Introduction
Cancer is one of the most common causes of fatality
and up to 90% of cancer-associated mortality results from
metastasis; however, the underlying mechanisms of metastasis remain
poorly understood (1). The initial
steps of local invasion include the activation of signaling
pathways that control cytoskeletal dynamics in tumor cells, and the
turnover of cell matrix and cell-cell junctions, followed by tumor
cell migration into the adjacent tissue (2).
Epithelial-to-mesenchymal transition (EMT) is a key
step during embryonic morphogenesis, but is also involved in the
progression of primary tumors toward metastasis. The EMT phenotype
is distinguished by the loss of cell-to-cell adhesion together with
the disassembly of tight, adherens and gap junctions, and
phenotypic changes in the cells from an epithelial to a motile,
fibroblast-like morphology (3).
Invasion and metastasis, mediated by EMT, are largely determined by
the loss of E-cadherin functionality, as E-cadherin is critical for
the maintenance of adherent junctions between neighboring cells and
it preserves physical integrity in epithelial cells (4). In addition, during tumorigenesis,
certain epithelial cells develop the ability of self-renewal, a
trait associated with cancer stem cells, and undergo EMT to
facilitate cancer metastasis and recurrence (5).
Histone acetylation and deacetylation are important
in the modulation of chromatin topology and the regulation of gene
transcription. Histone deacetylase inhibitors (HDACIs) are novel
anticancer drugs, which induce histone (hyper-) acetylation and
counteract aberrant gene repression. HDACI treatment may also
result in the inhibition of tumor cell proliferation, in culture
and in vivo, by inducing cell cycle arrest, differentiation
and/or apoptosis. HDACIs have been shown to induce non-histone
protein acetylation, which alters signaling networks relevant for
tumorigenesis (6). Several HDACIs
have been used in phase I and II clinical trials for the treatment
of a number of hematological malignancies and solid tumors
(7).
Previous studies have suggested that HDACIs act as
EMT suppressors in normal epithelial and cancer cells (8,9).
However, HDACIs have also been reported to induce significant
morphological changes, resembling EMT in prostate cancer cells, in
addition to the ability to upregulate the expression of mesenchymal
cell markers, such as Vimentin, Snail, Slug and Twist (10). These findings provoke controversy
as to whether HDACIs are EMT suppressors or inducers, and the
implications of this for cancer therapy. To investigate these
questions, a panel of human cancer cells was treated with TSA and
its effect on cell morphology, molecular expression and migration
ability was evaluated.
Materials and methods
Cell culture and reagents
The BGC-823 human gastric cancer cell line was
purchased from the Institute of Biochemistry and Cell Biology,
Shanghai Institute for Biological Science, Chinese Academy of
Sciences (Shanghai, China). The MCF-7 human breast cancer and
KYSE-510 human esophageal squamous cancer cell lines were obtained
from the American Type Culture Collection (Rockville, MD, USA). The
cells were maintained in RPMI 1640 medium containing penicillin
(100 U/ml), streptomycin (100 mg/ml) and 10% fetal bovine serum at
37°C in a humidified incubator supplemented with 5% CO2
in air. Cell morphology was observed with an Olympus IX71 inverted
microscope (Olympus Corporation, Tokyo, Japan). TSA was purchased
from Beyotime Institute of Biotechnology (Haimen, China) and
dissolved in dimethylsulfoxide (6.62 mm) for storage. The TSA was
diluted to the appropriate concentrations with phosphate-buffered
saline (PBS) prior to use. The primary antibodies used in this
study were a rabbit monoclonal antibody to E-cadherin (Cell
Signaling Technology, Inc., Danvers, MA, USA), a goat polyclonal
antibody to β-catenin (Santa Cruz Biotechnology, Inc., Dalls, TX,
USA) and a mouse monoclonal antibody to β-actin (Abmart, Shanghai,
China). The secondary antibodies were HRP-conjugated anti-rabbit,
mouse or goat secondary antibodies (Cell Signaling Technology,
Inc.).
Immunocytochemistry assay
The cells were seeded on 96-well plates and allowed
to adhere overnight. Following treatment with TSA for 24 h, the
cells were washed with cold PBS and fixed in 4% paraformaldehyde
for 15 min, followed by permeabilization in 0.1% Triton X-100 for 5
min. The cells were then blocked with 5% goat serum for 30 min,
followed by incubation with primary antibody overnight at 4°C.
Following careful washing, the cells were incubated with
horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h
at room temperature, then stained with 3,3′-diaminobenzidine
subsequent to additional thorough washing.
RNA isolation and quantitative polymerase
chain reaction (qPCR)
Total RNA was isolated using TRIzol reagent
(Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s
instructions and quantified by spectrophotometric measurement
(Molecular Devices, Sunnyvale, CA, USA). RNA (2 μg) was
reverse-transcribed to cDNA using the RevertAid first strand cDNA
synthesis kit (Thermo Fisher Scientific, Rockford, IL, USA)
according to the manufacturer’s instructions. qPCR analysis was
performed using SYBR Green-based detection on a StepOnePlus
real-time PCR instrument (Applied Biosystems, Inc., Foster City,
CA, USA). The relative mRNA expression levels of the genes of
interest were normalized to those of GAPDH and calculated using the
2−ΔΔCt method. The following primers were used: Sense:
5′-GAC CGC ACA CAG CAA GGC GAT-3′ and antisense: 5′-GCT GTC CCG CCG
ATT GAG GG-3′ for Vimentin; sense: 5′-TCA GCA GGG CCG GAG ACC TA-3′
and antisense: 5′-TCC ACG GGC CTG TCT CGC TT-3′ for Twist; sense:
5′-CGC CCC CAT ACC AGA ACC TCG-3′ and antisense: 5′-GTC CAG TTG GCA
CTC GCC CC-3′ for E-cadherin; sense: 5′-TGG TGC CCA GGG AGA ACC
CC-3′ and antisense: 5′-TGT CAC CTG GAG GCA GCC CA-3′ for
β-catenin; and sense: 5′-CTG GGC TAC ACT GAG CAC C-3′ and
antisense: 5′-AAG TGG TCG TTG AGG GCA ATG-3′ for GAPDH.
Western blot analysis
To prepare the whole-cell extract, the cells were
washed with cold PBS once and harvested by scraping in
radioimmunoprecipitation assay lysis buffer. The protein content
was determined by the Bradford assay (Beyotime Institute of
Biotechnology, Shanghai, China). The extracted proteins were
separated by SDS polyacrylamide gel electrophoresis and transferred
to polyvinylidene fluoride membranes (Beyotime Institute of
Biotechnolgy, Haimen, China). The membranes were first blocked with
5% (w/v) non-fat dry milk in Tris-buffered saline with Tween-20 and
then probed with the indicated primary antibodies with gentle
agitation at 4°C overnight. Subsequent to washing four times, the
membranes were incubated with HRP-conjugated secondary antibodies 1
h. The signals were detected using an enhanced chemiluminescence
detection kit (Thermo Fisher Scientific).
Wound-healing assay
A total of 1×106 cells were plated on
6-well plates. When the monolayer reached ~70% confluence, a scrape
was inflicted using sterile pipette tips. The wound-healing rate
was evaluated by capturing images of the cells after 24 h with an
Olympus IX71 inverted microscope (Olympus Corporation).
Colony forming assay
The cells were seeded on 96-well plates at 10 cells
per well and allowed to adhere overnight. Fresh medium either with
or without 200 nm TSA was then added to each well. After three
days, the numbers of colonies in each well with a diameter >100
μm were counted using an an Olympus IX71 inverted microscope
(Olympus Corporation).
Statistical analysis
The data are presented as the mean ± standard error
of the mean. Statistical analyses were performed using Student’s
t-test. P<0.05 was considered to indicate a statistically
significant difference.
Results
TSA induces mesenchymal-like
morphological changes in human cancer cells
TSA is known as a potent HDAC inhibitor, which binds
to the zinc-containing catalytic domain of the class I and II
HDACs, including HDACs 1 to 10 (11). To determine whether TSA induces EMT
in cancer cells, BGC-823 human gastric cancer cells were treated
with different concentrations of TSA. Untreated BGC-823 cells
exhibited a cobblestone-like epithelial morphology. As shown in
Fig. 1A, after 24 h treatment with
200 nm TSA, the cells exhibited a spindle-like shape similar to
mesenchymal cell characteristics, but 50 nm TSA did not exert these
effects (Fig. 1A). TSA also
stimulated mesenchymal-like morphological changes in MCF-7 human
breast cancer cells in a concentration-dependent manner (Fig. 1B). Following three days culture
with 200 nM TSA, BGC-823 and MCF-7 cells all reached ~100%
confluence and the mesenchymal cell morphology disappeared,
indicating that 200 nM TSA induced no marked cytotoxicity in
BGC-823 or MCF-7 cells (Fig.
1A).
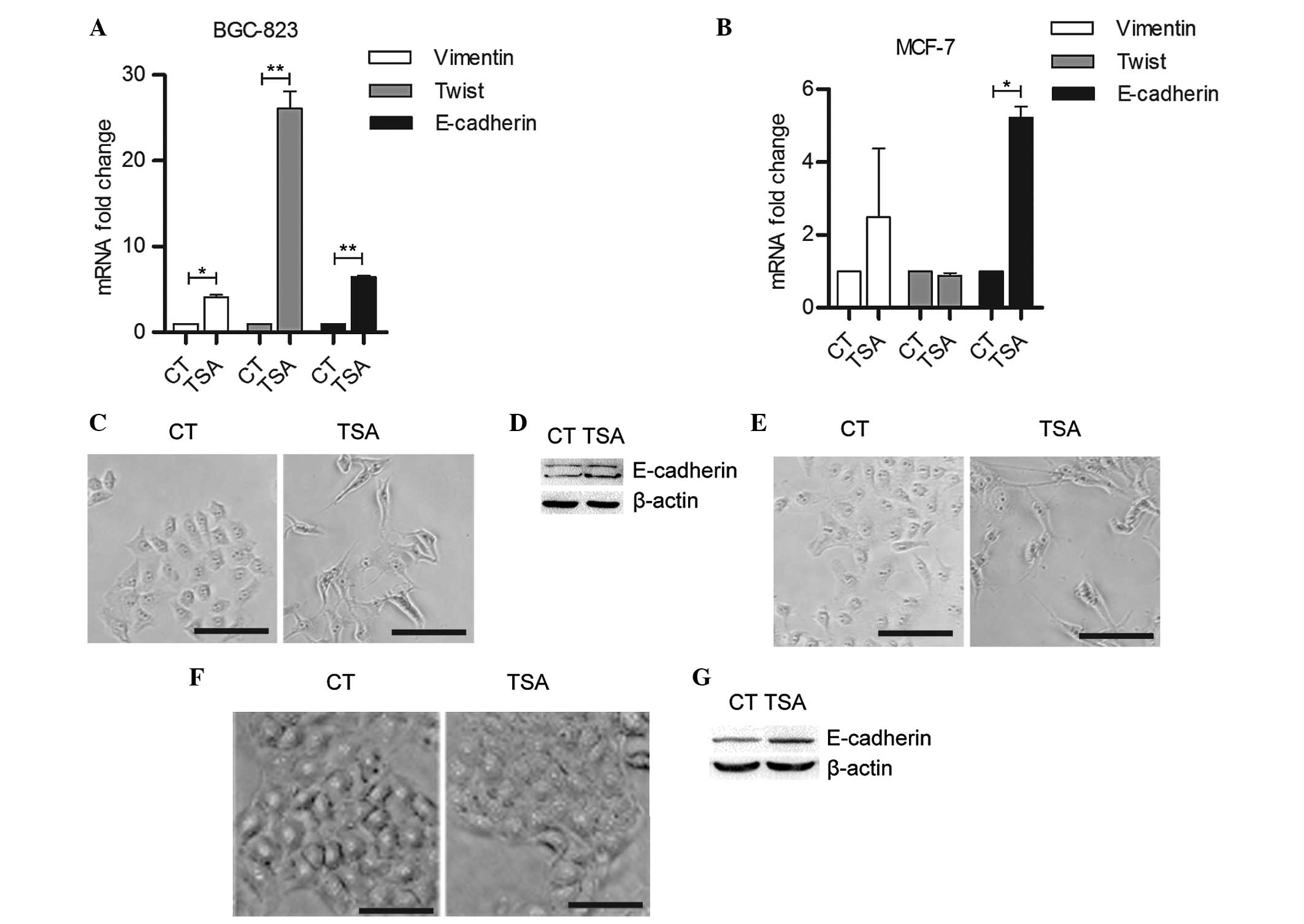
TSA increases the expression levels of
mesenchymal markers and E-cadherin
To confirm whether TSA functions as an inducer of
EMT, the effect of TSA on the expression levels of mesenchymal and
epithelial markers was determined by qPCR. As shown in Fig. 2A, 200 nm TSA treatment
significantly upregulated Vimentin and Twist mRNA transcription in
BGC-823 cells. In MCF-7 cells, the Vimentin mRNA expression levels
were increased but the Twist expression levels were marginally
reduced in response to TSA treatment (Fig. 2B). However, the E-cadherin
transcription levels were significantly increased in the two cell
lines following TSA treatment (Fig. 2A
and B). Immunocytochemistry and western blotting assays further
confirmed that TSA induced E-cadherin expression (Fig. 2C–E). The KYSE-510 human esophageal
squamous cancer cells exhibited no marked morphological change in
response to TSA treatment (Fig.
2F); however, E-cadherin expression levels were increased
following TSA treatment, indicating that the upregulation of
E-cadherin was independent of TSA-induced mesenchymal-like
cytoskeleton remolding (Fig.
2G).
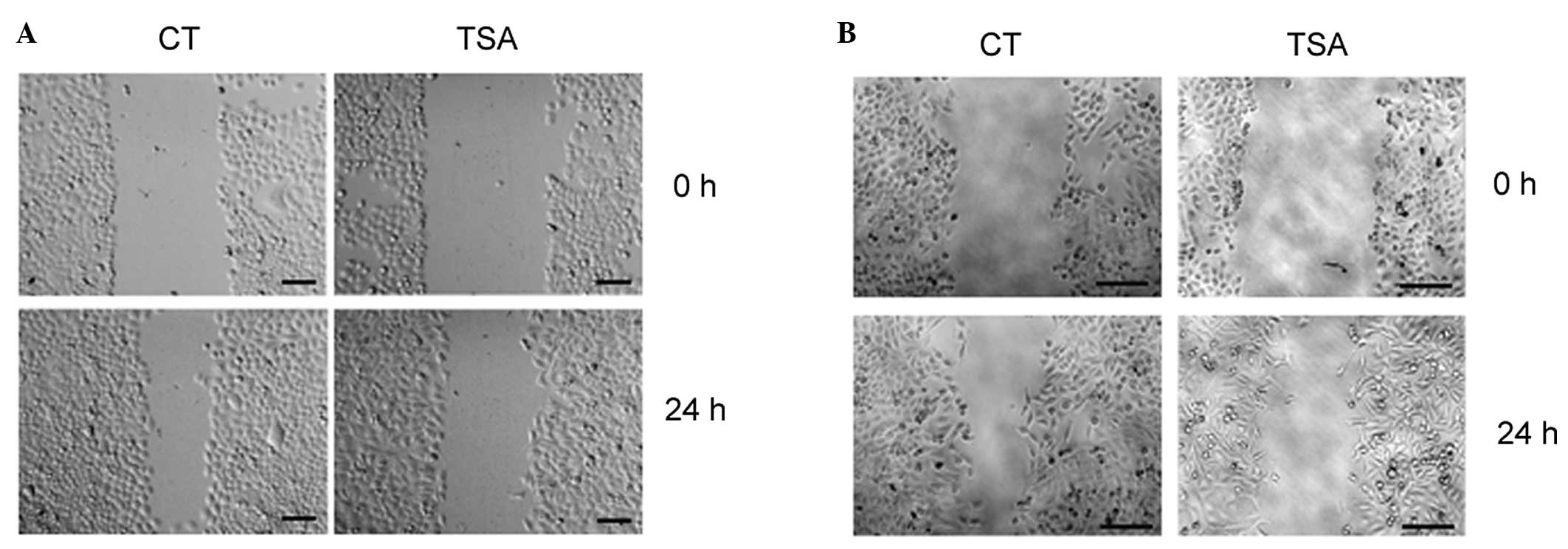
TSA treatment reduces cancer cell
mobility
The cytoskeletal remodeling during EMT induces
spindle-like cancer cell morphology, which facilitates cell
mobility. A wound-healing assay was performed to examine the effect
of TSA on cancer cell migratory ability. As shown in Fig. 3A and B, 200 nm TSA attenuated the
wound-healing process in BGC-823 and MCF-7 cells.
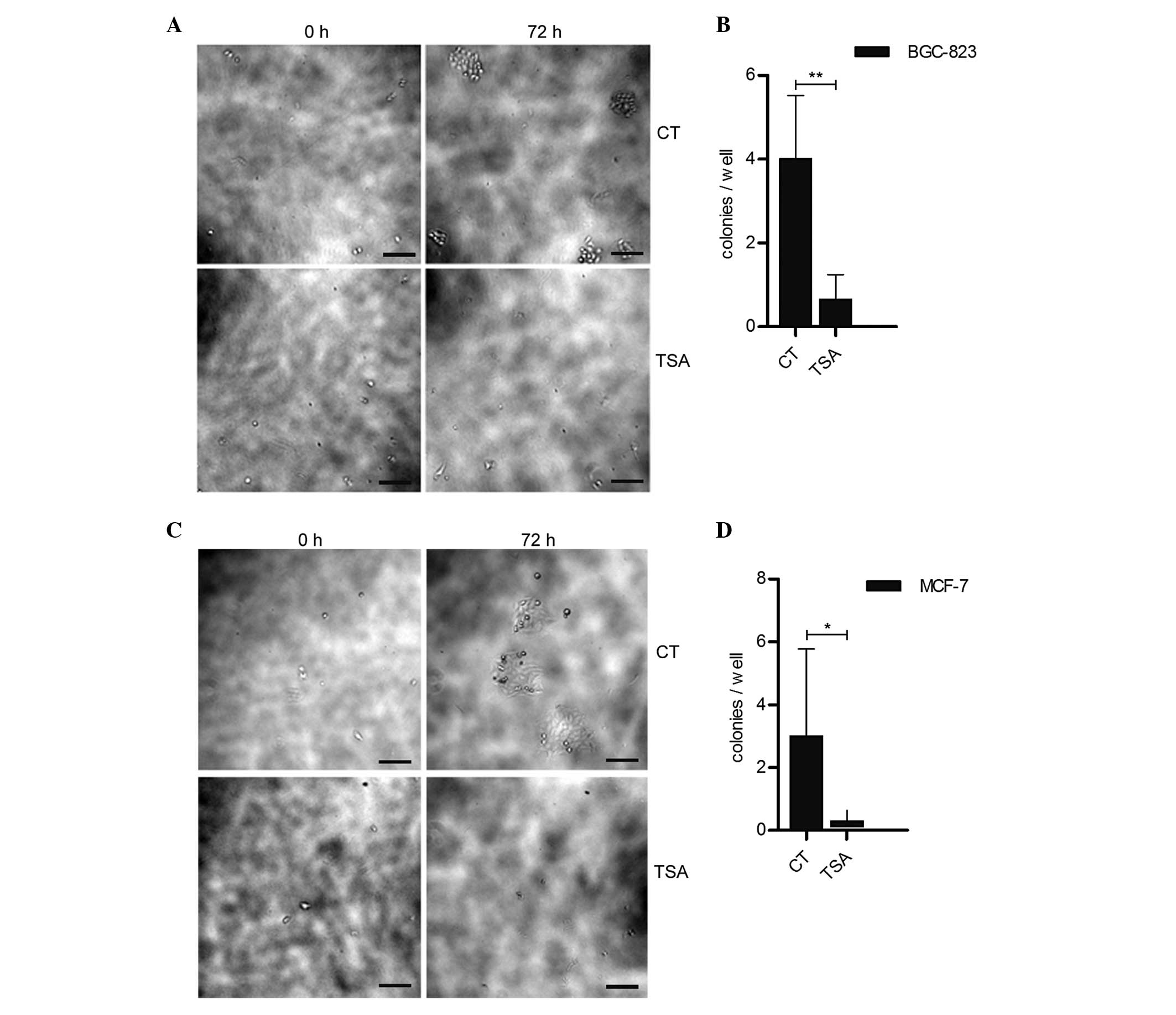
TSA treatment reduces cancer cell colony
formation
Cells that have undergone EMT have been demonstrated
to be the source of cancer stem-like cells (5). High colony-forming efficiency is one
of the hallmarks of cancer stem cells. It was determined whether
TSA enhances the cancer cell colony-forming ability. Fig. 4 indicates that cancer cell colony
formation was significantly inhibited in the TSA treatment groups
compared with the control groups. Since BGC-823 cells are able to
achieve confluency in 200 nm TSA (Fig.
1A), the reduced colony-forming ability in BGC-823 and MCF-7
was not due to the cytotoxic effect of TSA.
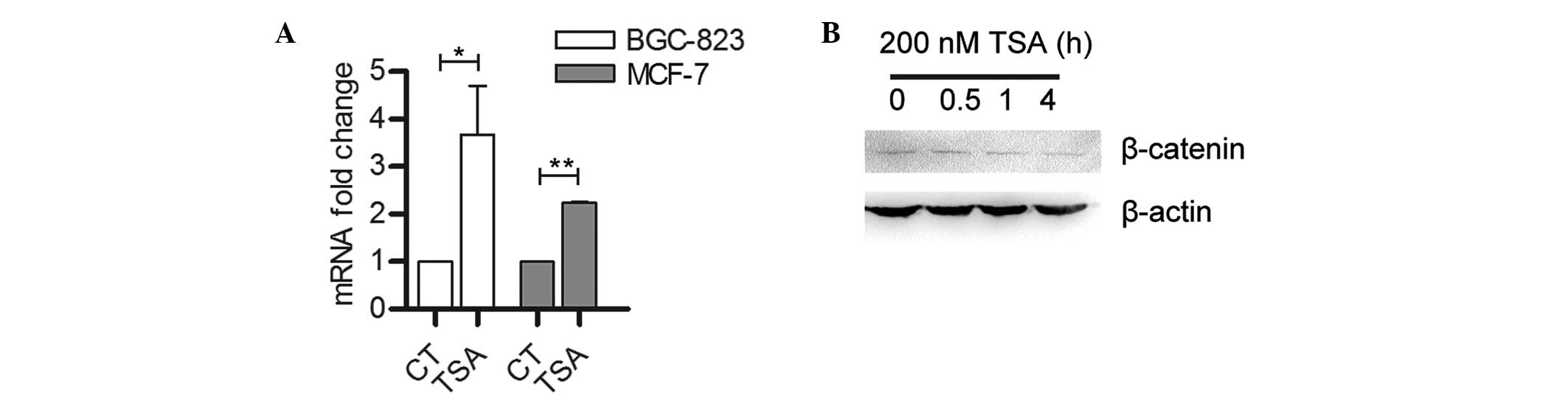
β-catenin is inhibited by TSA
treatment
Diverse extra- and intracellular signals, including
those of the Wnt/β-catenin signaling pathway, have been reported to
induce and/or maintain EMT in various cell types (12). In the present study, β-catenin mRNA
expression levels were found to be significantly upregulated in
BGC-823 and MCF-7 cells following TSA treatment (Fig. 5A). However, β-catenin protein
expression levels in BGC-823 cells were reduced following TSA
treatment (Fig. 5B), suggesting
that TSA is involved in either translational or post-translational
regulation of β-catenin expression.
Discussion
The status of histone acetylation is dependent on
the balance between histone acetyltransferase (HAT) and HDAC
activity. The opposing activity of HATs and HDACs tightly regulate
gene expression through chromatin modification (13). HATs transfer acetyl groups to
N-terminal lysine residues in histones, which induces increases in
local chromatin and elevates accessibility of regulatory proteins
to DNA, whereas HDACs catalyze the removal of acetyl groups,
resulting in chromatin condensation and transcriptional repression.
The identification of increased HDAC expression levels and activity
in cancer tissues has resulted in the rational design of HDACIs as
potential therapeutic agents for cancer therapy (11,14).
Studies have demonstrated that HDACIs increase
E-cadherin expression levels and reverse EMT in normal human cells
and human cancer cells (8,9). By contrast, other studies have shown
that HDACIs are cytoskeleton remodelers, and induce mesenchymal
markers and molecules mediating drug resistance, cell mobility and
self-renewal, supporting the hypothesis that HDACIs act as EMT
inducers in cancer cells (10). In
the present study, TSA was found to induce epithelial-mesenchymal
morphological transition in BGC-823 and MCF-7 cells in a
dose-dependent manner (Fig. 1).
TSA treatment enhanced the transcription levels of the mesenchymal
markers Vimentin and Twist, but also induced the expression of
E-cadherin in BGC-823, MCF-7 and KYSE-510 cells (Fig. 2). Loss of the epithelial homotypic
adhesion molecule E-cadherin is a hallmark of EMT (4). E-cadherin is required for the
formation of stable adherens junctions, and reduced expression
levels of E-cadherin have been reported in various cancer cells,
being associated with tumor progression and metastasis (15). In the present study, in accordance
with the increased expression levels of E-cadherin, TSA treatment
was found to reduce cell migration in the wound-healing assay
(Fig. 3). Furthermore, the
administration of TSA significantly reduced colony formation in
BGC-823 and MCF-7 cells (Fig. 4).
These results contradict those of previous studies that suggest
that TSA is an inducer of EMT in human cancer cells (10).
Certain non-histone proteins are also targets of
HDAC-catalyzed acetylation with varying functional effects. For
example, the gene regulatory activity of transcription factors,
such as p53 and nuclear factor-κB, is modulated through direct
acetylation and deacetylation by HATs and HDACs (16–17).
The Wnt/β-catenin signaling pathway is critical for spontaneous or
induced EMT in cancer cells (12).
In an unstimulated cell, β-catenin is phosphorylated and
subsequently degraded in the adenomatous polyposis coli/Axis
inhibitor/glycogen synthase kinase 3 complex. In the presence of
stimuli, such as induction by Wnt ligand, β-catenin phosphorylation
and degradation is inhibited. The accumulating β-catenin
translocates to the nucleus, where it interacts with lymphoid
enhancer/T cell transcription factors and regulates the expression
of target genes, including c-myc and cyclin D1. The activity of
β-catenin is tightly regulated by various modifications, including
phosphorylation, acetylation and ubiquitination (18,19).
In the present study, although TSA enhanced the transcription level
of β-catenin in BGC-823 and MCF-7 cells, the protein levels were
reduced following TSA treatment (Fig.
5), which was consistent with previous studies reporting that
HDAC6 is required for β-catenin activation in colon cancer cells
(20,21). Whether TSA-mediated EMT induction
and inhibition, elevation of β-catenin mRNA levels and inhibition
of β-catenin protein levels are associated with other signaling
pathways, such as the decorin-β-catenin-E-cadherin signaling
pathway (22), remains unclear and
is currently under investigation.
Acknowledgements
This study was supported in part by the Nature
Science Foundation of China (grant nos. 91229115 and 81272251) and
by the Xinxiang Medical University New Faculty Startup Fund.
Abbreviations:
|
TSA
|
trichostatin A
|
|
EMT
|
epithelial-mesenchymal transition
|
|
HDAC
|
histone deacetylase
|
|
HDACI
|
HDAC inhibitor
|
|
HAT
|
histone acetyltransferase
|
References
|
1
|
Chaffer CL and Weinberg RA: A perspective
on cancer cell metastasis. Science. 331:1559–1564. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sahai E: Illuminating the metastatic
process. Nat Rev Cancer. 7:737–749. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kang Y and Massagué J:
Epithelial-mesenchymal transitions: twist in development and
metastasis. Cell. 118:277–279. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lu J, Guo H, Treekitkarnmongkol W, et al:
14-3-3ζ cooperates with ErbB2 to promote progression of ductal
carcinoma in situ progression to invasive breast cancer by inducing
epithelial-mesenchymal transition. Cancer Cell. 16:195–207.
2009.
|
|
5
|
Mani SA, Guo W, Liao MJ, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Buchwald M, Krämer OH and Heinzel T: HDACi
- targets beyond chromatin. Cancer Lett. 280:160–167. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Acharya MR, Sparreboom A, Venitz J and
Figg WD: Rational development of histone deacetylase inhibitors as
anticancer agents: a review. Mol Pharmacol. 68:917–932. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yoshikawa M, Hishikawa K, Marumo T and
Fujita T: Inhibition of histone deacetylase activity suppresses
epithelial-to-mesenchymal transition induced by TGF-beta1 in human
renal epithelial cells. J Am Soc Nephrol. 18:58–65. 2007.
View Article : Google Scholar
|
|
9
|
Bruzzese F, Leone A, Rocco M, et al: HDAC
inhibitor vorinostat enhances the antitumor effect of gefitinib in
squamous cell carcinoma of head and neck by modulating ErbB
receptor expression and reverting EMT. J Cell Physiol.
226:2378–2390. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kong D, Ahmad A, Bao B, et al: Histone
deacetylase inhibitors induce epithelial-to-mesenchymal transition
in prostate cancer cells. PloS One. 7:e450452012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bolden JE, Peart MJ and Johnstone RW:
Anticancer activities of histone deacetylase inhibitors. Nat Rev
Drug Discov. 5:769–784. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Scheel C, Eaton EN, Li SH, et al:
Paracrine and autocrine signals induce and maintain mesenchymal and
stem cell states in the breast. Cell. 145:926–940. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Roth SY, Denu JM and Allis CD: Histone
acetyltransferases. Annu Rev Biochem. 70:81–120. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dokmanovic M, Clarke C and Marks PA:
Histone deacetylase inhibitors: overview and perspectives. Mol
Cancer Res. 5:981–989. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Juan LJ, Shia WJ, Chen MH, et al: Histone
deacetylases specifically down-regulate p53-dependent gene
activation. J Biol Chem. 275:20436–20443. 2000. View Article : Google Scholar
|
|
17
|
Ashburner BP, Westerheide SD and Baldwin
AS Jr: The p65 (RelA) subunit of NF-kappaB interacts with the
histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to
negatively regulate gene expression. Mol Cell Biol. 21:7065–7077.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Winer IS, Bommer GT, Gonik N and Fearon
ER: Lysine residues Lys-19 and Lys-49 of beta-catenin regulate its
levels and function in T cell factor transcriptional activation and
neoplastic transformation. J Biol Chem. 281:26181–26187. 2006.
View Article : Google Scholar
|
|
19
|
Li VS, Ng SS, Boersema PJ, et al: Wnt
signaling through inhibition of beta-catenin degradation in an
intact Axin1 complex. Cell. 149:1245–1256. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li Y, Zhang X, Polakiewicz RD, et al:
HDAC6 is required for epidermal growth factor-induced beta-catenin
nuclear localization. J Biol Chem. 283:12686–12690. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mak AB, Nixon AM, Kittanakom S, et al:
Regulation of CD133 by HDAC6 promotes β-catenin signaling to
suppress cancer cell differentiation. Cell Rep. 2:951–963.
2012.PubMed/NCBI
|
|
22
|
Bi X, Pohl N, Qian Z, et al:
Decorin-mediated inhibition of colorectal cancer growth and
migration is associated with E-cadherin in vitro and in mice.
Carcinogenesis. 33:326–330. 2012. View Article : Google Scholar : PubMed/NCBI
|