Introduction
Long noncoding RNAs (lncRNAs), which are a type of
noncoding RNA (ncRNA) varying in size from 200 bp to >100 kb,
are transcribed by RNA polymerase II, and are often spliced and
polyadenylated (1–4). They have been identified by a variety
of methods and a growing number of specific lncRNAs have been
demonstrated to affect genomic functions, including imprinting,
enhancer function, X-chromosome inactivation, chromatin structure
(including the lncRNA HOTAIR, which served as a scaffold to
assemble and target Polycomb Raepressive Complex 2 and
LSD1/CoREST/REST complexes to the HOXD locus and co-ordinated H3K27
methylation and H3K4 demethylation for affecting chromatin
structure) and genomic rearrangements during the generation of
antibody diversity (4). Multiple
studies have demonstrated that significant numbers of lncRNAs are
regulated during development, exhibit cell type-specific
expression, localize to specific subcellular compartments and are
associated with human diseases (1). Certain studies have revealed that
lncRNAs are widely expressed in the mammalian nervous system and a
large amount are likely to be important in neuronal development and
activity (5,6). Furthermore, lncRNAs are now being
implicated in neurodegenerative processes, including Huntington’s
disease (HD), amyotrophic lateral sclerosis (ALS) and Alzheimer’s
disease (AD) (5,6). Previous studies demonstrated that
lncRNA caused increases in levels of taurine upregulated gene 1 and
nuclear enriched abundant transcript 1 in the HD caudate, while
maternally expressed 3 was downregulated (5). Furthermore, in ALS, fused in
sarcoma/translocated in sarcoma (FUS/TLS) protein acts as an RNA
binding protein that is able to be recruited by a lncRNA to the
genomic locus encoding cyclin D1, where it represses cyclin D1
transcription. However, mutations in the FUS/TLS gene
caused an lncRNA-mediated abnormality in cyclin D1 transcription
regulation in a subset of ALS cases (5). In addition, the abnormal expression
of certain lncRNAs, including ATXN8OS and the antisense transcript
for β-secretase-1 (BACE1-AS) are closely correlated with AD.
Certain observations suggested that the mutant lncRNA ATXN8OS
transcript contributes to the pathogenesis of spinocerebellar
ataxia type 8 by altering the activity of the
MBNL/cellobiose-6-phosphate hydrolase alternative splicing protein
in AD (5). By contrast, Faghihi
et al identified a lncRNA conserved noncoding BACE1-AS that
regulates the mRNA and protein expression of β-secretase-1 (BACE1)
in the brain in an AD mouse model (7). Previous studies indicated that BACE1
is a crucial enzyme in AD pathophysiology (7,8).
Sequential cleavage of amyloid precursor protein (APP) by the
β-site cleaving enzyme BACE1, which is essential for amyloid
β-protein (Aβ) 1–42 and Aβ1–40 biosynthesis, and
secretase, initiates the ‘amyloid cascade’ that is central to the
pathophysiology of AD (7,8). Furthermore, Aβ1–42
oligomers produced by BACE1 affect key aspects of AD (7–9). The
results of the study by Faghihi et al demonstrated that
lncRNA BACE1-AS is elevated in AD and drives the rapid feed-forward
regulation of β-secretase (7).
Although the functions of lncRNAs remain to be fully elucidated,
lncRNA network changes in neurodegenerative processes may be
important in understanding and treating the associated diseases.
Based on previous evidence, the present study hypothesized that the
inhibition of endogenous lncRNA BACE1-AS by RNAi silencing
technology may attenuate the ability of BACE1 to cleave APP, thus
delaying the production of Aβ1–42 oligomers. Therefore,
the present study aimed to investigate this hypothesis in an in
vitro senile plaque (SP) AD cell model using synthetic
Aβ1–42-treated SH-SY5Y cells transfected with
siRNA-BACE1-AS or siRNA-mock expression plasmid DNA.
Materials and methods
Cell culture and Aβ1–42
treatment
The AD SP cell model was generated as previously
described (8). The SH-SY5Y cell
lines were seeded in a six-well plate in Dulbecco’s modified
Eagle’s medium supplemented with 10% fetal calf serum, penicillin
(100 U/ml) and glutamine (0.3 mg/ml; all ingredients were purchased
from Invitrogen Life Technologies, Grand Island, NY, USA) and
incubated in a humidified tissue culture incubator containing 5%
CO2 at 37°C until 80% confluence was achieved. Then, 10
μmol/l large aggregates of synthetic Aβ1–42
(Sigma-Aldrich, St. Louis, MO, USA) were added to the cultures.
Following 24 h, the drug-containing medium was replaced with fresh
normal cell medium for continued culture.
MTT assay for cell proliferation
Each group of SH-SY5Y cells was seeded at
2×103 cells per well in a 96-well plate until 85%
confluent. MTT (Sigma-Aldrich) reagent (5 mg/ml) was added to the
maintenance cell medium at different time-points and incubated at
37°C for an additional 4 h. The reaction was terminated with 150 μl
dimethylsulfoxide (Sigma-Aldrich) per well, the cells were lysed
for 15 min, and the plates were gently agitated for 5 min. The
absorbance values were determined using an ELISA reader (Model 680;
Bio-Rad, Hercules, CA, USA) at 490 nm.
RNA extraction and analysis by
quantitative polymerase chain reaction (qPCR)
Total RNA from each group was isolated with TRIzol
reagent (Invitrogen Life Technologies), according to the
manufacturer’s instructions. The RNA samples were treated with
DNase I (Sigma-Aldrich), quantified, and reverse-transcribed into
cDNA with the ReverTra Ace-α First Strand cDNA Synthesis kit
[Toyobo (Shanghai) Biotech Co., Ltd., Shanghai, China]. qPCR was
conducted using a RealPlex4 real-time PCR detection system from
Eppendorf AG (Barkhausenweg, Hamburg, Germany), with SYBR-Green
Real-time PCR Master mix [Toyobo (Shanghai) Biotech Co., Ltd.] as
the detection dye. qPCR amplification was performed for >40
cycles with denaturation at 95°C for 15 sec and annealing at 57°C
for 45 sec. Target cDNA was quantified with the Eppendorf
BioSpectrometer (Eppendorf AG). A comparative threshold cycle (Ct)
was used to determine gene expression relative to a control
(calibrator), and steady-state mRNA levels are reported as an
n-fold difference relative to the calibrator. For each sample, the
marker gene Ct values were normalized using the following formula:
ΔCt = Ct_genes − Ct_18S RNA. To determine relative expression
levels, the following formula was used: ΔΔCt = ΔCt_samplegroups −
ΔCt_controlgroup. The values used to plot the relative expression
of the markers were calculated using the 2−ΔΔCt method.
The mRNA levels were calibrated on the basis of levels of 18S rRNA.
The cDNA of each gene was amplified with primers as previously
described (7). The following
primers were used: BACE1, forward 5′-GCAGGGCTACTACGTGGAGA-3′ and
reverse 5′-CAGCACCCACTGCAAAGTTA-3′; APP, forward
5′-TTTGGCACTGCTCCTGCT-3′ and reverse 5′-CCACAGAACATGGCAATCTG-3′;
Ki67, forward 5′-TGGGTCTGTTATTGATGAGCC-3′ and reverse
5′-TGACTTCCTTCCATTCTGAAGAC-3′; 18s rRNA, forward
5′-CAGCCACCCGAGATTGAGCA-3′ and reverse
5′-TAGTAGCGACGGGCGGTGTG-3′.
Western blot analysis
The cells were lysed using a 2X loading lysis buffer
(Beyotime Institute of Biotechnology, Shanghai, China). The total
amount of proteins from the cultured cells was subjected to 12%
SDS-PAGE and transferred onto a hybrid polyvinylidene difluoride
(PVDF) membrane (Millipore, Bedford, MA, USA). Following inhibition
with 5% (w/v) non-fat dried milk in Tris-buffered saline with
Tween-20 (TBST; Beyotime Institute of Biotechnology), the PVDF
membranes were washed four times (15 min each) with TBST at room
temperature and incubated with primary antibodies, including rabbit
anti-human Ki67 antibody (Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA), rabbit anti-human BACE1, Aβ1–40, Aβ1–42 and GAPDH
antibodies (Cell Signaling Technology, Inc., Beverly, MA, USA).
Following extensive washing, the membranes were incubated with
horseradish peroxidase (HRP)-conjugated goat anti-rabbit
immunoglobulin (Ig) G secondary antibody (1:1,000; Santa Cruz
Biotechnology, Inc.) for 1 h. Following washing four times (15 min
each) with TBST at room temperature, the immunoreactivity was
visualized using an enhanced chemiluminescence kit from Perkin
Elmer, Inc. (Norwalk, CT, USA).
Immunofluorescence (IF) staining
The cultured cells were washed three times with
phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde
(Sigma-Aldrich) for 30 min. Following inhibition, the cells were
initially incubated with primary antibody overnight at 4°C, and
then with fluorescein isothiocyanate- or Cy3-conjugated goat
anti-rabbit IgG antibody (1:200; Sigma-Aldrich) and 5 μg/ml DAPI
(Sigma-Aldrich) at room temperature for 30 min. Then, the cells
were thoroughly washed with TBST and viewed through a fluorescence
microscope (DMI3000; Leica, Allendale, NJ, USA).
ELISA assay
The Aβ1–42 ELISA kit (Hermes Criterion
Biotechnology, Vancouver, BC, Canada) was used according to the
manufacturer’s instructions. Briefly, all the cells and
supernatants were harvested and dissociated in 0.1 M Tris (pH 7.4)
containing 1% Triton X-100 (Sigma-Aldrich) and 5 mM
MgCl2 by sonication. The concentration of
Aβ1–42 was measured and the data were normalized against
the protein concentration and expressed as a nanogram of
Aβ1–42 per milligram of total protein. All the samples
were added to anti-Aβ1–42 antibody-precoated microtest
wells and incubated for 60 min. Following washing three times, the
HRP-conjugated detection antibodies were then added followed by the
addition of the substrate solution. The absorbance was determined
at a wavelength of 450 nm.
RNA extraction and northern blot
analysis
Northern blotting was performed as previously
described (10–12). For all the groups, 20 μg of good
quality total RNA was analyzed on a 7.5 M urea 12% polyacrylamide
denaturing gel and transferred onto a Hybond N+ nylon
membrane (Amersham, Freiburg, Germany). The membranes were
crosslinked using ultraviolet light for 30 sec at 1,200
mjoule/cm2. Hybridization was performed with the
antisense starfire probe to detect the lncRNA
BACE1-AS fragments according to the manufacturer’s
instructions (7). Following
washing, the membranes were exposed for 20–40 h to Kodak XAR-5
films (Sigma-Aldrich). As a positive control, all the membranes
were hybridized with a human U6 snRNA probe. The sequence was as
follows: Human U6 snRNA, 5′-GCAGGGGCCATGCTAATCTTCTCTGTATCG-3′. The
exposure times for the U6 control probe varied between 15 and 30
min.
Ribonuclease protection assay (RPA)
As previously described (7), each RNA sample was treated with
ribonuclease A+T (Sigma-Aldrich), which digests single stranded
RNAs but not RNA duplexes. The RNA samples were incubated at 37°C
for 60 min prior to treatment with an RNAse A+T cocktail
(Sigma-Aldrich). Subsequently, the samples were incubated at 37°C
for 30 min after addition of the RNAse cocktail, and treated with
proteinase K. The RPA assay was then used to detect BACE1 and
BACE1-AS employing two sets of probes by northern blotting. The
first set of probes were designed to target the overlapping region
of the BACE1 sense and antisense transcripts and the second set to
target the non-overlapping region of these transcripts.
siRNA and cell transfection
An siRNA targeted lncRNA BACE1-AS expression plasmid
was constructed as previously described (7). SH-SY5Y cells were transfected with
0.3 μg siRNA-BACE1-AS or an siRNA-mock vector using Lipofectamine
2000 (Invitrogen Life Technologies), according to the
manufacturer’s instructions.
Statistical analysis
Each experiment was performed as least three times
and the data are expressed as the mean ± standard error. The
differences were evaluated using Student’s t-test. P<0.05 was
considered to indicate a statistically significant difference. All
statistical analyses were performed using the SPSS 10.0 statistical
software package (SPSS, Inc., Chicago, IL, USA).
Results
Exogenous Aβ1–42 suppresses
SH-SY5Y cell proliferation and induces AD relative protein
expression
Firstly, the MTT assay was used to evaluate whether
exogenous Aβ1–42 was able to suppress SH-SY5Y cell
proliferation. Large aggregates of synthetic Aβ1–42
suppressed the proliferation of SH-SY5Y cells in a time-dependent
manner (Fig. 1A). Next, the
ability of exogenous Aβ1–42 to induce SP formation was
assessed by qPCR, immunofluorescence (IF) staining and western blot
analysis of the expression levels of APP-related factors. The qPCR
results demonstrated that the expression of APP mRNA in the
Aβ1–42-treated group was markedly elevated compared with
that in the untreated (WT) and DMSO-treated control groups, while
Ki67 expression was decreased on day six of Aβ1–42
treatment (Fig. 1). However, no
significant difference in mRNA expression levels of APP and Ki67
was identified between the Aβ1–42-treated group and the
control groups on day zero. Furthermore, western blot analysis
confirmed that Aβ1–42 and Aβ1–40 protein
expression was significantly increased in the Aβ1–42
treated group compared with the control groups, while Ki67
expression was markedly decreased on day six (Fig. 1). Additionally, IF staining
confirmed the accumulation of Aβ1–42 and Aβ1–40
proteins, but not Ki67, in the Aβ1–42 treated group
compared with the WT and DMSO-treated control groups on day six
(Fig. 1). These data indicated
that exogenous Aβ1–42 inhibited SH-SY5Y cell
proliferation and induced the expression of APP-related factors and
SP formation.
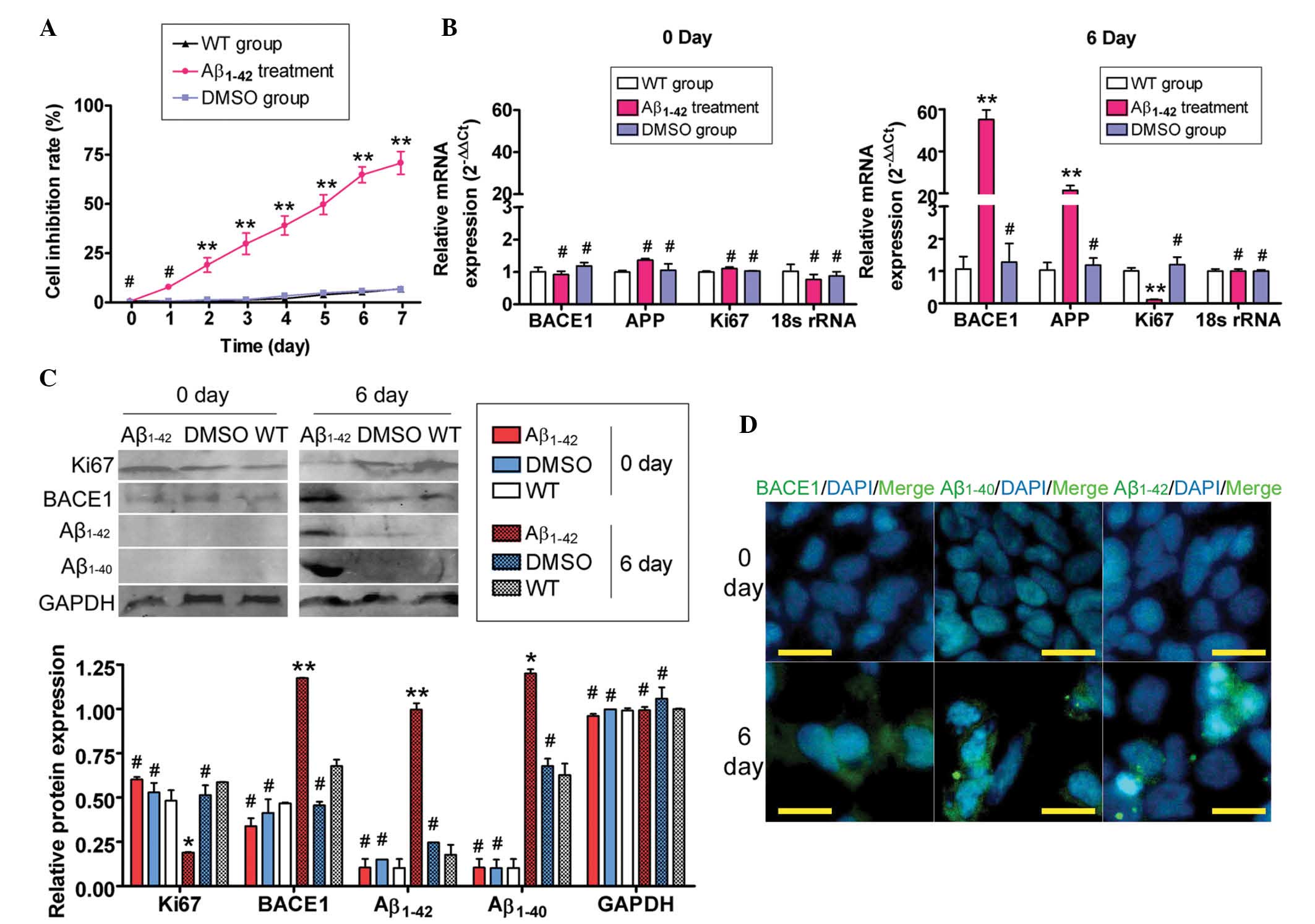 | Figure 1Exogenous Aβ1–42 affected SH-SY5Y cell
proliferation and gene expression. (A) MTT assays demonstrated that
large aggregates of synthetic Aβ1–42 inhibited SH-SY5Y cell
proliferation in a time-dependent manner (**P<0.01
and #P>0.05 vs. WT group; n=3). (B) Results of
quantitative polymerase chain reaction analysis demonstrated that
the mRNA expression of BACE1 and APP in the Aβ1–42 treatment group
was markedly elevated, while the Ki67 expression in this group was
markedly decreased compared with that in the other two groups on
day six. However, no significant differences in the mRNA expression
levels (normalized against 18S rRNA levels) of BACE1, APP and Ki67
were identified between the Aβ1–42-, the WT- and the DMSO- treated
groups on day 0 (**P<0.01 and #P>0.05
vs. WT group; n=3). (C) Western blot analysis confirmed that the
expression of the BACE1, Aβ1–42 and Aβ1–40 proteins was
significantly increased in the Aβ1–42 treatment group, compared
with the WT- and DMSO-treated groups, while the expression of Ki67
in this group was markedly decreased on day six. GAPDH was used as
a loading control (**P<0.01, *P<0.05
and #P>0.05 vs. WT group; n=3). (D) Immunofluorescent
staining confirmed that the expression of the BACE1, Aβ1–42 and
Aβ1–40 proteins was significantly increased in the Aβ1–42-treated
group on day six, while the expression of these proteins was not
detected on day zero (original magnification, ×200). Aβ, amyloid
β-protein; WT, untreated group; APP, amyloid precursor protein;
BACE1, β-secretase-1; DMSO, dimethylsulfoxide. |
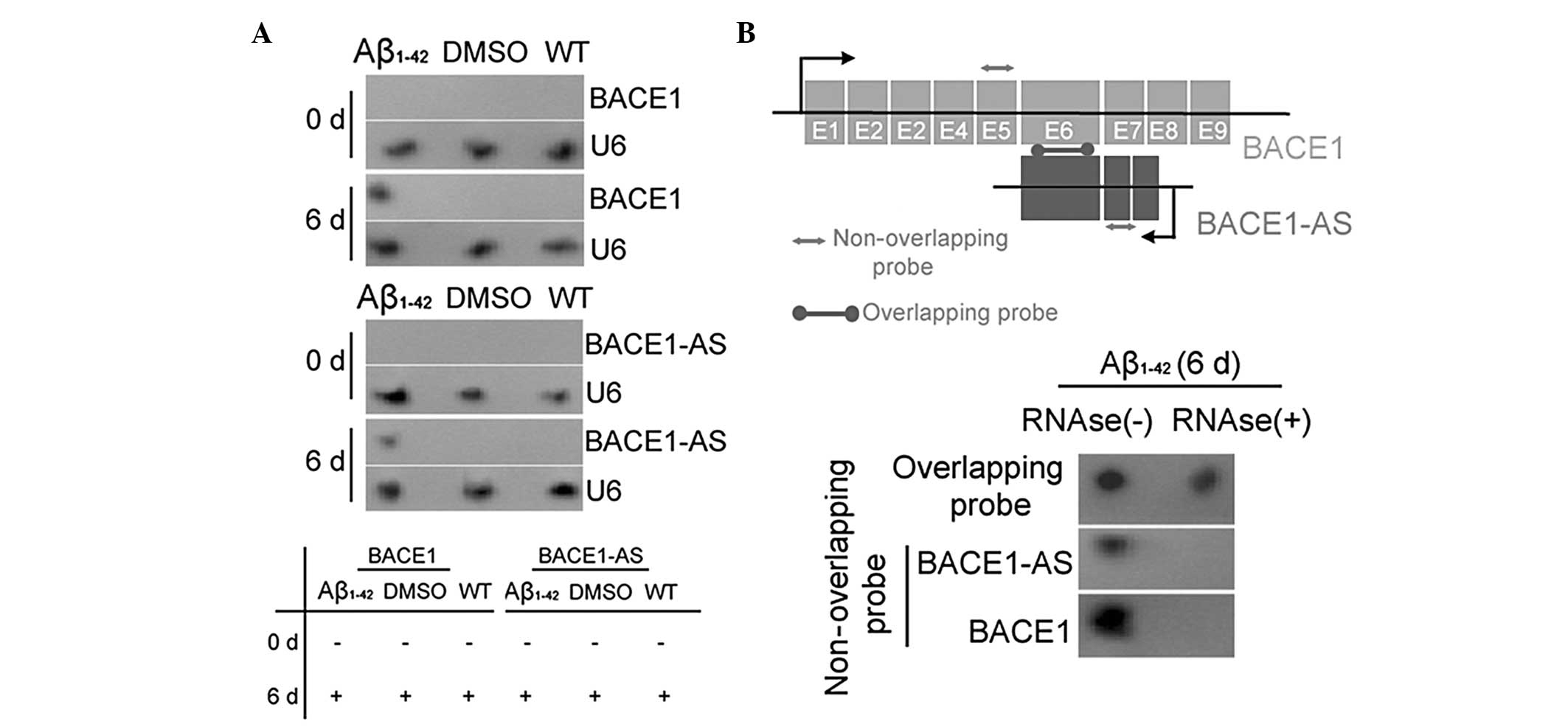
Exogenous Aβ1–42 induces BACE1
and lncRNA BACE1-AS expression
The expression of the enzyme BACE1 in SH-SY5Y cells,
which is closely associated with Aβ1–42 processing, was
investigated prior to (day zero) and following (day six)
Aβ1–42 treatment using qPCR, IF staining and western
blot analysis. The qPCR analysis demonstrated that the expression
of BACE1 mRNA in the Aβ1–42-treated group was significantly
elevated compared with that in the two control groups on day six
(Fig. ). However, no significant differences in the mRNA expression
levels of BACE1 in the three groups at day zero were identified.
Western blotting confirmed that the BACE1 protein was expressed at
significantly higher levels in the Aβ1–42 treated group than in the
WT and DMSO-treated control groups on day six (Fig. 1). This pattern of expression was
confirmed by IF staining of the BACE1 enzyme expression in SH-SY5Y
cells on day six (Fig. 1). These
data indicated that exogenous Aβ1–42 induced the expression of the
APP-related processing enzyme BACE1. By contrast, northern blot
analysis indicated that BACE1 mRNA and lncRNA BACE1-AS
hybridization signals were higher in the extracts of Aβ1–42-treated
cells than in those of the two control groups (Fig. 2A). RNase protection assays (RPA)
were performed using BACE1 mRNA from each group to determine RNA
duplex formation. Northern blotting revealed that non-overlapping
probe hybridization signals were weaker than overlapping probe
hybridization signals in SH-SY5Y cells following RNase treatment
(Fig. 2B). This demonstrated that
the overlapping part of BACE1 mRNA and lncRNA BACE1-AS transcripts
were protected from degradation, thus, indicating that BACE1 and
BACE1-AS indeed form an RNA duplex. These data indicated that
exogenous Aβ1–42 not only promoted the expression of the
APP-cleaving enzyme BACE1, but also induced lncRNA BACE1-AS
expression. Furthermore, lncRNA BACE1-AS formed RNA duplexes with,
and increased the stability of, BACE1 mRNA.
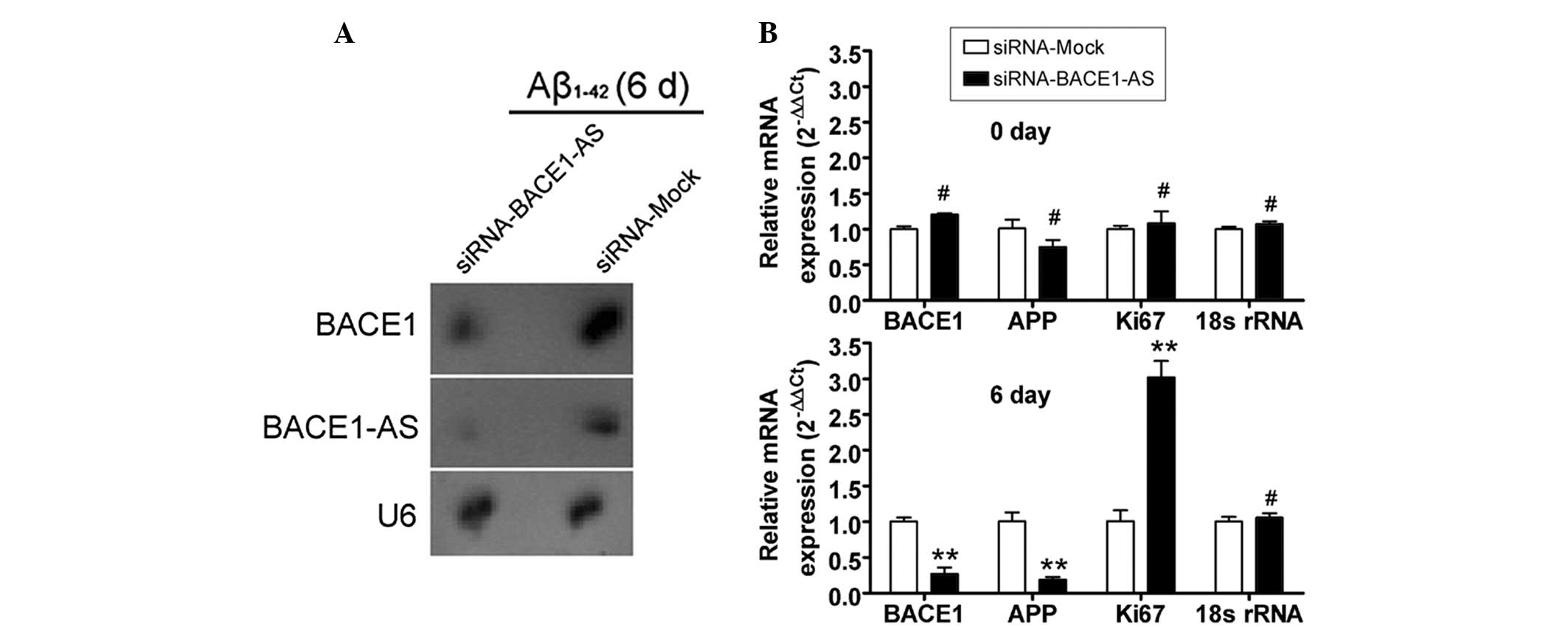
Attenuation of the ability of BACE1 to
cleave APP by siRNA silencing of lncRNA BACE1-AS expression
The potential of siRNA silencing of lncRNA BACE1-AS
expression to reduce the stability of BACE1 mRNA and to attenuate
the ability of BACE1 to cleave APP was then investigated in SH-SY5Y
cells. Northern blot analysis indicated that the lncRNA BACE1-AS
hybridization signal was weaker in the siRNA-BACE1-AS-transfected
cell group than that in the siRNA mock-transfected group (Fig. 3). Furthermore, a strong BACE1
hybridization signal was detected in the siRNA mock-transfected
group, however, not in the siRNA-BACE1-AS-transfected group. In
addition, qPCR analysis demonstrated that in
siRNA-BACE1-AS-transfected cells, the expression levels of APP and
BACE1 mRNA, however, not those of Ki67 mRNA, were significantly
lower than those in the siRNA mock-transfected group on day six of
Aβ1–42 treatment (Fig. 3).
However, no significant differences in the expression levels of
APP, BACE1 and Ki67 mRNA between the two groups were observed at
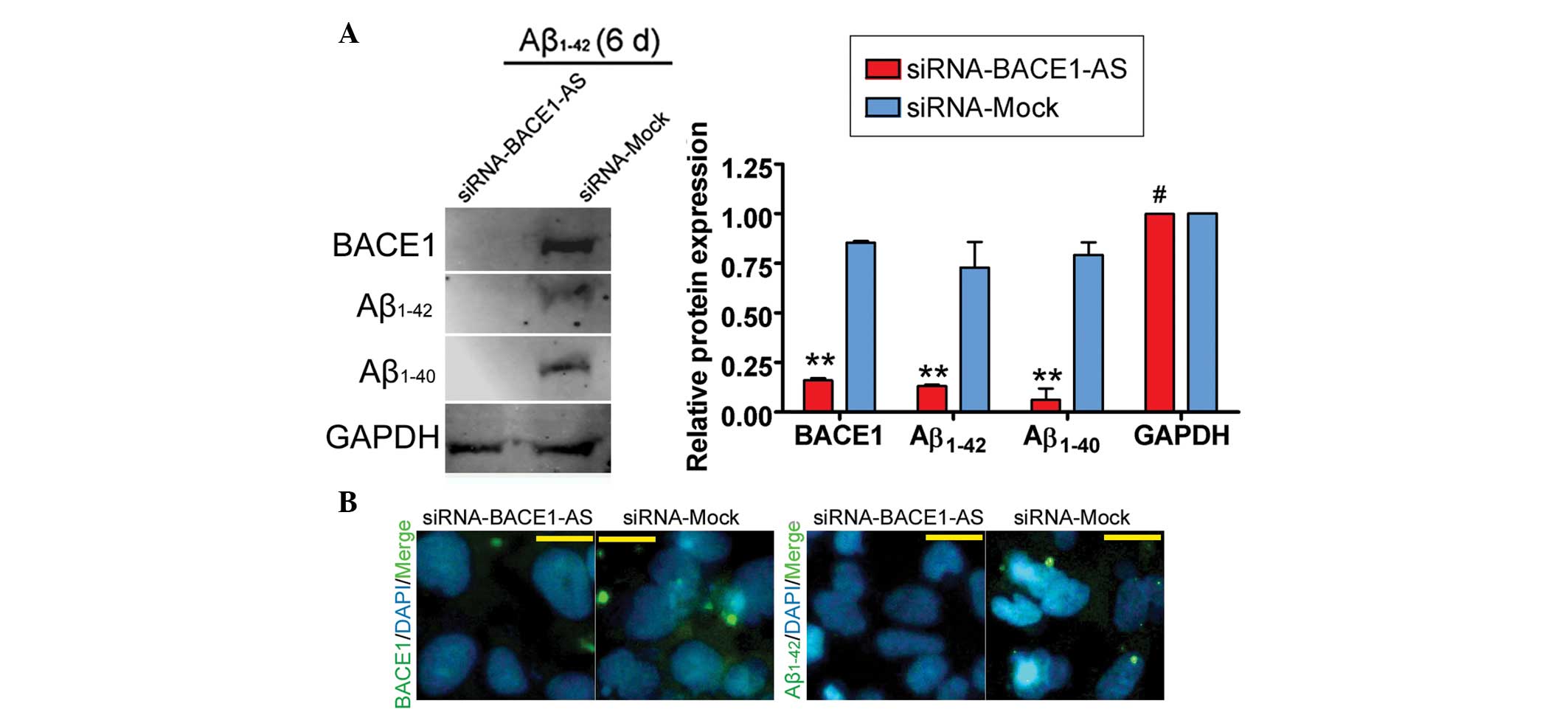
day zero. Furthermore, IF staining and western blot analysis
confirmed that the expression of BACE1, Aβ1–42 and Aβ1–40 proteins
was significantly decreased in the siRNA-BACE1-AS-transfected group
compared with that in the siRNA mock-transfected group (Fig. 4). These results indicated that the
ability of BACE1 to cleave APP was decreased in SH-SY5Y cells by
siRNA-mediated silencing of lncRNA BACE1-AS downregulation. These
results also indicated that the stability of BACE1 in SH-SY5Y cells
was closely associated with lncRNA BACE1-AS expression.
Discussion
The nature and functions of ncRNAs appear to be
numerous and varied. A range of small ncRNAs, including siRNAs,
microRNAs and piRNAs, have been implicated in a host of roles,
including transcriptional regulation, control of chromatin
structure, heterochromatin formation and proteomic status (4). However, accumulating evidence
indicated the existence in mammals of a specific class of ncRNA,
namely lncRNAs, which vary in size from 200 bp to >1,000 bp,
which is much larger than the variety of small ncRNAs that have
been identified. Several studies have reported difficulty in
cloning the full length of various lncRNAs, possibly due to the
increased complexity in their structure compared with that of most
small ncRNAs. By contrast, lncRNAs have a wide variety of sources
and are involved in numerous processing and regulatory pathways.
LncRNAs are transcribed by RNA polymerase II, and are often spliced
and polyadenylated (4). They have
been identified by a variety of methods and the number of specific
lncRNAs demonstrated to affect genomic function is growing. These
include lncRNAs with roles in imprinting, enhancer function, X
chromosome inactivation, chromatin structure and genomic
rearrangements during the generation of antibody diversity
(4). Despite associations with a
number of disorders, lncRNAs remain a relatively unexamined area in
the study of diseases, and may represent a source of new
therapeutic targets (1). To date,
the majority of studies have indicated that lncRNAs act as negative
regulators of their target genes. However, Faghihi et al
(7) identified an lncRNA that
acted as a positive regulator of its target gene in a study of the
pathogenesis of AD (1,7). The study identified an lncRNA
BACE1-AS gene, which generates amyloid β (Aβ). The lncRNA BACE1-AS
increased the stability of the BACE1 mRNA, thus leading to the
amplified production of Aβ peptides and the deleterious
feed-forward cycles of disease progression (7). Based on these observations, the
present study hypothesized that silencing the expression of
endogenous lncRNA BACE1-AS diminishes Aβ formation and neuronal
damage as a consequence. The present study demonstrated that
β-secretase expression was significantly reduced at the mRNA and
protein levels in SH-SY5Y cells as a result of siRNA-mediated
silencing of lncRNA BACE1-AS expression. Furthermore, exogenous
Aβ1–42 did not stimulate the formation of endogenous Aβ (1–40/1–42)
in siRNA-BACE1-AS-transfected SH-SY5Y cells. These data indicated
that the inhibition of the expression of lncRNA BACE1-AS
effectively inhibited the endogenous production of Aβ peptides. By
contrast, when the expression of lncRNA BACE1-AS was silenced in
transfected SH-SY5Y cells, treatment with exogenous Aβ peptides had
a significantly reduced cytotoxic effect and these cells maintained
their normal state. Therefore, lncRNA BACE1-AS is likely to be an
important factor in the formation of mature Aβ peptides. The
ability of BACE1 to cleave APP was attenuated via silencing the
expression of lncRNA BACE1-AS.
Acknowledgements
This study was supported by a grant from the
National Natural Science Foundation of China (no. 81202811),
Shanghai Municipal Health Bureau Fund (no. 20124320) and Project
funded by the China Postdoctoral Science Foundation (no.
2014M550250) to Te Liu. In addition, this study was supported by
the Budget Program of Shanghai Municipal Education Commission (no.
2011JW64) to Zhihua Yu.
References
|
1
|
Wilusz JE, Sunwoo H and Spector DL: Long
noncoding RNAs: functional surprises from the RNA world. Genes Dev.
23:1494–1504. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Huarte M: LncRNAs have a say in protein
translation. Cell Res. 23:449–451. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tsai MC, Manor O, Wan Y, et al: Long
noncoding RNA as modular scaffold of histone modification
complexes. Science. 329:689–693. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Caley DP, Pink RC, Trujillano D and Carter
DR: Long noncoding RNAs, chromatin, and development. Scientific
World Journal. 10:90–102. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Johnson R: Long non-coding RNAs in
Huntington’s disease neurodegeneration. Neurobiol Dis. 46:245–254.
2012.
|
|
6
|
Qureshi IA, Mattick JS and Mehler MF: Long
non-coding RNAs in nervous system function and disease. Brain Res.
1338:20–35. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Faghihi MA, Modarresi F, Khalil AM, et al:
Expression of a noncoding RNA is elevated in Alzheimer’s disease
and drives rapid feed-forward regulation of beta-secretase. Nat
Med. 14:723–730. 2008.
|
|
8
|
Isobe I, Yanagisawa K and Michikawa M: A
possible model of senile plaques using synthetic amyloid
beta-protein and rat glial culture. Exp Neurol. 162:51–60. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Querfurth HW and LaFerla FM: Alzheimer’s
disease. N Engl J Med. 362:329–344. 2010.
|
|
10
|
Liu T, Shen D, Xing S, et al: Attenuation
of exogenous angiotensin II stress-induced damage and apoptosis in
human vascular endothelial cells via microRNA-155 expression. Int J
Mol Med. 31:188–196. 2013.
|
|
11
|
Liu T, Cheng W, Huang Y, Huang Q, Jiang L
and Guo L: Human amniotic epithelial cell feeder layers maintain
human iPS cell pluripotency via inhibited endogenous microRNA-145
and increased Sox2 expression. Exp Cell Res. 318:424–434. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cheng W, Liu T, Jiang F, et al:
microRNA-155 regulates angiotensin II type 1 receptor expression in
umbilical vein endothelial cells from severely pre-eclamptic
pregnant women. Int J Mol Med. 27:393–399. 2011.PubMed/NCBI
|