroprotective
effects of high-density lipoproteins. Annu Rev Med. 54:321–341.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li D and Mehta JL: Oxidized LDL, a
critical factor in atherogenesis. Cardiovasc Res. 68:353–354. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mehta JL: Oxidized or native low-density
lipoprotein cholesterol: which is more important in atherogenesis?
J Am Coll Cardiol. 48:980–982. 2006.
|
|
11
|
Itabe H: Oxidative modification of LDL:
its pathological role in atherosclerosis. Clin Rev Allergy Immunol.
37:4–11. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kalayoglu MV and Byrne GI: Induction of
macrophage foam cell formation by Chlamydia pneumoniae. J
Infect Dis. 177:725–729. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kalayoglu MV, Hoerneman B, LaVerda D,
Morrison SG, Morrison RP and Byrne GI: Cellular oxidation of
low-density lipoprotein by Chlamydia pneumoniae. J Infect
Dis. 180:780–790. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
He P, Mei C, Cheng B, Liu W, Wang Y and
Wan J: Chlamydia pneumoniae induces macrophage-derived foam
cell formation by up-regulating acyl-coenzyme A: cholesterol
acyltransferase 1. Microbes Infect. 11:157–163. 2009. View Article : Google Scholar
|
|
15
|
O‘Connell BJ, Denis M and Genest J:
Cellular physiology of cholesterol efflux in vascular endothelial
cells. Circulation. 110:2881–2888. 2004.
|
|
16
|
Collot-Teixeira S, Martin J, McDermott-Roe
C, Poston R and McGregor JL: CD36 and macrophages in
atherosclerosis. Cardiovasc Res. 75:468–477. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chang TY, Chang CC and Cheng D:
Acyl-coenzyme A: cholesterol acyltransferase. Annu Rev Biochem.
66:613–638. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Caldwell HD, Kromhout J and Schachter J:
Purification and partial characterization of the major outer
membrane protein of Chlamydia trachomatis. Infect Immun.
31:1161–1176. 1981.PubMed/NCBI
|
|
19
|
Redgrave TG, Roberts DC and West CE:
Separation of plasma lipoproteins by density-gradient
ultracentrifugation. Anal Biochem. 65:42–49. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu Y, Lin JH, Liao HL, Friedli O Jr,
Verna L, Marten NW, Straus DS and Stemerman MB: LDL induces
transcription factor activator protein-1 in human endothelial
cells. Arterioscler Thromb Vasc Biol. 18:473–480. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Draper HH and Hadley M: Malondialdehyde
determination as index of lipid peroxidation. Methods Enzymol.
186:421–431. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Scoccia AE, Molinuevo MS, McCarthy AD and
Cortizo AM: A simple method to assess the oxidative susceptibility
of low density lipoproteins. BMC Clin Pathol. 1:12001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gamble W, Vaughan M, Kruth HS and Avigan
J: Procedure for determination of free and total cholesterol in
micro- or nanogram amounts suitable for studies with cultured
cells. J Lipid Res. 19:1068–1070. 1978.PubMed/NCBI
|
|
24
|
Liu W, He P, Cheng B, et al: Chlamydia
pneumoniae disturbs cholesterol homeostasis in human THP-1
macrophages via JNK-PPARγ dependent signal transduction pathways.
Microbes Infect. 12:1226–1235. 2010. View Article : Google Scholar
|
|
25
|
Kunjathoor VV, Febbraio M, Podrez EA, et
al: Scavenger receptors class A-I/II and CD36 are the principal
receptors responsible for the uptake of modified low density
lipoprotein leading to lipid loading in macrophages. J Biol Chem.
277:49982–49988. 2002. View Article : Google Scholar
|
|
26
|
Cominacini L, Rigoni A, Pasini AF, Garbin
U, Davoli A, Campagnola M, Pastorino AM, Lo Cascio V and Sawamura
T: The binding of oxidized low density lipoprotein (ox-LDL) to
ox-LDL receptor-1 reduces the intracellular concentration of nitric
oxide in endothelial cells through an increased production of
superoxide. J Biol Chem. 276:13750–13755. 2001.
|
|
27
|
Roy Chowdhury SK, Sangle GV, Xie X,
Stelmack GL, Halayko AJ and Shen GX: Effect of extensively oxidized
low-density lipoprotein on mitochondrial function and reactive
oxygen species in porcine aortic endothelial cells. Am J Physiol
Enolocrinol Metab. 298:E89–E98. 2010.PubMed/NCBI
|
|
28
|
Chen H, Li D, Saldeen T and Mehta JL:
Transforming growth factor-beta(1) modulates oxidatively modified
LDL-induced expression of adhesion molecules: role of LOX-1. Circ
Res. 89:1155–1160. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lin TM, Campbell LA, Rosenfeld ME and Kuo
CC: Monocyte-endothelial cell coculture enhances infection of
endothelial cells with Chlamydia pneumoniae. J Infect Dis.
181:1096–1100. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kalayoglu MV, Perkins BN and Byrne GI:
Chlamydia pneumoniae-infected monocytes exhibit increased
adherence to human aortic endothelial cells. Microbes Infect.
3:963–969. 2001. View Article : Google Scholar
|
|
31
|
Vielma SA, Mironova M, Ku JR and
Lopes-Virella MF: Oxidized LDL further enhances expression of
adhesion molecules in Chlamydophila pneumoniae-infected
endothelial cells. J Lipid Res. 45:873–880. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Price DT and Loscalzo J: Cellular adhesion
molecules and atherogenesis. Am J Med. 107:85–97. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hassan HH, Denis M, Krimbou L, Marcil M
and Genest J: Cellular cholesterol homeostasis in vascular
endothelial cells. Can J Cardiol. 22(Suppl B): 35B–40B. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Apostolov EO, Shah SV, Ray D and Basnakian
AG: Scavenger receptors of endothelial cells mediate the uptake and
cellular proatherogenic effects of carbamylated LDL. Arterioscler
Thromb Vasc Biol. 29:1622–1630. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yoshida T, Koide N, Mori I, Ito H and
Yokochi T: Chlamydia pneumoniae infection enhances
lectin-like oxidized low-density lipoprotein receptor (LOX-1)
expression on human endothelial cells. FEMS Microbiol Lett.
260:17–22. 2006. View Article : Google Scholar
|
|
36
|
O‘Connell BJ and Genest J Jr: High-density
lipoproteins and endothelial function. Circulation. 104:1978–1983.
2001.
|
|
37
|
Zhu Y, Liao H, Xie X, et al: Oxidized LDL
downregulates ATP-binding cassette transporter-1 in human vascular
endothelial cells via inhibiting liver X receptor (LXR). Cardiovasc
Res. 68:425–432. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sata M and Walsh K: Oxidized LDL activates
fas-mediated endothelial cell apoptosis. J Clin Invest.
102:1682–1689. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zock PL, Mensink RP, Harryvan J, de Vries
JH and Katan MB: Fatty acids in serum cholesteryl esters as
quantitative biomarkers of dietary intake in humans. Am J
Epidemiol. 145:1114–1122. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chang TY, Li BL, Chang CC and Urano Y:
Acyl-coenzyme A: cholesterol acyltransferases. Am J Physiol
Endocrinol Metab. 297:E1–E9. 2009. View Article : Google Scholar : PubMed/NCBI
|