Introduction
Helicobacter pylori, a Gram-negative,
spiral-shaped, microaerophilic bacterium found in the human
stomach, is recognized as a major risk factor for chronic
gastritis, peptic ulcers and gastric cancer (1). H. pylori infects approximately
half of the world’s population and has been classified as a
carcinogen by the World Health Organization and the International
Agency for Research on Cancer (2,3).
Gastric cancer is the second leading cause of cancer-associated
mortality worldwide (4). Although
considerable research has shown that H. pylori infection is
closely associated with gastric cancer, the molecular mechanisms of
gastric tumor initiation and development induced by H.
pylori infection remain poorly understood (5).
Cyclooxygenase (COX), also known as
prostaglandin-endoperoxide synthase, is a key rate-limiting enzyme
responsible for the formation of prostanoids and thromboxanes
(6,7). There are three COX isoenzymes, COX-1,
COX-2 and COX-3 (a splice variant of COX-1). COX-1 is considered a
constitutive enzyme that is expressed in nearly all mammalian
cells, whilst COX-2 is an inducible enzyme that is undetectable in
the majority of normal tissues (8,9).
However, COX-2 expression is elevated in many types of cancer,
including gastric cancer, and is closely correlated with the
clinical outcome (10–15). Furthermore, a previous study
demonstrated that COX-2 expression is associated with H.
pylori infection in human gastric cancer, and the elevation of
COX-2 expression may be mediated by the p38 mitogen-activated
protein kinases (MAPK) pathway (16).
Prostaglandin E2 (PGE2) is the main
product generated from arachidonic acid catalyzed by COX-2, and it
exerts a wide range of pathological effects via receptors on the
cell and nuclear membranes (17–19).
Enhanced expression of PGE2 is found in several types of
cancer and is associated with tumor growth and angiogenesis
(20,21). PGE2 receptors (EPs) are
types of G protein-coupled receptor (GPCR), and exist in at least
four isoforms: EP1, EP2, EP3 and EP4 (22–25).
PGE2 binds to EPs to promote the expression of vascular
endothelial growth factor (VEGF) in human prostate PC3 and gastric
MKN28 cancer cells (20,26). However, whether EPs mediate VEGF
expression in gastric cancer cells following H. pylori
infection, and the type of EP involved in such regulation, have yet
to be elucidated.
The MAPK family of proteins are involved in cell
differentiation, migration, apoptosis and autophagy (27). p38 MAPK is a key member of this
family and it is part of a signaling cascade that modulates
cellular responses to cytokines and stresses, including
inflammatory cytokines, osmotic shock, lipopolysaccharide,
ultraviolet light and growth factors (26). Previous studies have demonstrated
that p38 MAPK is also activated following H. pylori
infection, and promotes COX-2 expression in MKN45 gastric cancer
cells, which has been found to be involved in mediating H.
pylori-induced gastric tumorigenesis (16,28–30).
Several studies have investigated the oncogenic potential of the
p38 MAPK pathway, since p38 MAPK also has a critical role in
regulating VEGF expression leading to angiogenesis (27,31).
However, whether p38 MAPK is involved in regulating VEGF expression
in H. pylori-infected gastric cells is yet to be
elucidated.
To investigate the mechanism of H.
pylori-induced gastric cancer, VEGF expression was analyzed in
MKN45 gastric cells following H. pylori infection. It was
found that p38 MAPK has a critical role in regulating VEGF
expression in H. pylori-infected MKN45 cells, and this
effect may be mediated via the COX-2-EP2/EP4 pathway.
Materials and methods
Cell culture and reagents
MKN45 cells were obtained from the American Type
Culture Collection (Manassas, VA, USA) and cultured in RPMI-1640
(Invitrogen™ Life Technologies, Carlsbad, CA, USA) containing 10%
(v/v) bovine serum albumin (BSA; Invitrogen Life Technologies)
supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin
(Invitrogen Life Technologies). Cells were cultured in a humidified
incubator containing 5% CO2 at 37°C. The p38 MAPK
inhibitor SB203580, the COX-2 inhibitor
N-[2-(cyclohexyloxy)-4-nitrophenyl] methanesulfonamide (NS-398),
the EP2 inhibitor AH6089 and the EP4 inhibitor AH23848 were all
obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA).
For the inhibition treatment, confluent cells were treated with 20
μM SB203580, 50 μM NS-398, 50 μM AH6089 or 50 μM AH23848 for the
indicated times.
H. pylori culture
The cagA- and vacA-positive H. pylori strain
(NCTC11637) was acquired and cultured as previously described
(15). In brief, H. pylori
were cultured on Columbia Agar plates (Oxoid, Thermo Fisher
Scientific, Basingstoke, UK) containing 10% sheep blood and
incubated at 37°C for 48–72 h, with 5% O2, 10%
CO2 and 85% N2. Prior to use, H.
pylori were identified using Gram staining, colony morphology
and positive oxidase, catalase and urease reactions. To prepare
H. pylori for infection, the cells were suspended in
phosphate-buffered saline (PBS) and cell density was determined
using spectrophotometry (Eppendorf BioSpectrometer; Eppendorf,
Hamburg, Germany). Confluent MKN45 cells were then incubated with
H. pylori at a quantity of 100 bacteria per cell for the
indicated times.
RNA isolation and quantitative polymerase
chain reaction (qPCR)
Total RNA was isolated from MKN45 cells using
RNAisol Reagent kit (Takara Bio, Inc., Shiga, Japan) in accordance
with the manufacturer’s instructions. RNA quality was verified
using spectrophotometry at an absorbance ratio of A260/280. Total
RNA (1 μg) was used for reverse transcription into cDNA using
Prime-Script™ RT-PCR kit (Takara Bio, Inc.) under the following
conditions: 37°C for 15 min and 85°C for 5 sec. A total of 1 μl
cDNA was then used for qPCR amplification using the ABI 7300
Real-Time PCR system (Applied Biosystems, Foster City, CA, USA)
under the following conditions: 95°C for 10 sec, 95°C for 5 sec,
and 60°C for 31 sec, run for 40 cycles. The forward and reverse
primers for VEGF, COX-2 and GAPDH were used at a final
concentration of 200 nM and the sequences were as follows: VEGF,
5′-GGCCTCCGAAACCATGAACT-3′ (forward) and 5′-CACTTGGCATGGTGGAGGTA-3′
(reverse); COX-2, 5′-AATGAGTACCGCAAACGCTTCT-3′ (forward) and
5′-TTCTGCAGCCATTTCCTTCTC-3′ (reverse); GAPDH,
5′-CCACTCCTCCACCTTTGAC-3′ (forward) and 5′-ACCCTGTTGCTGTAGCCA-3′
(reverse). The TaqMan® probes (Invitrogen Life
Technologies) used were as follows: 5′-TGTCTTGGGTGCATTGGAGC-3′ for
VEGF, 5′-CCTGAAGCCGTACACATCATTTG-3′ for COX-2 and
5′-TTGCCCTCAACGACCACTTTGTC-3′ for GAPDH.
Lenti-virus RNA interference (RNAi)
plasmid construction and virus infection
The plasmids of lenti-virus RNAi system were
obtained from Shanghai Genechem Co., Ltd. (Shanghai, China). Four
small interfering RNAs (siRNAs) against human COX-2 mRNA [National
Center for Biotechnology Information (NCBI) GenBank, NM-000963.2]
were designed using the siRNA Target Finder from GenScript
(Piscataway, NJ, USA). The target sequences used were as follows:
Clone 1, 5′-GCT GAATTTAACACCCTCTAT-3′ (1230–1250 bp); clone 2,
5′-CCATTCTCCTTGAAAGGACTT-3′ (1677–1697 bp); clone 3,
5′-GCAGATGAAATACCAGTCTTT-3′ (1463–1483 bp); and clone 4,
5′-CATTCCCTTCCTTCGAAAT-3′ (407–425 bp). The clones were then
inserted into a green fluorescent protein (GFP)-expressing
pFU-GW-RNAi vector in accordance with the manufacturer’s
instructions. The pFU-GW-RNAi vector was co-transfected with
pHelper 1.0 and pHelper 2.0 vectors into 293T cells using
Lipofectamine® 2000 Transfection Reagent (Invitrogen
Life Technologies). The virus was subsequently analyzed and
amplified in 293T cells. The appropriate amount of virus was then
used to infect MKN45 cells for 72 h in order to suppress the
endogenous COX-2 expression.
Western blot analysis
Cells were lysed using lysis buffer solution (50 mM
Tris-HCl, pH 7.5; 150 mM NaCl) containing 1% nonyl
phenoxypolyethoxylethanol-40, 0.5% sodium deoxycholate, 0.1% SDS, 1
mM phenylmethanesulfonylfluoride, 10 nM microcystin, 1 μg/ml
aprotinin and 1 μg/ml leupeptin (all constituents of the lysis
buffer were purchased from Sangon Biotech Shanghai Co., Ltd.,
Shanghai, China). Following centrifugation at 14,000 × g for 20
min, the protein in the supernatant was quantified using the BCA
Protein Assay Reagent (Merck Millipore, Billerica, MA, USA) and
equal amounts of protein were separated by 10% SDS-PAGE (Beyotime
Institute of Biotechnology, Shanghai, China), prior to being
transferred onto a polyvinylidene fluoride membrane (Bio-Rad,
Hercules, CA, USA). Following blocking with 5% fat-free milk in
Tris-buffered saline in 0.05% Tween 20 (TBST; Sangon Biotech
Shanghai Co., Ltd.) for 1 h at room temperature, the membranes were
then separately incubated overnight at 4°C with the following
antibodies: Rabbit anti-human COX-2 monoclonal antibody and rabbit
anti-human β-actin monoclonal antibody (1:1,000; Cell Signaling
Technology, Inc., Beverly, MA, USA), rabbit anti-human EP-2
polyclonal antibody and rabbit anti-human EP-4 polyclonal antibody
(1:1,000; Abcam, Cambridge, UK). Membranes were rinsed three times
with TBST (5 min each time) and then incubated with horseradish
peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch,
West Grove, PA, USA) for 1 h at room temperature prior to
visualization using the Pierce enhanced chemiluminescence kit
(Pierce Biotechnology, Inc., Rockford, IL, USA). Results were
analyzed using ImageJ software (National Institutes of Health,
Bethesda, MD, USA).
ELISA
The cell culture medium was centrifuged at 3,000 × g
for 5 min, and the supernatant was then used for further analysis.
ELISA was performed in accordance with the manufacturer’s
instructions (Invitrogen™ Life Technologies). Briefly, the
microtiter plates were incubated with 100-μl samples at 37°C for
120 min. The plates were washed five times with 10 mM PBS, prior to
incubation with 100 μl VEGF and PGE2 primary antibodies
labeled with biotin at 37°C for 60 min. The plates were then rinsed
five times with 10 mM PBS and 100 μl avidin-biotin-peroxidase
complex was added, prior to the plates being cultured at 37°C for
30 min. Following extensive rinsing, the plates were then filled
with 100 μl TMB Microwell substrate and incubated in darkness at
37°C for 15 min. The reaction was terminated using 100 μl TMB stop
solution and the optical density (OD) values were analyzed within
30 min using a microplate reader (Multiskan Spectrum; Thermo Fisher
Scientific, Waltham, MA, USA) at 450 nm. The OD values were
subsequently converted into concentrations deduced from a
calibration curve.
Statistical analysis
Statistical analysis was performed using the SPSS
19.0 software package (SPSS, Inc., Chicago, IL, USA). Statistical
significance was determined using a one-way analysis of variance
(ANOVA) followed by a Newman-Keuls test. All results are presented
as the mean ± standard error (SE) for three independent
experiments. P<0.05 was considered to indicate a statistically
significant difference.
Results
H. pylori infection enhances expression
levels of VEGF in MKN45 cells
To determine whether H. pylori induced VEGF
expression, confluent MKN45 cells were co-cultured with H.
pylori for 0, 6, 12 and 24 h. Cells were then harvested to
examine the relative expression levels of VEGF mRNA compared with
GAPDH mRNA expression levels using qPCR. It was found that VEGF
mRNA levels in cells treated with H. pylori for 6 h
(P<0.05), 12 h (P<0.01) and 24 h (P<0.01) were
significantly elevated compared with cells not exposed to H.
pylori (Fig. 1A). The VEGF
mRNA expression was highest in cells treated with H. pylori
for 12 h. VEGF mRNA expression in cells treated with H.
pylori for 12 h was significantly higher than that in cells
treated for 6 h (P<0.01); however, expression appeared to
decline after 24 h (Fig. 1A). The
VEGF protein expression levels were analyzed using ELISA. MKN45
cells were incubated with H. pylori for 0, 12, 24, 36 and 48
h, and the supernatant was harvested and analyzed. It was found
that VEGF protein levels increased with the length of incubation,
and were significantly elevated in cells cultured with H.
pylori for 36 h (P<0.05) and 48 h (P<0.01) (Fig. 1B).
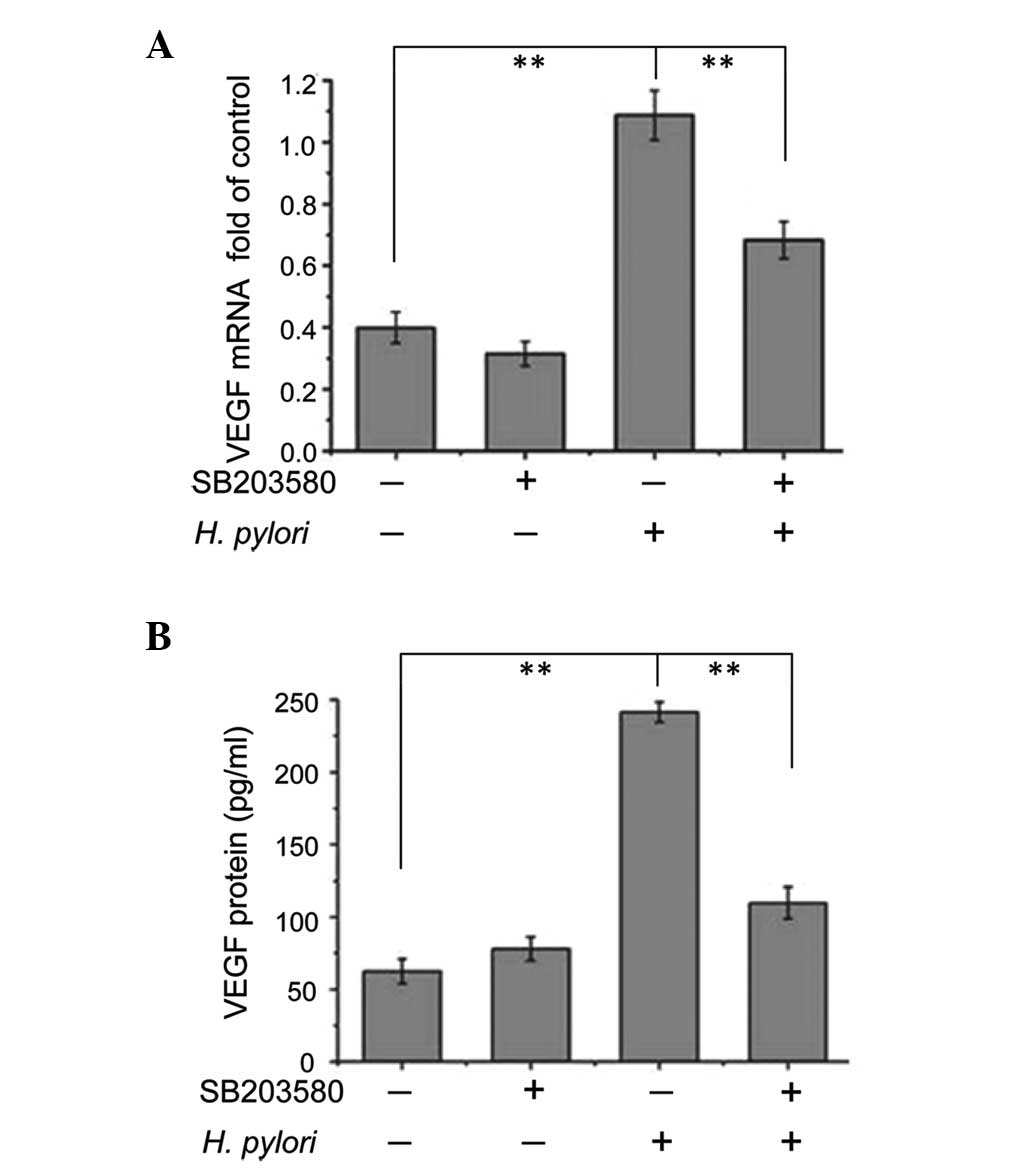
Inhibition of p38 MAPK attenuates the
effects of H. pylori on VEGF expression
To examine whether p38 MAPK modulated VEGF
expression, confluent MKN45 cells were pretreated with p38 MAPK
inhibitor SB203580 for 2 h prior to incubation with or without
H. pylori for 12 or 48 h. Cells were then harvested and the
levels of VEGF mRNA expression relative to GAPDH mRNA expression
were analyzed using qPCR (cells treated with H. pylori for
12 h), while VEGF protein expression levels were analyzed using
ELISA (cells treated with H. pylori for 48 h). In cells that
were not exposed to H. pylori, treatment with SB203580 did
not affect the VEGF mRNA or protein expression levels (Fig. 2A and B). Without SB203580
pretreatment, VEGF mRNA and protein levels in H.
pylori-treated cells were significantly increased compared with
cells not treated with H. pylori (P<0.01) (Fig. 2A and B). However, when pretreated
with SB203580, VEGF mRNA and protein expression levels in H.
pylori-treated cells significantly decreased compared with
H. pylori-treated cells without incubation with SB203580
(P<0.01) (Fig. 2A and B).
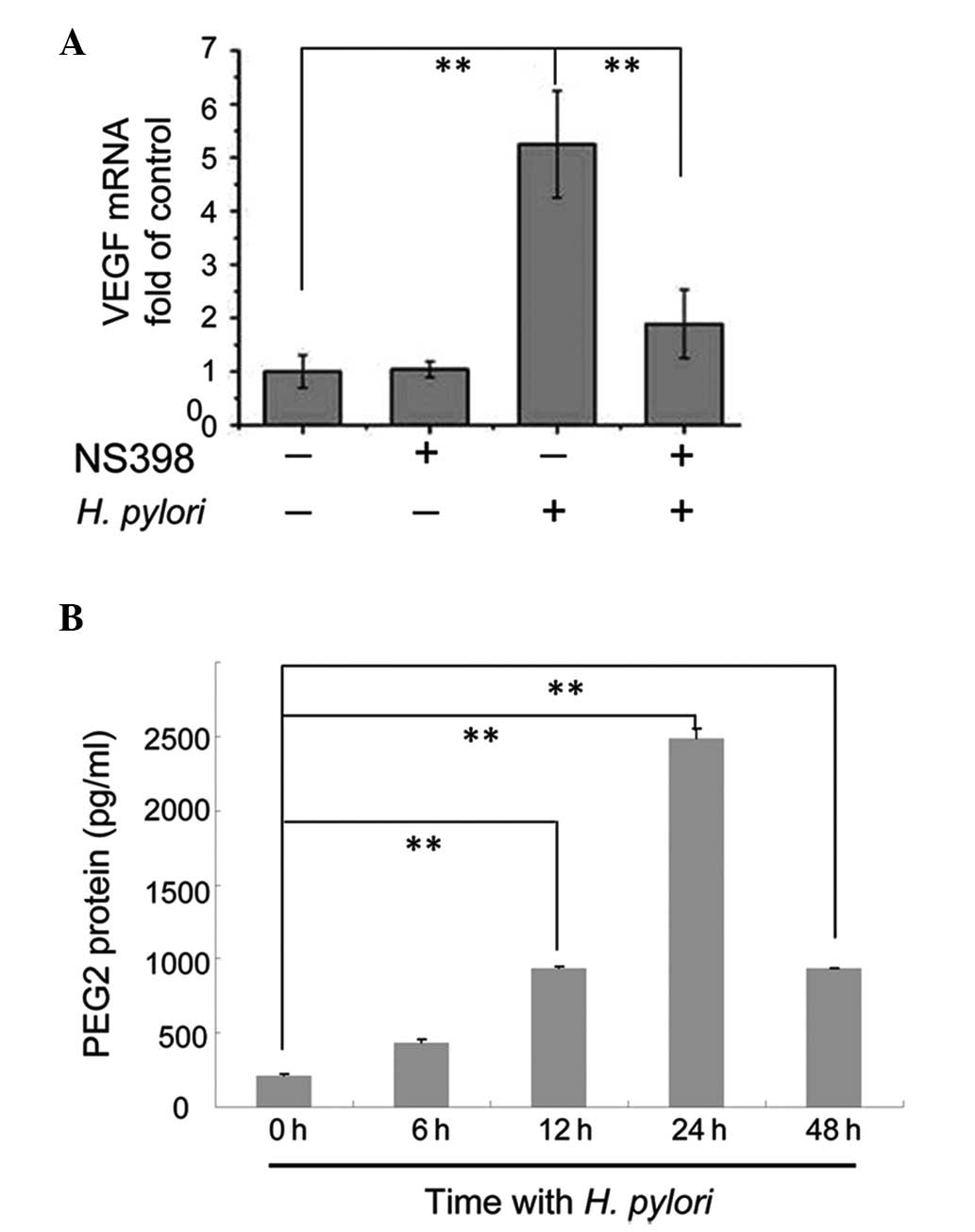
Blocking COX-2 with the inhibitor NS-398
attenuates the effects of H. pylori on VEGF expression
It was demonstrated in our previous study that p38
MAPK activity was essential for increased expression of COX-2 in
MKN45 cells following H. pylori infection (15); therefore, in the present study, it
was investigated whether COX-2 was involved in the p38
MAPK-mediated upregulation of VEGF expression. Confluent MKN45
cells were pretreated with the COX-2 inhibitor NS-398 for 2 h prior
to incubation with or without H. pylori for 12 h. The cells
were then harvested to measure the expression levels of VEGF mRNA
compared with GAPDH mRNA using qPCR. In cells not exposed to H.
pylori, treatment with NS-398 did not affect the VEGF mRNA
expression levels (Fig. 3A).
Consistent with our previous results, VEGF mRNA levels in H.
pylori-treated cells increased significantly (P<0.01)
(Fig. 3A). However, in H.
pylori-infected cells pretreated with NS-398, VEGF mRNA
expression levels were downregulated compared with H.
pylori-infected cells not incubated with NS-398 (P<0.01)
(Fig. 3A). Our previous studies
demonstrated that H. pylori increased COX-2 expression in
MKN45 cells (16); therefore, it
was hypothesized that the downstream products of COX-2 may have
been elevated. The present study analyzed the protein expression
levels of PGE2, one such downstream product of COX-2, in
MKN45 cells incubated with H. pylori for 0, 6, 12, 24 and 48
h using ELISA. PGE2 protein levels in cells treated with
H. pylori for 12, 24 and 48 h were significantly higher
compared with cells not exposed to H. pylori (P <0.01),
with the highest expression observed in in cells treated with H.
pylori for 24 h (Fig. 3B).
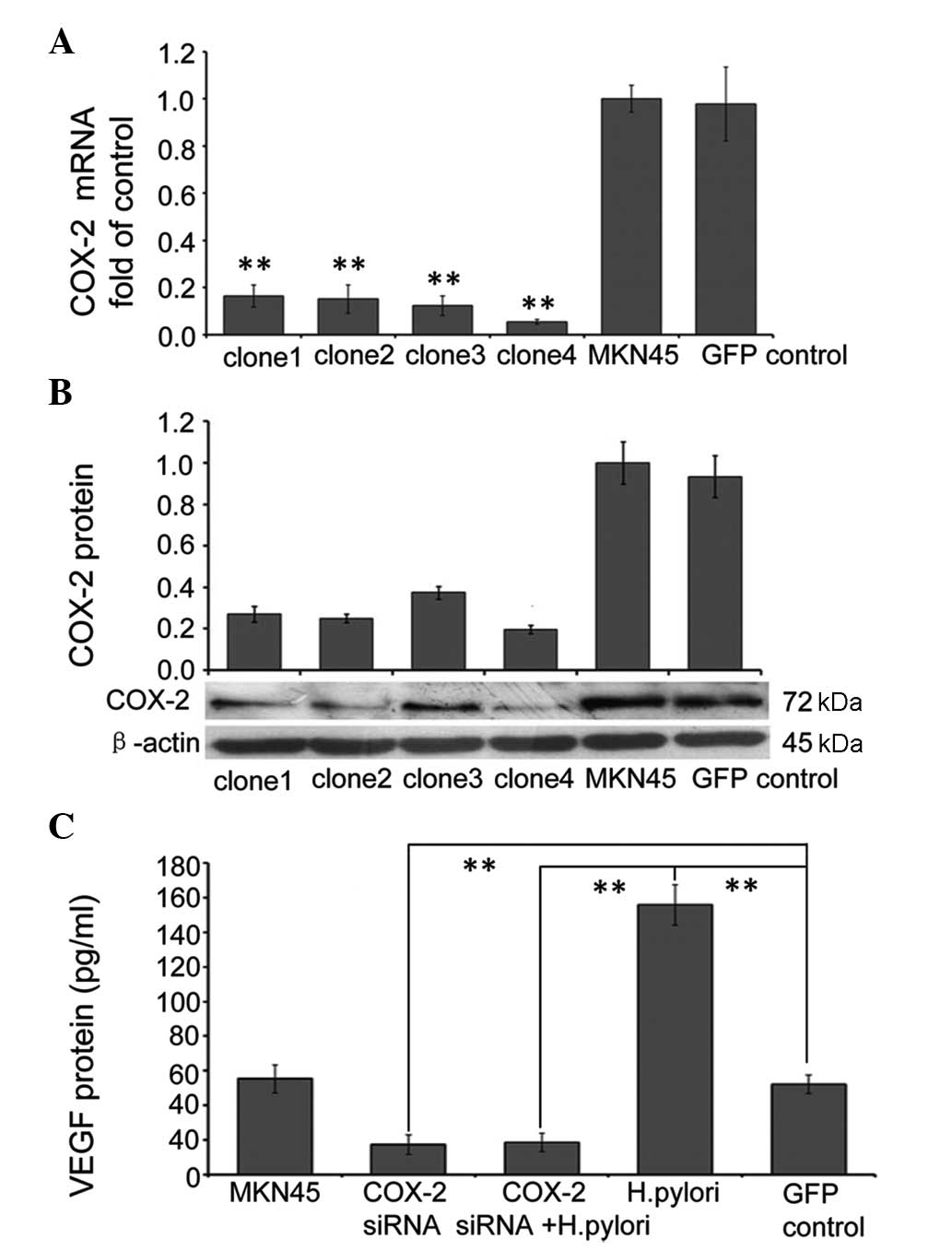
RNAi-mediated suppression of COX-2
attenuates the effects of H. pylori on VEGF expression
In order to specifically suppress endogenous COX-2
expression, four lentiviral based RNAi clones targeted to different
parts of the COX-2 gene were designed, and the inhibitory effect
was analyzed by infecting MKN45 cells for 72 h with each clone
separately. The cells were then harvested and the expression levels
of COX-2 mRNA relative to GAPDH mRNA expression were analyzed using
qPCR, while COX-2 protein expression was measured using western
blot analysis. All four of the clones of lenti-viral RNAi targeted
to COX-2 efficiently suppressed mRNA and protein expression levels
of COX-2; however, clone 4 suppressed COX-2 mRNA and protein
expression levels by ~95 and ~81%, respectively and, therefore, was
used in the following experiments (Fig. 4A and B). To determine the effects
of COX-2 RNAi on VEGF expression, confluent MKN45 cells were
infected with control RNAi (GFP control), or COX-2 RNAi for 72 h,
and then cells were incubated with or without H. pylori for
48 h. The supernatant was harvested to measure the protein
expression of VEGF using ELISA. The results showed that background
expression of VEGF in MKN45 cells was significantly reduced in
cells infected with COX-2 siRNA (P<0.01), and the same effect
was observed in cells also treated with H. pylori. This
indicates that RNAi-mediated suppression of COX-2 inhibits the
upregulation of VEGF expression in MKN45 cells following H.
pylori treatment (Fig.
4C).
EP2/EP4-mediated regulation of VEGF
expression upon H. pylori infection
Since it was found that COX-2 promotes VEGF
expression in H. pylori-treated MKN45 cells and that the
downstream product of COX-2, PGE2, was upregulated,
further studies were performed to determine whether the
PGE2 receptors EP2/EP4 were also associated with
COX-2-mediated upregulation of VEGF. Confluent MKN45 cells were
incubated with H. pylori for 0, 2, 6, 12, 24 and 48 h and
then harvested. Western blot analysis was performed to determine
the protein expression levels of EP2 and EP4, using β-actin as the
internal control. As shown in Fig.
5A, no significant difference was observed in the protein
expression levels of EP2 or EP4 during the continual induction of
H. pylori (Fig. 5A). To
examine whether EP2/EP4 expression was associated with VEGF
expression in MKN45 cells, confluent cells were pretreated with
specific inhibitors against EP2 and EP4, AH6089 and AH23848,
respectively, for 2 h prior to incubation with or without H.
pylori for 12 or 48 h. The cells were then harvested and the
expression levels of VEGF mRNA relative to GAPDH mRNA were analyzed
using qPCR (cells treated with H. pylori for 12 h), while
expression levels of VEGF protein were analyzed using ELISA (cells
treated with H. pylori for 48 h). In cells not exposed to
H. pylori, treatment with AH6089 or AH23848 did not affect
VEGF mRNA or protein expression levels (P>0.05) (Fig. 5B and C). In cells not treated with
AH0689 or AH2348, VEGF mRNA and protein levels in H.
pylori-infected cells increased significantly compared with
cells not infected with H. pylori (P<0.01) (Fig. 5B and C). However, when cells were
pretreated with AH6089, VEGF mRNA (P<0.05) and protein
(P<0.01) expression levels in H. pylori-infected cells
significantly decreased compared with H. pylori-infected
cells without incubation of AH6089 (Fig. 5B and C). A similar result was
observed in cells pretreated with the inhibitor AH23848: VEGF mRNA
and protein expression levels in H. pylori-infected cells
significantly decreased compared with H. pylori-infected
cells without incubation of AH23848 (P<0.01) (Fig. 5B and C).
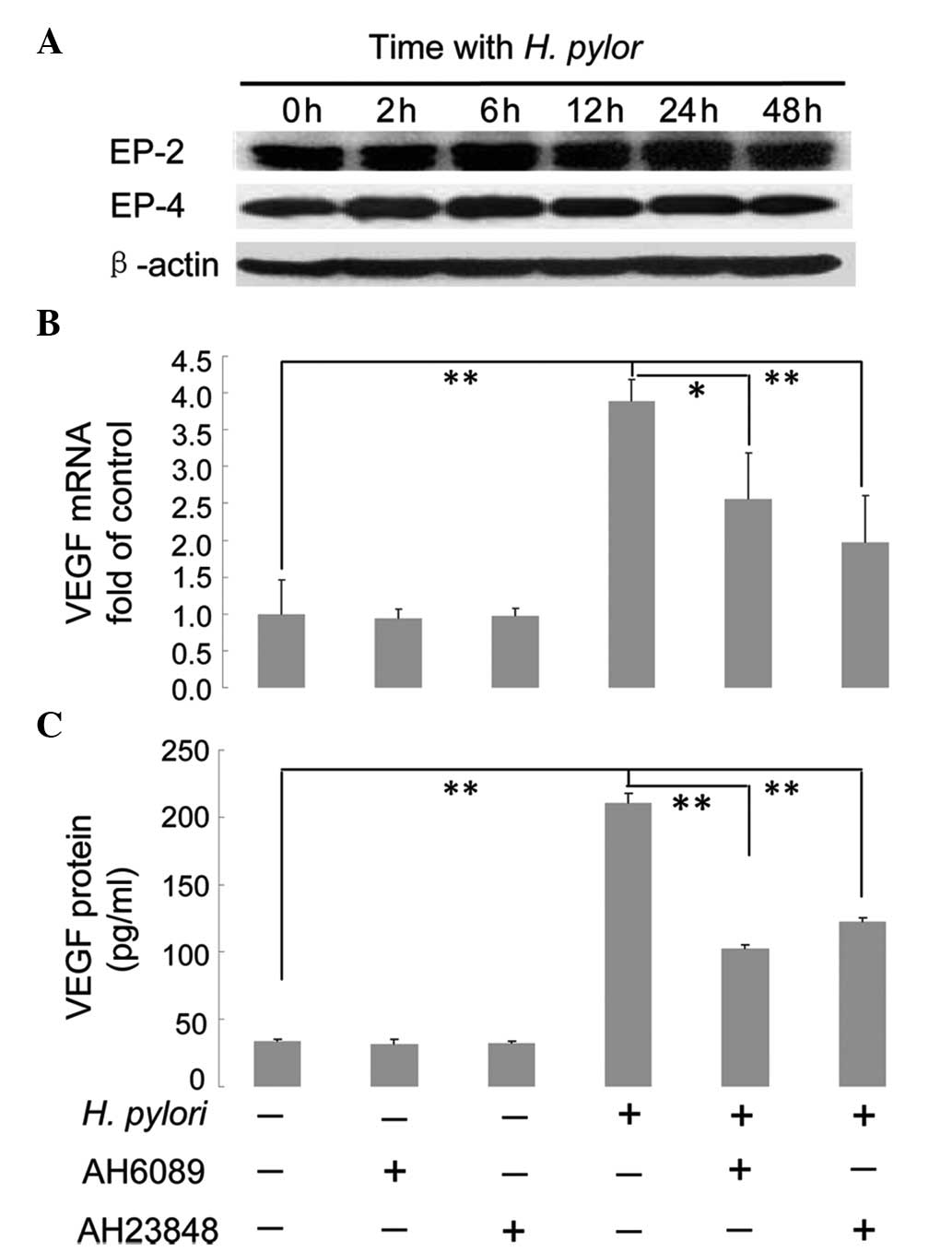 | Figure 5Effect of EP2/EP4 inhibitors on H.
pylori-induced VEGF expression in MKN45 cells. (A) Protein
expression of EP2 and EP4 does not change in H.
pylori-treated MKN45 cells. Confluent MKN45 cells were
co-cultured with H. pylori for 0, 2, 6, 12, 24 and 48 h,
prior to western blot analysis to measure the expression of EP2 and
EP4. β-actin was used as an internal control. (B) The inhibitors
AH6089 and AH23848 attenuate the effects of H. pylori on
VEGF mRNA expression. Confluent cells were pretreated for 2 h with
50 μM specific inhibitors against EP2 or EP4, AH6089 and AH23848,
respectively. Cells were then incubated with or without H.
pylori for 12 h, and harvested to measure the relative
expression levels of VEGF mRNA compared with GAPDH using
quantitative polymerase chain reaction. (C) The inhibitors AH6089
and AH23848 attenuate the effects of H. pylori on VEGF
protein expression. Confluent cells were pretreated for 2 h with 50
μM specific inhibitors against EP2 or EP4, AH6089 and AH23848,
respectively. Cells were then incubated with or without H.
pylori for 48 h, and harvested to measure the expression levels
of VEGF protein using ELISA. *P<0.05 and
**P<0.01. EP, prostaglandin E2 receptor; VEGF,
vascular endothelial growth factor. |
Discussion
VEGF, an oncogenic marker in cancer diagnosis, has
potent angiogenic activity on endothelial cells and promotes tumor
growth (32). Enhanced expression
of VEGF is often observed in malignant tumors and is frequently
used as a therapeutic target (32). A previous study demonstrated that
H. pylori infection promoted gastric cancer cell invasion
via upregulation of VEGF expression (33). However, the mechanism by which
H. pylori induces VEGF expression in gastric cancer has yet
to be elucidated. The p38 MAPK pathway has been found to be
involved in tumor growth and metastasis through regulation of the
production of VEGF in cancer. p38 MAPK activity also has a key role
in increasing H. pylori-induced COX-2 expression in gastric
cancer cells (16,27,31).
Therefore, in the present study, the role of p38 MAPK in the
regulation of VEGF expression in gastric cancer cells exposed to
H. pylori was investigated. It was found that mRNA and
protein expression levels of VEGF were significantly increased
following H. pylori infection; however, the p38
MAPK-specific inhibitor, SB203580, significantly attenuated this
effect, indicating that p38 MAPK was involved in promoting VEGF
expression in H. pylori-infected MKN45 cells (Figs. 1 and 2).
COX-2 activity is associated with H. pylori
infection in gastric cells, and may promote the production of
PGE2 (16,26). In a previous study, we demonstrated
that H. pylori infection upregulates the expression of COX-2
via the p38 MAPK/activating transcription factor 2 pathway
(16). In this present study, the
expression of PGE2 in H. pylori-infected cells
was analyzed and it was found that PGE2 levels were
significantly increased, suggesting that COX-2 activity was
upregulated following H. pylori infection (Fig. 3B). To investigate whether the p38
MAPK-associated upregulation of VEGF expression was mediated by
COX-2, the COX-2 specific inhibitor, NS-398, was used in cells
exposed to H. pylori. The results show that VEGF expression
significantly decreased in NS-398-treated cells (Fig. 3A). Endogenous COX-2 expression was
also downregulated using RNAi and it was found that background
expression levels of VEGF in MKN45 cells were significantly
reduced, and the same effect was observed in H.
pylori-treated cells, indicating that RNAi-mediated inhibition
of COX-2 suppressed the upregulation of VEGF expression in MKN45
cells following H. pylori infection (Fig. 4C).
The results of the present study suggest that COX-2
is involved in the regulation of VEGF expression, downstream of p38
MAPK. EP2 and EP4 are PGE2 receptors, and have been
shown to have an important role in modulating VEGF production in
prostate cancer cells (20,26).
In the present study, the roles of EP2/EP4 in VEGF production in
gastric cancer cells following H. pylori infection were
investigated. The results demonstrated that inhibition of EP2 and
EP4 with the specific inhibitors AH6089 and AH23848 significantly
decreased H. pylori-induced VEGF levels in cells, indicating
that EP2/EP4 mediate the upregulation of VEGF expression in H.
pylori-infected gastric cancer cells (Fig. 5B and C). The protein expression of
EP2/EP4 was then analyzed, and it was found that H. pylori
infection did not alter the EP2/EP4 protein levels. This suggests
that the EP2/EP4-associated upregulation of VEGF is not mediated by
EP2/EP4 protein levels, but by increased levels of PGE2
product, induced by enhanced COX-2 activity (Fig. 5A).
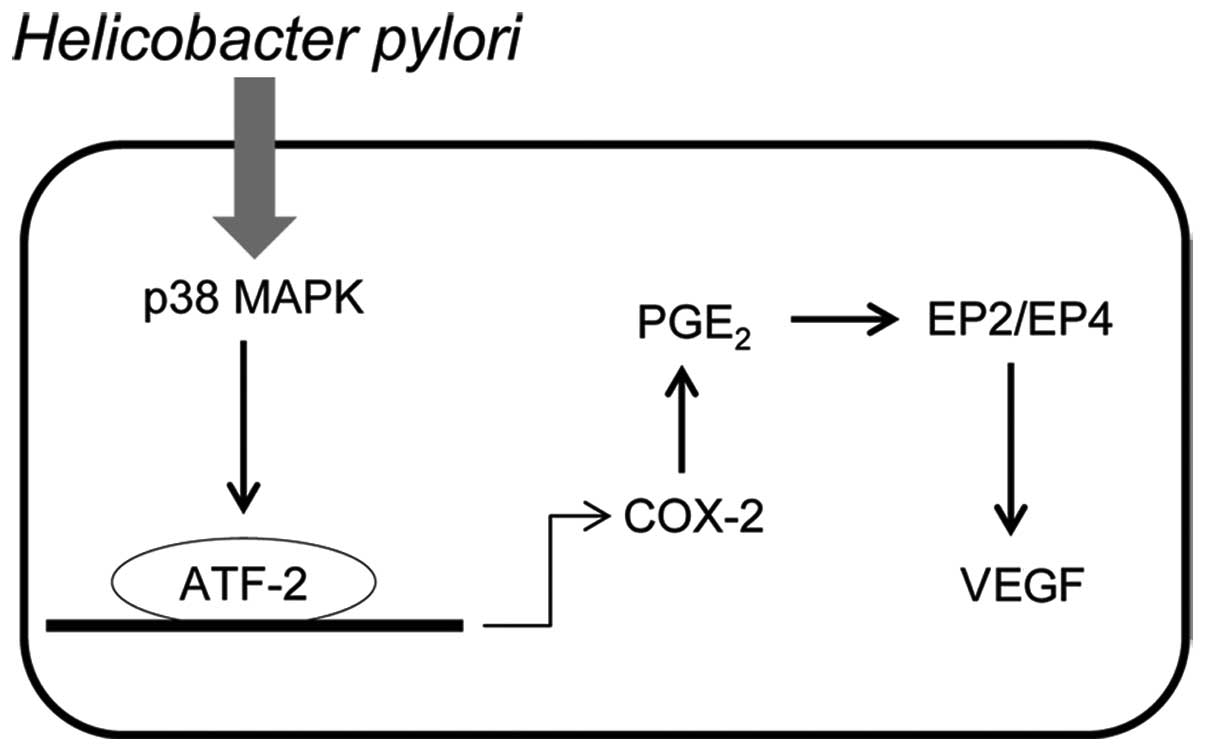
In combination, these results suggest a novel
pathway of p38 MAPK-COX-2-PGE2-EP2/EP4 for the
regulation of VEGF expression in H. pylori-infected gastric
cells (Fig. 6). Following H.
pylori infection, p38 MAPK is activated and COX-2 expression
levels are upregulated, the level of PGE2 is therefore
increased. PGE2 then binds to EP2/EP4 to promote VEGF
expression. In a previous study we demonstrated that Jianpi Jiedu,
a formulation used in traditional Chinese medicine, was
demonstrated to downregulate COX-2 expression via inhibition of the
H. pylori-induced p38 MAPK pathway (34). Therefore, further studies may be
performed to examine whether the Jianpi Jiedu recipe regulates VEGF
expression via modulation of this pathway, as shown in Fig. 6.
In conclusion, the present study elucidated a p38
MAPK-mediated signaling pathway that regulates VEGF expression in
H. pylori-infected gastric cancer cells. This contributes to
the investigation into the pathogenesis of H. pylori-induced
gastric cancer. Furthermore, this study may provide novel
therapeutic targets for H. pylori-induced gastric
cancer.
Acknowledgements
This study was funded and supported by the National
Natural Science Foundation of China (nos. 81072955, 81273958 and
81202663), the Program of Shanghai Municipal Education Commission
(12ZZ118), the Science and Technology Commission of Shanghai
Municipality (12ZR1449300, 1214090250), the Shanghai Municipal
Health Bureau (2010019, XBR2011061, 2010044) and the Major Program
of Technology Innovation, Putuo District, Shanghai
(2009PTKW001).
References
|
1
|
Peek RM Jr and Crabtree JE: Helicobacter
infection and gastric neoplasia. J Pathol. 208:233–248. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ernst PB, Peura DA and Crowe SE: The
translation of Helicobacter pylori basic research to patient
care. Gastroenterology. 130:188–206; quiz 212–213. 2006.
|
|
3
|
No authors listed. IARC working group on
the evaluation of carcinogenic risks to humans: some industrial
chemicals; Lyon. 15–22 February 1994; IARC Monogr Eval Carcinog
Risks Hum. 60. pp. 1–560. 1994
|
|
4
|
Wroblewski LE, Peek RM Jr and Wilson KT:
Helicobacter pylori and gastric cancer: factors that
modulate disease risk. Clin Microbiol Rev. 23:713–739. 2010.
View Article : Google Scholar
|
|
5
|
Conteduca V, Sansonno D, Lauletta G, Russi
S, Ingravallo G and Dammacco F: H. pylori infection and
gastric cancer: state of the art (review). Int J Oncol. 42:5–18.
2013.
|
|
6
|
de Vries EF: Imaging of cyclooxygenase-2
(COX-2) expression: potential use in diagnosis and drug evaluation.
Curr Pharm Des. 12:3847–3856. 2006.PubMed/NCBI
|
|
7
|
Greenhough A, Smartt HJ, Moore AE, et al:
The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and
adaptation to the tumour microenvironment. Carcinogenesis.
30:377–386. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zidar N, Dolenc-Strazar Z, Jeruc J, et al:
Expression of cyclooxygenase-1 and cyclooxygenase-2 in the normal
human heart and in myocardial infarction. Cardiovasc Pathol.
16:300–304. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zidar N, Odar K, Glavac D, Jerse M, Zupanc
T and Stajer D: Cyclooxygenase in normal human tissues - is COX-1
really a constitutive isoform, and COX-2 an inducible isoform? J
Cell Mol Med. 13:3753–3763. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Buskens CJ, Van Rees BP, Sivula A, et al:
Prognostic significance of elevated cyclooxygenase 2 expression in
patients with adenocarcinoma of the esophagus. Gastroenterology.
122:1800–1807. 2002. View Article : Google Scholar
|
|
11
|
Erkinheimo TL, Lassus H, Sivula A, et al:
Cytoplasmic HuR expression correlates with poor outcome and with
cyclooxygenase 2 expression in serous ovarian carcinoma. Cancer
Res. 63:7591–7594. 2003.PubMed/NCBI
|
|
12
|
Juuti A, Louhimo J, Nordling S, Ristimäki
A and Haglund C: Cyclooxygenase-2 expression correlates with poor
prognosis in pancreatic cancer. J Clin Pathol. 59:382–386. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Menter DG, Schilsky RL and DuBois RN:
Cyclooxygenase-2 and cancer treatment: understanding the risk
should be worth the reward. Clin Cancer Res. 16:1384–1390. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ristimäki A, Sivula A, Lundin J, et al:
Prognostic significance of elevated cyclooxygenase-2 expression in
breast cancer. Cancer Res. 62:632–635. 2002.PubMed/NCBI
|
|
15
|
Thiel A, Mrena J and Ristimäki A:
Cyclooxygenase-2 and gastric cancer. Cancer Metastasis Rev.
30:387–395. 2011. View Article : Google Scholar
|
|
16
|
Li Q, Liu N, Shen B, et al:
Helicobacter pylori enhances cyclooxygenase 2 expression via
p38MAPK/ATF-2 signaling pathway in MKN45 cells. Cancer Lett.
278:97–103. 2009. View Article : Google Scholar
|
|
17
|
Fujino H, Xu W and Regan JW: Prostaglandin
E2 induced functional expression of early growth response factor-1
by EP4, but not EP2, prostanoid receptors via the
phosphatidylinositol 3-kinase and extracellular signal-regulated
kinases. J Biol Chem. 278:12151–12156. 2003. View Article : Google Scholar
|
|
18
|
Golijanin D, Tan JY, Kazior A, et al:
Cyclooxygenase-2 and microsomal prostaglandin E synthase-1 are
overexpressed in squamous cell carcinoma of the penis. Clin Cancer
Res. 10:1024–1031. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sheng H, Shao J, Washington MK and DuBois
RN: Prostaglandin E2 increases growth and motility of colorectal
carcinoma cells. J Biol Chem. 276:18075–18081. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ding YB, Shi RH, Tong JD, et al: PGE2
up-regulates vascular endothelial growth factor expression in MKN28
gastric cancer cells via epidermal growth factor receptor signaling
system. Exp Oncol. 27:108–113. 2005.
|
|
21
|
Pai R, Szabo IL, Soreghan BA, Atay S,
Kawanaka H and Tarnawski AS: PGE(2) stimulates VEGF expression in
endothelial cells via ERK2/JNK1 signaling pathways. Biochem Biophys
Res Commun. 286:923–928. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Araki H, Ukawa H, Sugawa Y, Yagi K, Suzuki
K and Takeuchi K: The roles of prostaglandin E receptor subtypes in
the cytoprotective action of prostaglandin E2 in rat stomach.
Aliment Pharmacol Ther. 14(Suppl 1): 116–124. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Breyer RM, Emeson RB, Tarng JL, et al:
Alternative splicing generates multiple isoforms of a rabbit
prostaglandin E2 receptor. J Biol Chem. 269:6163–6169.
1994.PubMed/NCBI
|
|
24
|
Pang L and Knox AJ: Bradykinin stimulates
IL-8 production in cultured human airway smooth muscle cells: role
of cyclooxygenase products. J Immunol. 161:2509–2515.
1998.PubMed/NCBI
|
|
25
|
Takeuchi K, Araki H, Umeda M, Komoike Y
and Suzuki K: Adaptive gastric cytoprotection is mediated by
prostaglandin EP1 receptors: a study using rats and knockout mice.
J Pharmacol Exp Ther. 297:1160–1165. 2001.PubMed/NCBI
|
|
26
|
Jain S, Chakraborty G, Raja R, Kale S and
Kundu GC: Prostaglandin E2 regulates tumor angiogenesis in prostate
cancer. Cancer Res. 68:7750–7759. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cuenda A and Rousseau S: p38 MAP-kinases
pathway regulation, function and role in human diseases. Biochim
Biophys Acta. 1773:1358–1375. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Choi IJ, Kim JS, Kim JM, Jung HC and Song
IS: Effect of inhibition of extracellular signal-regulated kinase 1
and 2 pathway on apoptosis and bcl-2 expression in Helicobacter
pylori-infected AGS cells. Infection Immun. 71:830–837. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim H, Seo JH and Kim KH: The effect of
p38 mitogen-activated protein kinase on mucin gene expression and
apoptosis in Helicobacter pylori-infected gastric epithelial
cells. Ann NY Acad Sci. 1010:90–94. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Seo JH, Lim JW, Kim H and Kim KH:
Helicobacter pylori in a Korean isolate activates
mitogen-activated protein kinases, AP-1, and NF-kappaB and induces
chemokine expression in gastric epithelial AGS cells. Lab Invest.
84:49–62. 2004. View Article : Google Scholar
|
|
31
|
Kim JH, Studer RK, Vo NV, Sowa GA and Kang
JD: p38 MAPK inhibition selectively mitigates inflammatory
mediators and VEGF production in AF cells co-cultured with
activated macrophage-like THP-1 cells. Osteoarthritis Cartilage.
17:1662–1669. 2009. View Article : Google Scholar
|
|
32
|
Goel HL and Mercurio AM: VEGF targets the
tumour cell. Nat Rev Cancer. 13:871–882. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu CY, Wang CJ, Tseng CC, et al:
Helicobacter pylori promote gastric cancer cells invasion
through a NF-kappaB and COX-2-mediated pathway. World J
Gastroenterol. 11:3197–3203. 2005. View Article : Google Scholar
|
|
34
|
Liu NN, Wang Y and Wu Q: Jianpi jiedu
recipe inhibited Helicobacter pylori-induced the expression
of cyclooxygenase-2 via p38MAPK/ATF-2 signal transduction pathway
in human gastric cancer cells. Zhongguo Zhong Xi Yi Jie He Za Zhi.
31:926–931. 2011.(In Chinese).
|