Introduction
Insulin resistance is a condition of impaired or
diminished insulin sensitivity, in which the normal expression
levels of insulin fail to induce the normal insulin response in the
liver, adipose tissue and muscle cells. Insulin acts as a critical
factor in the pathogenesis of type 2 diabetes and metabolic
syndrome. Hepatic glucose metabolism is tightly controlled by
circulating insulin, due to its inhibitory effects on
gluconeogenesis and stimulatory effects on glycogenesis in the
liver. In insulin resistant states, hepatic glycogen synthesis is
impaired, which results in increased glucose production (1).
Insulin signaling is essential for the maintenance
of hepatic glucose homeostasis. In stable states, glycogensynthase
(GS) is phosphorylated by activated glycogen synthase kinase 3
(GSK-3), which causes the inhibition of glycogen synthesis. The
phosphoinositide-3 kinase (PI3K)/AKT pathway is activated when
hepatocytes respond to insulin, leading to the phosphorylation of
GSK-3 and inactivation of GSK. This results in the generation of
glycogen synthesis through activated GS (2–4).
Hyperglycemia is regarded as a consequence of insufficient insulin
secretion and insulin resistance. Several studies have identified
that hyperglycemia can lead to an apparent improvement of insulin
resistance in the liver (5).
Glucotoxicity is a major cause of β-cell dysfunction that can lead
to impaired insulin signaling action or insulin resistance in the
liver, consistent with decreased glycogen levels in
hepatocytes.
MicroRNAs (miRNAs) are a class of non-coding 18–25
nucleotide endogenous RNA molecules, which act as specific gene
silencers to regulate the target gene expression at the
posttranscriptional level by base pairing to the 3′ untranslated
region of the target mRNA. Numerous studies have indicated that
miRNAs have a critical regulatory role in various metabolic
diseases, including diabetes mellitus, obesity and metabolic
syndrome. miR-375, miR-29, miR-9 and Let-7 have been previously
associated with regulating insulin secretion (6). miR-375 and miR-124a have additionally
been shown to participate in pancreatic islet development (7) and β-cell differentiation (8). Although it has been reported that
miR-181 may participate in the development of insulin resistance by
the regulation of Sirtuin 1 expression at the translational level
in hepatocytes, the mechanisms of miRNAs involved in hepatic
insulin resistance remain unknown (9).
Decreased levels of miR-152 have been identified to
accelerate the tumor growth of certain types of tumors (10). It has been reported that DNA
methyltransferase-1 (DNMT-1) is a target of miR-152, and functions
in the maintenance of DNA methylation (8,11,12).
In cancer cells, miR-152 was shown to directly modulate the
expression of DNMT-1, which in turn could modulate the expression
of specific oncogenes and tumor suppressor genes, leading to
enhanced carcinoma growth. There is little known regarding the role
of miR-152 in the regulation of hepatic insulin resistance and
glucose metabolism. This study has provided for the first time, to
the best of our knowledge, novel experimental evidence showing that
high glucose levels impaired the activation of the AKT/GSK pathway
and the synthesis of glycogen in hepatocytes, at least in part
through the downregulation of miR-152.
Materials and methods
Cell culture
NCTC 1469 cells were derived from mouse liver cells
(American Type Culture Collection, Manassas, VA, USA) and cultured
in low-glucose Dulbecco’s modified Eagle’s medium (5 mmol/l
glucose; Gibco-BRL, Carlsbad, CA, USA) supplemented with 10% horse
serum (HyClone, Logan, UT, USA), 100 U/ml penicillin (Gibco-BRL)
and 0.1 mg/ml streptomycin (Gibco-BRL) at 37°C in a humidified
atmosphere of 95% O2 and 5% CO2.
Isolation of mouse primary
hepatocytes
Male C57BL/6J mice (age, 12 weeks) were provided by
Peking University Health Science Center (originally purchased from
Jackson Laboratory, Bar Harbor, ME, USA). All animal procedures
were performed in accordance with the National Institutes of Health
Animal Care and Use Guidelines. All animal protocols were approved
by the Animal Ethics Committee at the Beijing Institute of
Geriatrics (Beijing, China). Primary hepatocytes were isolated by a
two-step collagenase perfusion [0.2 mg/ml type IV collagenase
(Sigma, St. Louis, MO, USA) in Hanks’ balanced salt solution], as
described previously (13). The
hepatocytes were collected by centrifugation at 430 × g for 8 min.
Immediately after harvesting, the cells were suspended in
pre-warmed William’s E medium (Sigma) supplemented with 10% fetal
bovine serum, 20 ng/ml dexamethasone (Sigma), ITS (containing 5
mg/l insulin, 5 mg/l trasferrin and 5 μg/l sodium selenate; Sigma)
and 10 μg/ml gentamicin (Invitrogen Life Technologies, Carlsbad,
CA, USA). Hepatocytes were plated in collagen-coated
25-cm2 flasks at a density of 1×106 cells per
flask.
Transfection of miRNA mimic and
inhibitor
The mimic and inhibitor of miR-152 were purchased
from Genepharma (Shanghai, China). miRNA mimic and inhibitor
controls were used as negative controls, respectively. Hiperfect
Transfection Reagent (Qiagen, Hilden, Germany) was used for the
transfection of the miR-152 mimic and inhibitor. The expression of
miR-152 was detected by quantitative polymerase chain reaction
(qPCR), 48 h after transfection.
RNA isolation and qPCR
Enriched miRNA was isolated using an miRNA isolation
kit (Takara Bio Inc., Shiga, Japan). Stem-loop reverse
transcription-PCR was performed on the samples to detect and
quantify mature miRNA, using a stem-loop antisense primer mix
(Tables I and II) and AMV transcriptase (Takara). The
cDNA was routinely tested by qPCR based on the SYBR Green I method,
according to the manufacturer’s instructions (Takara).
Amplification and detection of the specific products were performed
according to the manufacturer’s instructions using an ABI PRISM
7500 system (Applied Biosystems®, Invitrogen Life
Technologies). The U6 small nucleolar RNA was used as the
house-keeping small RNA reference gene. The relative gene
expression was normalized to the U6 small nucleolar RNA. Each
reaction was performed in triplicate, and analysis was performed
using the 2−ΔΔCT method.
 | Table ISequences of primers used for reverse
transcription. |
Table I
Sequences of primers used for reverse
transcription.
|
Reverse-transcriptional primer
(5′-3′) |
|---|
| U6 |
GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAAAATATG |
| miR-152 |
GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCCAAGT |
| Table IINucleotide of primers used for
qPCR. |
Table II
Nucleotide of primers used for
qPCR.
| Forward primer
(5′-3′) |
|---|
| Universal primer | GTGCAGGGTCCGAGGT |
| U6 |
GCGCGTCGTGAAGCGTTC |
| miR-152 | TCAGTGCATGACAGA |
Western blot analysis
Cell lysates (15–30 μg of protein) were separated by
10% SDS-PAGE, then transferred to a polyvinylidene fluoride
membrane (Millipore, Billerica, MA, USA). The membranes were then
blocked with 8% non-fat dry milk and probed with the primary
antibodies at 4°C overnight. The blots were incubated with
horseradish peroxidase-conjugated anti-IgG, followed by detection
with enhanced chemiluminescence (Millipore). Antibodies against
AKT, phosphorylated AKT (Ser473), glycogen synthase
kinase (GSK), and phosphorylated GSK (Ser9) were
purchased from Cell Signaling Technology, Inc., (Beverly, MA, USA).
Antibodies to β-actin were obtained from Santa Cruz (Santa Cruz
Biotechnology Inc., Santa Cruz, CA, USA).
Glycogen content measurement
Glycogen levels were measured in cells incubated
with 1 nmol/l insulin (United States Biological, Salem, MA, USA),
for 3 h, using a Glycogen Assay kit (BioVision, Mountain View, CA,
USA).
Statistical analysis
All values are represented as the mean ± standard
error of the mean, of the indicated number of measurements. One-way
analysis of variance was used to determine statistical
significance. P<0.05 considered to indicate a statistically
significant difference.
Results
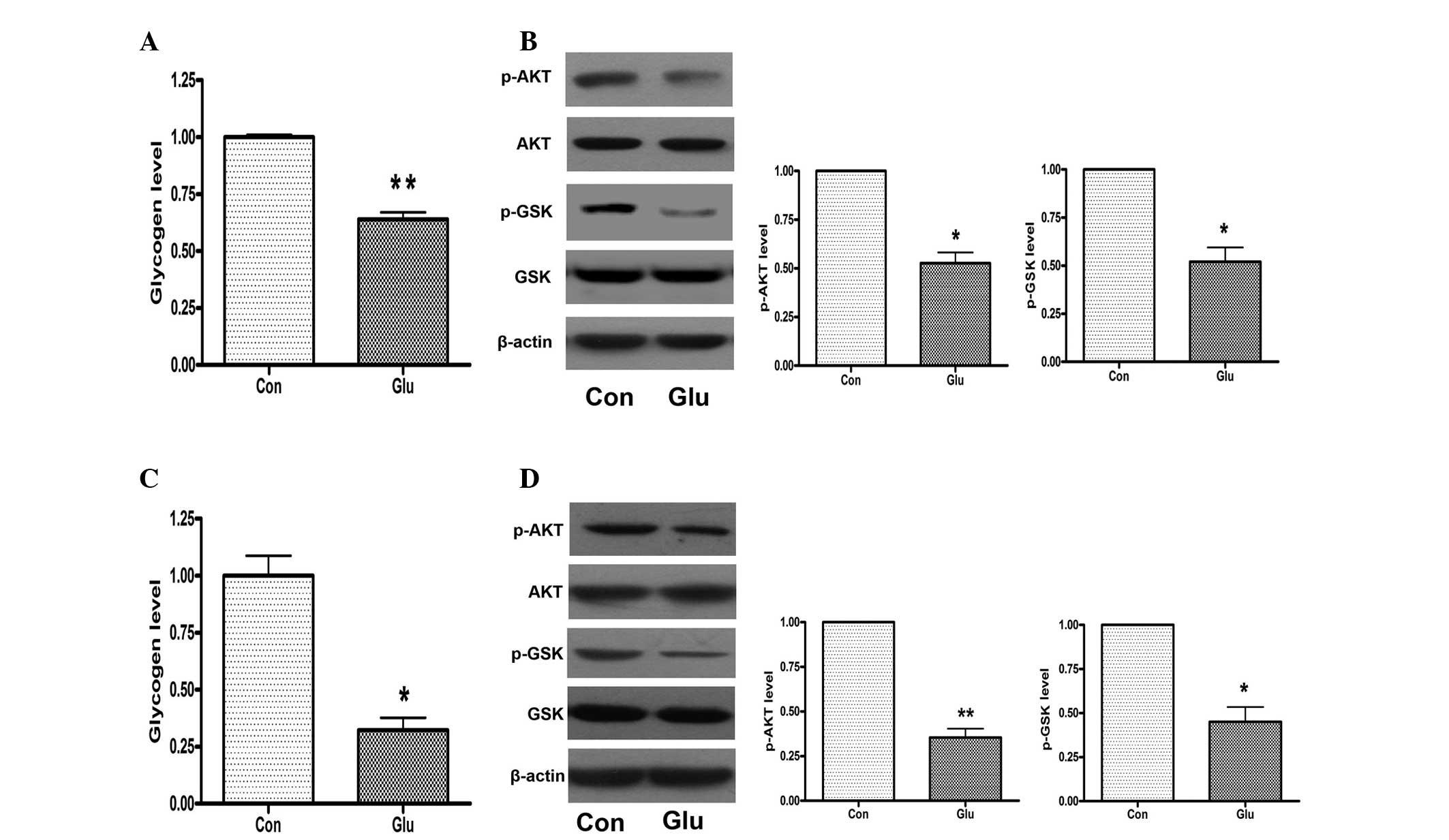
High glucose levels induce a reduction of
glycogen synthesis in hepatocytes through impairing phosphorylation
of AKT and GSK
Mouse NCTC 1469 hepatocytes were treated with 25 mM
glucose for 48 h and the glycogen levels were measured. As shown in
Fig. 1A, the high glucose
treatment significantly decreased the expression levels of glycogen
in NCTC 1469 cells. Furthermore, the phosphorylation of AKT and GSK
was significantly reduced in the NCTC 1469 cells treated with 25 mM
glucose for 48 h (Fig. 1B). In
order to further assess the effects of high glucose on glycogen
synthesis, mouse primary hepatocytes were also treated with 25 mM
glucose for 48 h. The results indicated that glycogen levels and
phosphorylation of AKT and GSK were reduced in mouse primary
hepatocytes in response to high glucose treatment (Fig. 1C and D), indicating that high
glucose induced a reduction of glycogen synthesis in the
hepatocytes, through impairing the phosphorylation of AKT and
GSK.
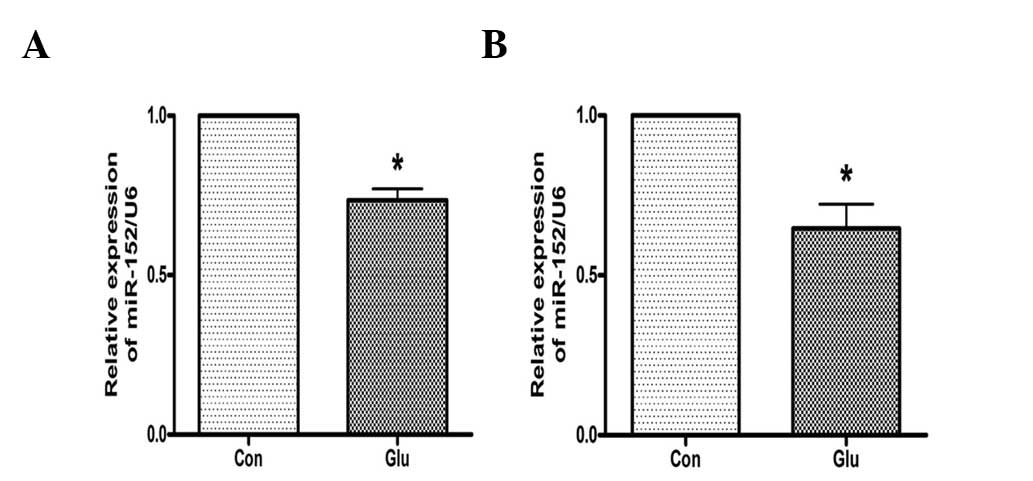
High glucose suppresses the expression of
miR-152 in hepatocytes
The effects of high glucose on the expression of
miR-152 were investigated. As analyzed by qPCR, the expression of
miR-152 was downregulated in NCTC 1469 cells treated with 25 mM
glucose for 48 h (Fig. 2A). High
glucose additionally suppressed the expression of miR-152 in mouse
primary hepatocytes (Fig. 2B).
These data suggest that miR-152 may be involved in glucose-induced
insulin resistance.
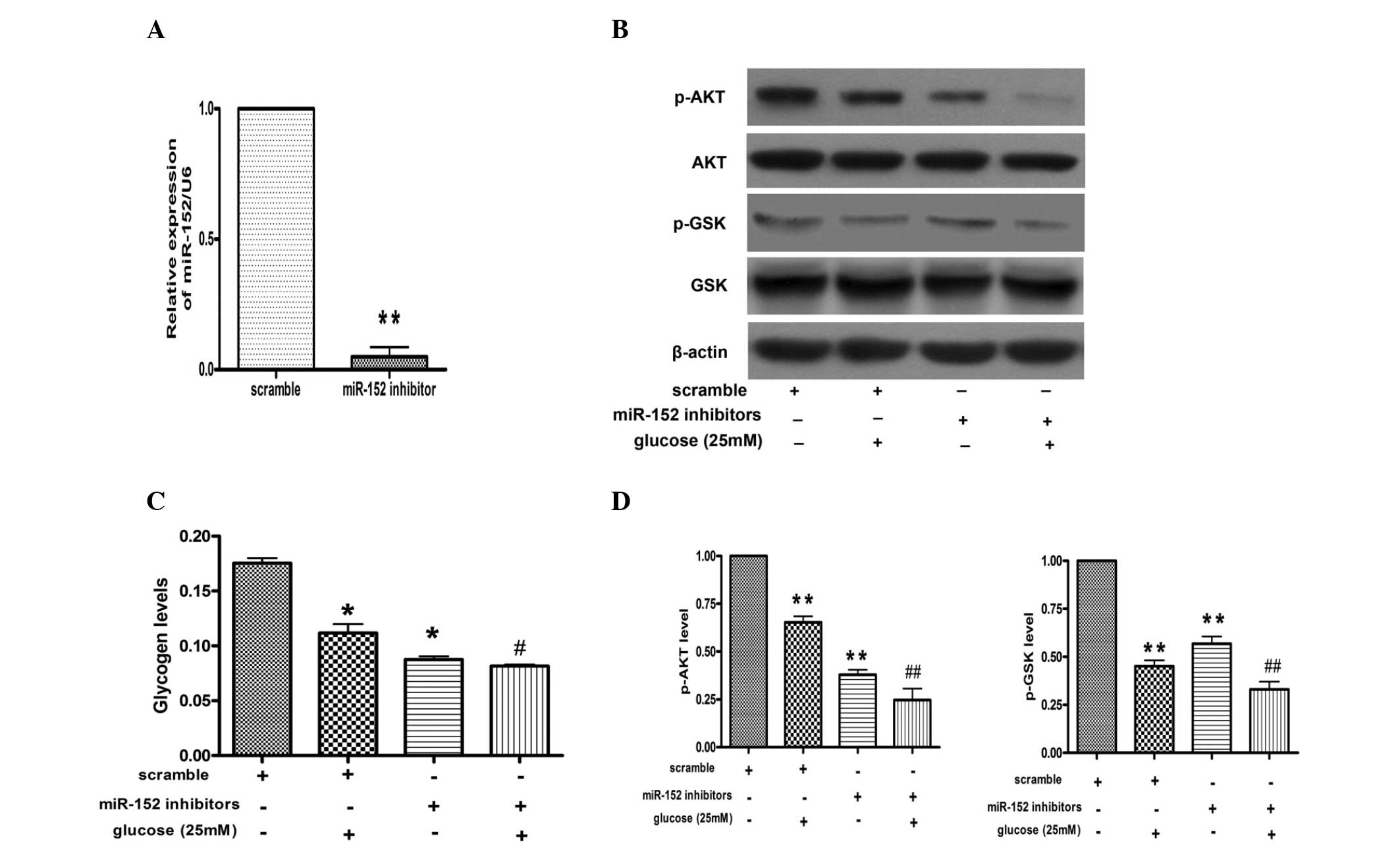
miR-152 inhibitor promotes reduction of
glycogen synthesis and impairment of AKT and GSK phosphorylation in
hepatocytes
In order to further investigate the effects of
miR-152 on glucose-induced reduction of glycogen synthesis, miR-152
inhibitor was transfected into the NCTC 1469 cells. As shown in
Fig. 3A, miR-152 levels were
decreased to ~10% in the NCTC 1469 cells transfected with the
miR-152 inhibitor, as compared with those transfected with
scrambled miRNA. Furthermore, downregulation of miR-152 inhibited
phosphorylation of AKT and GSK in NCTC 1469 cells treated with or
without glucose (Fig. 3B).
Furthermore, the transfection of miR-152 inhibitor decreased the
production of glycogen in NCTC 1469 cells treated with or without
glucose (Fig. 3C).
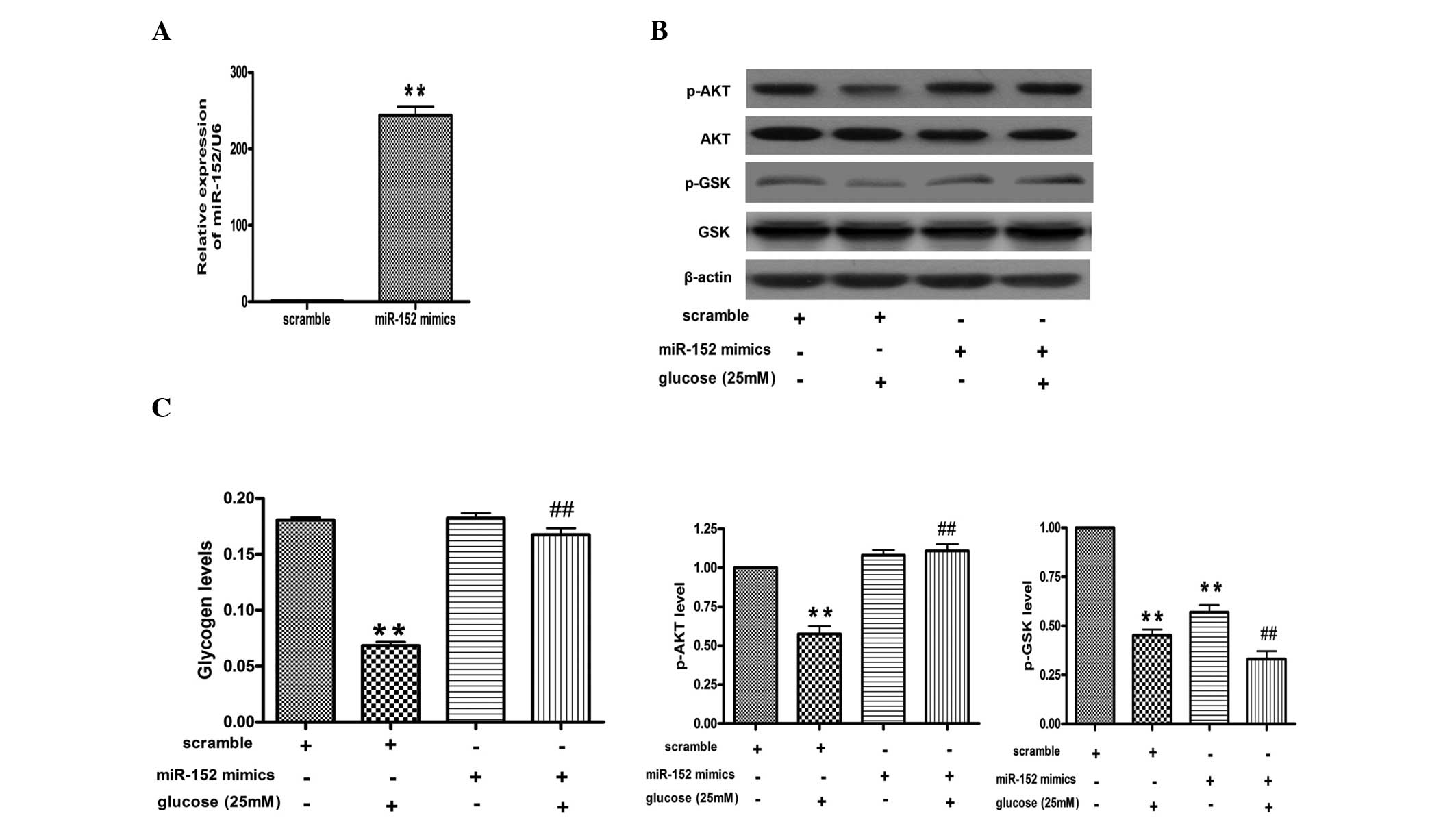
Upregulation of miR-152 reverses the
glucose-induced decrease in glycogen synthesis and AKT and GSK
phosphorylation in hepatocytes
miR-152 mimic was transfected into the NCTC 1469
cells for 48 h and then analyzed by qPCR. qPCR indicated that the
levels of miR-152 were increased by ~200 fold in the NCTC 1469
cells transfected with the miR-152 mimic, as compared with those
transfected with the scrambled miRNA (Fig. 4A). Furthermore, the transfection of
the miR-152 mimic increased the phosphorylation levels of AKT and
GSK, and rescued the effects of high glucose on the activation of
the AKT/GSK pathway (Fig. 4B).
Finally, it was identified that upregulation of miR-152 could
reverse the glucose-induced decrease in glycogen synthesis in
hepatocytes (Fig. 4C).
Discussion
Increasing evidence has indicated that miRNA is
involved in the pathogenesis of type 2 diabetes and insulin
resistance (14). In the present
study, it was identified that the activation of AKT and GSK, and
the levels of glycogen were inhibited in NCTC 1469 cells and mouse
primary hepatocytes, following exposure to 25 mM glucose for 48 h.
Furthermore, to the best of our knowledge this study demonstrated
for the first time, that high glucose levels suppressed the
expression of miR-152 in hepatocytes. In order to further assess
the effects of miR-152 on the glucose-induced reduction of glycogen
synthesis and activation of AKT and GSK, the miR-152 mimic and
inhibitor were transfected into the NCTC 1469 cells, respectively.
The results suggested that miR-152 could regulate the activation of
AKT and GSK, and subsequently modulate glycogen synthesis in the
NCTC 1469 cells treated with 25 mM glucose for 48 h.
Insulin resistance is a diminished capacity for
insulin to stimulate glucose uptake and glycogen synthesis in
peripheral tissues, including skeletal muscle, adipose tissue and
liver. It is a critical factor in the pathogenesis of type 2
diabetes. The liver has a central role in glucose and lipid
metabolism, and hepatic insulin resistance is a hallmark feature of
type 2 diabetes. It has been previously reported that high
glucose-induced oxidative stress is important in the development
and progression of hepatic insulin resistance. Furthermore, high
glucose has been shown to activate the protein kinase C and c-Jun
N-terminal kinase pathways, which act on the Ser307 phosphorylation
of insulin receptor substrate 1 and block the downstream activation
of the AKT pathway in the liver (11,12,15).
In the liver, the PI3K/AKT pathway functions in the insulin
signaling cascade, whereby activated AKT mediates the
phosphorylation and inactivation of GSK, which subsequently results
in the activation of GS and increased glycogen synthesis. The
resulting hyperglycemia is an important factor in the pathogenesis
of insulin resistance. Therefore, a high glucose-induced hepatic
insulin resistance cell model was used in the present study. Under
high glucose conditions, insulin fails to activate its signaling
pathway, resulting in an insulin resistant state. Hepatic insulin
resistance can be determined by measuring the insulin-mediated
phosphorylation of AKT and GSK, and expression levels of glycogen.
The present study identified that the levels of phosphorylation of
AKT and GSK were reduced, followed by impaired glycogen synthesis,
in NCTC 1469 cells and mouse primary hepatocytes, following
exposure to 25 mM glucose for 48 h. These results indicate that
high glucose levels induced a reduction in glycogen synthesis in
hepatocytes through impairing the phosphorylation of AKT and
GSK.
To investigate the mechanisms underlying the high
glucose-induced reduction of glycogen synthesis and impaired
phosphorylation of AKT and GSK in hepatocytes, the effects of high
glucose levels on the expression of miR-152 were investigated. The
results demonstrated that the expression of miR-152 was
downregulated in NCTC 1469 cells treated with 25 mM glucose for 48
h. Similarly, high glucose levels suppressed the expression of
miR-152 in mouse primary hepatocytes. These data indicate that
miR-152 is involved in glucose-induced insulin resistance.
It has been previously considered that miR-152 is
involved in various carcinomas. In human gastric and colorectal
cancer, expression levels of miR-152 were significantly lower, as
compared with matched non-tumor adjacent tissues (16). In cholangiocarcinoma, interleukin-6
was shown to regulate the expression of miR-152, thus linking
inflammation-associated cytokines with oncogenesis in
cholangiocarcinoma (17). Tsuruta
et al (18) reported that
miR-152 expression was decreased in human endometrial cancer, while
the restoration of miR-152 expression in endometrial cancer cell
lines was sufficient to inhibit tumor cell growth in vitro
and in vivo. Furthermore, it was identified that the DNMT-1,
E2F3, MET and Rictor genes were candidate targets of miR-152, and
the data further suggested that epigenetic silencing could drive
endometrial carcinogenesis (18).
DNMT-1 is the most abundant methyltransferase in mammalian cells,
and functions in the maintenance of DNA methylation.
Hypermethylation at promoter CpG islands and inactivation of
multiple tumor suppressor genes are common in carcinomas, and
contribute to tumor growth. DNMT-1 has a role in the establishment
and regulation of tissue-specific patterns of methylated cytosine
residues (19,20). In cancer cells, reduced expression
of miR-152 directly modulated the expression of DNMT-1 (21). Alteration in DNA methylation
modulates the expression of specific oncogenes and tumor suppressor
genes, leading to carcinoma growth (22,23).
However, the involvement of miR-152 in glycogen metabolism remained
unclear. To clarify this, miR-152 inhibitor and mimic were
transfected into NCTC 1469 cells, respectively. The transfection of
miR-152 inhibitor reduced the generation of glycogen, accompanied
by impaired phosphorylation of AKT and GSK in NCTC 1469 cells
treated with or without glucose. By contrast, upregulation of
miR-152 through transfection of miR-152 mimic could reverse the
glucose-induced decrease in glycogen synthesis and reduce AKT and
GSK phosphorylation in hepatocytes. These data indicate that high
glucose levels reduce hepatic glycogenesis by suppressing
miRNA-152, which modulates the AKT/GSK pathway, and in turn results
in insulin resistance, thereby miR-152 and the AKT-GSK pathway may
act as novel therapeutic targets in hepatic glycogenesis.
Acknowledgements
This study was supported by funding from the
National Basic Research Program of China (grant no. 2012CB517502)
and the National Natural Science Foundation of China (grant nos.
81270887 and 81070634).
References
|
1
|
Meshkani R and Adeli K: Hepatic insulin
resistance, metabolic syndrome and cardiovascular disease. Clin
Biochem. 42:1331–1346. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shearn CT, Fritz KS, Reigan P and Petersen
DR: Modification of Akt2 by 4-hydroxynonenal inhibits
insulin-dependent Akt signaling in HepG2 cells. Biochemistry.
50:3984–3996. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Henriksen EJ and Dokken BB: Role of
glycogen synthase kinase-3 in insulin resistance and type 2
diabetes. Curr Drug Targets. 7:1435–1441. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schinner S, Scherbaum WA, Bornstein SR and
Barthel A: Molecular mechanisms of insulin resistance. Diabet Med.
22:674–682. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nawano M, Oku A, Ueta K, et al:
Hyperglycemia contributes insulin resistance in hepatic and adipose
tissue but not skeletal muscle of ZDF rats. Am J Physiol Endocrinol
Metab. 278:E535–E543. 2000.PubMed/NCBI
|
|
6
|
Ramachandran D, Roy U, Garg S, et al:
Sirt1 and mir-9 expression is regulated during glucose-stimulated
insulin secretion in pancreatic β-islets. FEBS J. 278:1167–1174.
2011.PubMed/NCBI
|
|
7
|
Joglekar MV, Parekh VS and Hardikar AA:
Islet-specific microRNAs in pancreas development, regeneration and
diabetes. Indian J Exp Biol. 49:401–408. 2011.PubMed/NCBI
|
|
8
|
Poy MN, Hausser J, Trajkovski M, et al:
miR-375 maintains normal pancreatic alpha- and beta-cell mass. Proc
Natl Acad Sci USA. 106:5813–5818. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kornfeld JW, Baitzel C, Könner AC, et al:
Obesity-induced overexpression of miR-802 impairs glucose
metabolism through silencing of Hnf1b. Nature. 494:111–115. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen Y, Song Y, Wang Z, et al: Altered
expression of MiR-148a and MiR-152 in gastrointestinal cancers and
its clinical significance. J Gastrointest Surg. 14:1170–1179. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang WY, Lee JJ, Kim Y, et al:
Amelioration of insulin resistance by scopoletin in
high-glucose-induced, insulin-resistant HepG2 cells. Horm Metab
Res. 42:930–935. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nakatani Y, Kaneto H, Kawamori D, et al:
Modulation of the JNK pathway in liver affects insulin resistance
status. J Biol Chem. 279:45803–45809. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ramey G, Deschemin JC and Vaulont S:
Cross-talk between the mitogen activated protein kinase and bone
morphogenetic protein/hemojuvelin pathways is required for the
induction of hepcidin by holotransferrin in primary mouse
hepatocytes. Haematologica. 94:765–772. 2009. View Article : Google Scholar
|
|
14
|
Mao Y, Mohan R, Zhang S and Tang X:
MicroRNAs as pharmacological targets in diabetes. Pharmacol Res.
75:37–47. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nakajima K, Yamauchi K, Shigematsu S, et
al: Selective attenuation of metabolic branch of insulin receptor
down-signaling by high glucose in a hepatoma cell line, HepG2
cells. J Biol Chem. 275:20880–20886. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen Y, Song Y, Wang Z, et al: Altered
expression of MiR-148a and MiR-152 in gastrointestinal cancers and
its clinical significance. J Gastrointest Surg. 14:1170–1179. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Braconi C, Huang N and Patel T:
MicroRNA-dependent regulation of DNA methyltransferase-1 and tumor
suppressor gene expression by interleukin-6 in human malignant
cholangiocytes. Hepatology. 51:881–890. 2010.PubMed/NCBI
|
|
18
|
Tsuruta T, Kozaki K, Uesugi A, et al:
MiR-152 is a tumor suppressor microRNA that is silenced by DNA
hypermethylation in endometrial cancer. Cancer Res. 71:6450–6462.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hino R, Uozaki H, Murakami N, et al:
Activation of DNA methyltransferase 1 by EBV latent membrane
protein 2A leads to promoter hypermethylation of PTEN gene in
gastric carcinoma. Cancer Res. 69:2766–2774. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kang MY, Lee BB, Kim YH, et al:
Association of the SUV39H1 histone methyltransferase with the DNA
methyltransferase 1 at mRNA expression level in primary colorectal
cancer. Int J Cancer. 121:2192–2197. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ji W, Yang L, Yuan J, et al: MicroRNA-152
targets DNA methyltransferase 1 in NiS-transformed cells via a
feedback mechanism. Carcinogenesis. 34:446–453. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang J, Wang Y, Guo Y and Sun S:
Down-regulated microRNA-152 induces aberrant DNA methylation in
hepatitis B virus-related hepatocellular carcinoma by targeting DNA
methyltransferase 1. Hepatology. 52:60–70. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Braconi C, Huang N and Patel T:
MicroRNA-dependent regulation of DNA methyltransferase-1 and tumor
suppressor gene expression by interleukin-6 in human malignant
cholangiocytes. Hepatology. 51:881–890. 2010.PubMed/NCBI
|