Introduction
Nonalcoholic steatohepatitis (NASH) is a form of
metabolic liver disease. The features of NASH on liver biopsy
include steatosis, inflammation, liver cell injury and varying
degrees of fibrosis (1). Although
NASH has become a worldwide public health issue, the underlying
causes remain poorly understood. However, it is generally
hypothesized that lipid accumulation precedes hepatocellular injury
and liver inflammation (2).
Peroxisome proliferator-activated receptor (PPAR) α, a metabolic
nuclear receptor, regulates hepatic lipid disposal through direct
transcriptional control of genes involved in peroxisomal and
mitochondrial β-oxidation pathways, fatty acid uptake and
triglyceride catabolism (3). It is
hypothesized that PPARα is involved in the development of NASH, and
it has been shown that treatment with PPARα agonists improves liver
lipid accumulation and inflammation (4–6).
Aldose reductase (also termed AR, AKR1B1 and
EC1.1.1.21) catalyzes the rate-limiting reduction of glucose to
sorbitol with the aid of co-factor nicotinamide adenine
dinucleotide phosphate-oxidase (NADPH) (7). The role of AR in the development of
diabetic complications is well-established. However, AR expression
has been found to be induced in certain tissues in conditions other
than diabetes. Notably, hepatic AR is induced in several types of
liver disease, including alcoholic liver disease, hepatitis,
cirrhosis and hepatocellular carcinoma (8–10).
This indicates that AR is involved in the development of hepatitis
and fibrosis. We previously reported that AR regulated PPARα
phosphorylation and activity, thus influencing lipid homeostasis,
and that inhibition of AR improved hepatic steatosis in db/db
diabetic mice (11,12). This suggested that inhibition of AR
may curtail the development of NASH. The present study aimed to
investigate the effect of AR inhibition on the development of
nutrition-induced murine NASH in C57BL/6 mice fed a
methionine-choline-deficient (MCD) diet and to investigate the
mechanism underlying the effects of AR on the accumulation of lipid
in the liver.
Materials and methods
Animal experiments
All experiments were conducted according to
protocols and guidelines approved by Longyan University
Institutional Animal Care and Use Committee (Longyan, China).
C57BL/6 mice were obtained from The Shanghai Laboratory Animal
Center (Shanghai, China). All animals were maintained under a
12/12-h light/dark cycle. Male mice, 7–8 weeks of age, were
randomly divided into four experimental groups (each containing six
animals), namely, control diet-fed mice, control diet-fed mice +
zopolrestat (zopol), MCD diet-fed mice, MCD diet-fed mice + zopol.
The control diet was identical to the MCD diet (MP Biomedicals,
Aurora, OH) but supplemented with DL-methionine (3 g/kg) and
choline chloride (2 g/kg). Mice were administered with 50 mg/kg
body weight/day zopol as a single daily intraperitoneal injection
for 4 weeks. The same volumes of saline were also administered to
the other groups of mice as a control.
Quantitative analyses of mRNA expression
by quantitative polymerase chain reaction (qPCR)
Total RNA was isolated from tissues using the TRIzol
reagent (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer’s instructions. Complementary DNA (cDNA) was
synthesized from hepatic mRNA using RevertAid First Strand cDNA
synthesis kits (Fermentas, Vilnius, Lithuania). Hepatic PPARα, acyl
coenzyme A oxidase (ACO), liver fatty acid binding protein (L-FABP)
and carnitine palmitoyl transferase-1 (CPT-1) were analyzed with
the specific primers listed in Table
I. Quantitative PCRs were assayed using the FastStart Universal
SYBR Green Master mix (Rox; Roche Applied Science, Mannheim,
Germany) The reaction was run at 95°C for 10 min, followed by 40
cycles at 95°C for 15 sec, 55–58°C for 30 sec, 72°C for 30 sec and
a final extension at 72°C for 10 min. Each Ct value was normalized
to β-actin.
 | Table IPrimer sequences used for the
amplification of mRNA by quantitative polymerase chain reaction
(qPCR). |
Table I
Primer sequences used for the
amplification of mRNA by quantitative polymerase chain reaction
(qPCR).
| Gene | Forward primer | Reverse primer |
|---|
| PPARα |
5′-AAGAGGGCTGAGCGTAGGT-3′ |
5′-GGCCGGTTAAGACCAGACT-3′ |
| ACO |
5′-CCACATATGACCCCAAGACC-3′ |
5′-AGGCATGTAACCCGTAGCAC-3′ |
| CPT-1 |
5′-GTCAAGCCAGACGAAGAACA-3′ |
5′-CGAGAAGACCTTGACCATAG-3′ |
| L-FABP |
5′-GTGGTCCGCAATGAGTTCAC-3′ |
5′-GTATTGGTGATTGTGTCTCC-3′ |
| β-actin |
5′-CTATTGGCAACGAGCGGTTCC-3′ |
5′-GCACTGTGTTGGCATAGAGGTC-3′ |
Western blot analyses
Tissues were homogenized in ice-cold
radioimmunoprecipitation assay buffer (Beyotime, Haimen, China).
Each protein sample (40 μg) was loaded and separated on a 12%
SDS-polyacrylamide gel, and transferred to polyvinylidene
difluoride membranes (Millipore, Billerica, MA, USA). Blotted
membranes were then incubated with goat polyclonal anti-AR (Santa
Cruz Biotechnology, Santa Cruz, CA, USA; 1:500), rabbit polyclonal
anti-PPARα (Santa Cruz Biotechnology; 1:500) or rabbit polyclonal
anti-phospho-PPARα (Abcam, Cambridge, UK; 1:1000) in Tris-buffered
saline with 0.1% Tween-20 (TBST) and 5% non-fat milk at 4°C
overnight. After several washes, the membranes were incubated with
horseradish peroxidase-conjugated monoclonal anti-goat IgG or
anti-rabbit IgG (Sigma, St. Louis, MO, USA; 1:2000) in TBST and 5%
non-fat milk. Detection was achieved using the Supersignal
chemiluminescent substrate kit (Pierce, Rockford, IL, USA).
Histological examination
Formalin-fixed liver tissue was processed and
5-μm-thick paraffin sections were stained with hematoxylin and
eosin (H&E) for histological analyses. A hepatopathologist who
was blinded to the experimental conditions examined all sections
for steatosis and inflammation as previously described (13). Hepatic steatosis was graded
according to the percentage of lipid-laden hepatocytes as 0, 0%; 1,
1–33%; 2, 34–67%; or 3, 68–100%. Hepatic necroinflammation were
scored from 0 to 3, as follows: 0, no inflammatory foci; 1, mild;
2, moderate; and 3, severe.
Tissue and serum biochemical
measurements
Serum alanine aminotransferase (ALT) was measured
using an IDEXX analyzer (IDEXX Laboratories, Inc., Westbrook, ME,
USA). Total lipoperoxides were measured as thiobarbituric acid
reactive substances (TBARS) in 100 μl of liver homogenate using
Lipid Peroxidation Assay kits (Beyotime). TBARS were quantified
using malondialdehyde as a standard. Liver lipid was extracted
using chloroform/methanol. Briefly, pulverized liver was
homogenized in phosphate-buffered saline (PBS), then extracted with
chloroform/methanol (2:1), dried overnight and resuspended in a
solution of 60% butanol and 40% Triton X-114/methanol (2:1). The
liver triglyceride level was measured using colorimetric assays
(Sigma).
Statistical analysis
All data were processed and analyzed using the
GraphPad software Prism 5.0 (GraphPad Software, Inc., Chicago, IL,
USA) and are expressed as the mean ± standard error of the mean.
Student’s t-test was used for pair-wise comparisons and one-way
analysis of variance with Bonferroni’s Multiple Comparison test was
used for multi-group analyses. P<0.05 was considered to indicate
a statistically significant difference.
Results
Inhibition of AR attenuates diet-induced
hepatic steatosis and inflammation
Feeding mice with a lipogenic MCD diet was
previously shown to be capable of inducing a liver injury similar
to human NASH. MCD-fed mice are therefore a useful small animal
model of this disease (6,14). As an initial step in investigating
whether AR was involved in the development of MCD diet-induced
steatohepatitis, protein expression levels of hepatic AR in C57BL/6
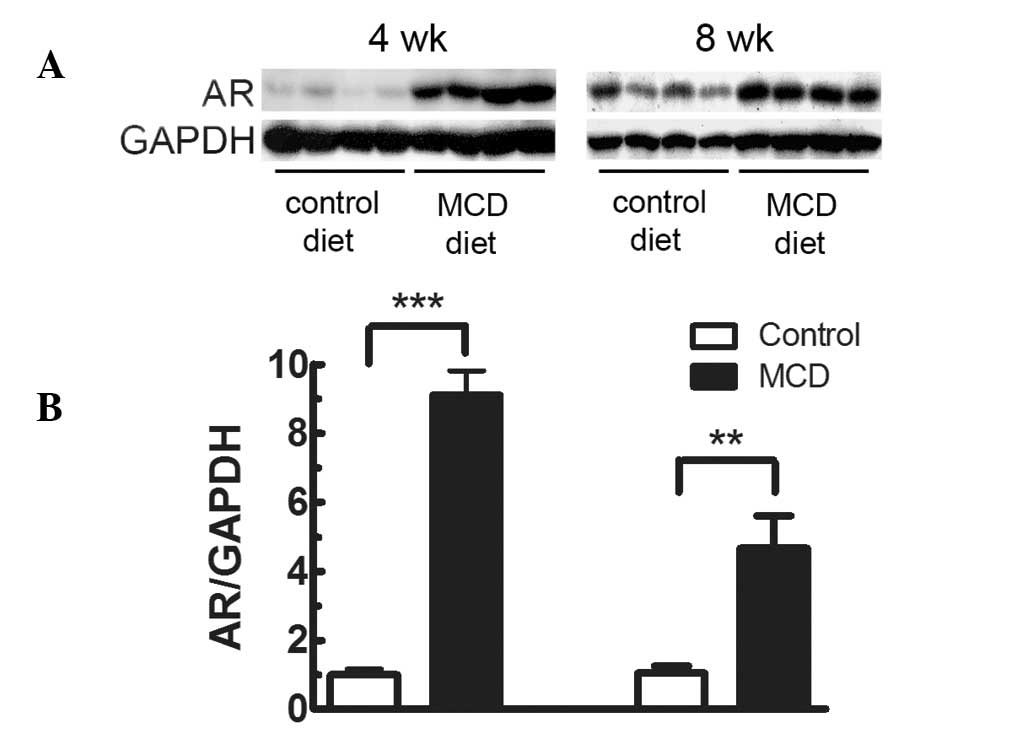
mice fed the MCD diet were measured. As shown in Fig. 1, hepatic AR protein expression in
mice fed the MCD diet for 4 weeks was 9.11-fold higher (P<0.001)
than that in mice fed the control diet. The elevation of hepatic AR
was also observed after feeding the MCD diet for 8 weeks
(4.68-fold; P<0.01). To further investigate the role of AR in
the development of diet-induced steatohepatitis, MCD diet-fed mice
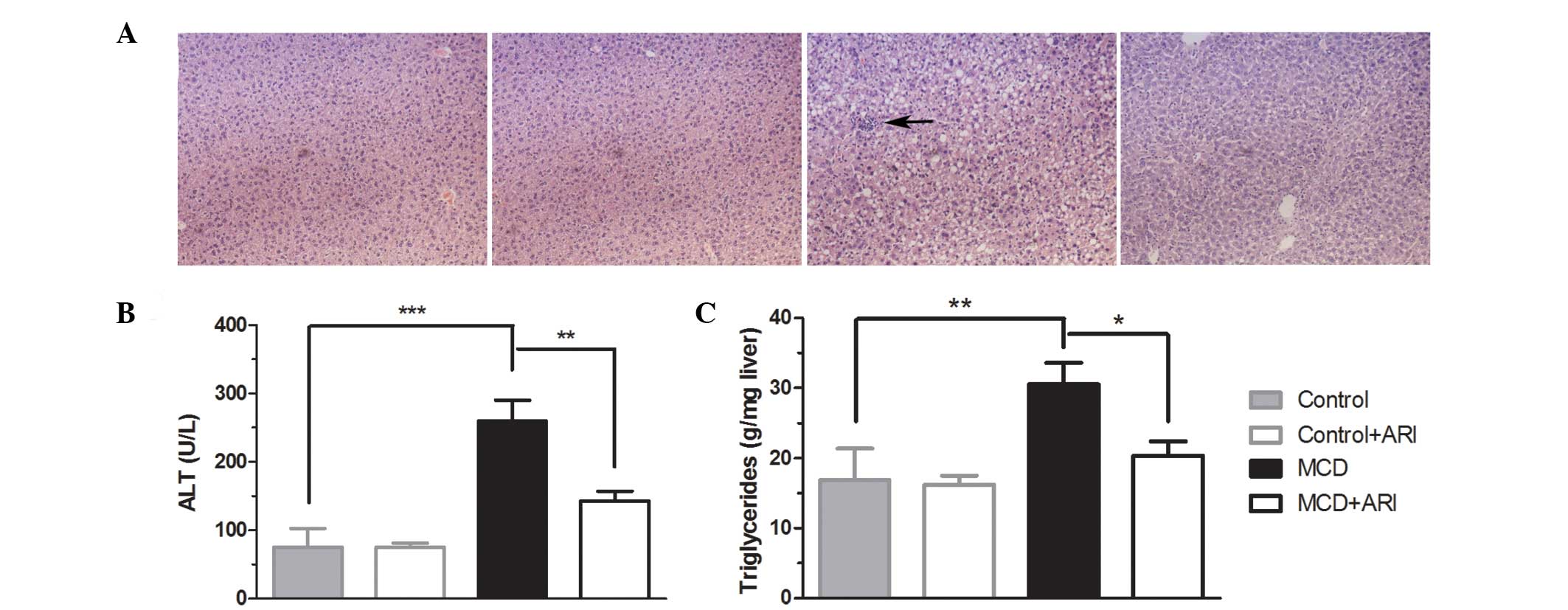
were treated with zopol, an inhibitor of AR, for 4 weeks. As shown
in Fig. 2A and Table II, examination of H&E-stained
sections demonstrated marked steatosis and lobular inflammation in
mice fed the MCD diet for 4 weeks, while mice fed the control diet
did not exhibit significant histological steatosis or inflammation.
Treating MCD diet-fed mice with zopol significantly improved the
levels of hepatic steatosis and inflammation. Consistent with these
histological findings, AR inhibition reduced serum ALT by 44.8%
(P<0.01; Fig. 2B) and reduced
liver triglycerides by 33.4% (P<0.05; Fig. 2C) compared with levels in mice on
the MCD diet. These data indicate that AR inhibitor treatment
ameliorates the development of MCD diet-induced steatohepatitis in
C57BL/6 mice.
| Table IIEffect of ARI treatment on scores for
hepatic steatosis and necroinflammation. |
Table II
Effect of ARI treatment on scores for
hepatic steatosis and necroinflammation.
| Measurement | Controls | Controls + ARI | MCD | MCD + ARI |
|---|
| Steatosis | 0.00±0.00 | 0.00±0.00 | 2.20±0.29 | 1.15±0.10a |
|
Necroinflammation | 0.00±0.00 | 0.00±0.00 | 0.95±0.19 | 0.19±0.10a |
This study also investigated the effect of AR
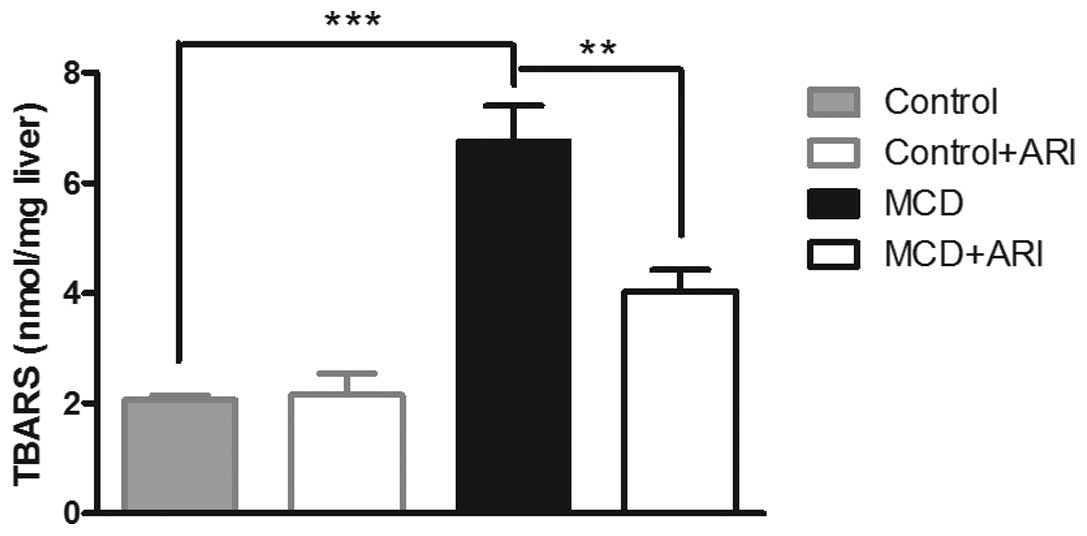
inhibition on hepatic lipoperoxide production. As shown in Fig. 3, intake of the MCD diet for 4 weeks
resulted in a prominent increase in the hepatic TBARS level,
compared with those in mice administered the control diet. This
increase was attenuated when the MCD-fed mice were also
administered zopol (P<0.01). These data suggest that hepatic AR
elevation may exacerbate the MCD diet-induced oxidative stress in
the livers of mice with steatohepatitis and that inhibition of AR
may reverse this process.
AR inhibition suppresses the MCD
diet-induced phosphorylation of hepatic PPARα and increases its
activity
PPARα is an important metabolic nuclear receptor
that regulates lipid metabolism (3). Activation of PPARα decreases hepatic
steatosis in MCD diet-fed C57BL/6 mice (5,6). To
clarify the mechanism whereby AR exacerbates the hepatic steatosis
in MCD diet-fed mice, the effect of AR inhibition on the activity
of PPARα was investigated. PPARα is a phosphoprotein and
phosphorylation is one of the most rapid and efficient mechanisms
through which its activity can be modulated. Firstly, the protein
expression level and the phosphorylation of PPARα was analyzed. As
shown in Fig. 4A, no significant
differences were identified in hepatic PPARα protein expression
amongst all four groups of mice. Western blot analysis using
antibodies that recognize phospho-PPARα at either serine 12 (S12)
or serine 21 (S21), two major phosphorylation sites located at the
A/B domain (AF-1) of PPARα, showed that the phosphorylation of
PPARα at these sites was markedly increased in the MCD diet-fed
mice compared with the levels in mice on the control diet.
Furthermore, this induction of PPARα phosphorylation was suppressed
in ARI-treated mice. In addition, with the phosphorylation of
PPARα, hepatic mRNA expression of ACO and L-FABP, two target genes
regulated by PPARα, were downregulated in mice fed the MCD diet.
Whilst the downregulation of ACO mRNA expression was reversed in
ARI-treated mice, the downregulation of L-FABP was not
significantly altered (Fig. 4B).
mRNA expression of CPT-1, another target gene regulated by PPARα,
was not significantly altered in all four groups of mice. These
data indicate that administration of an MCD diet resulted in the
phosphorylation of hepatic PPARα, which suppressed PPARα activity.
The results also suggest that AR inhibition attenuated the MCD
diet-induced phosphorylation of PPARα and thus suppression of its
activity.
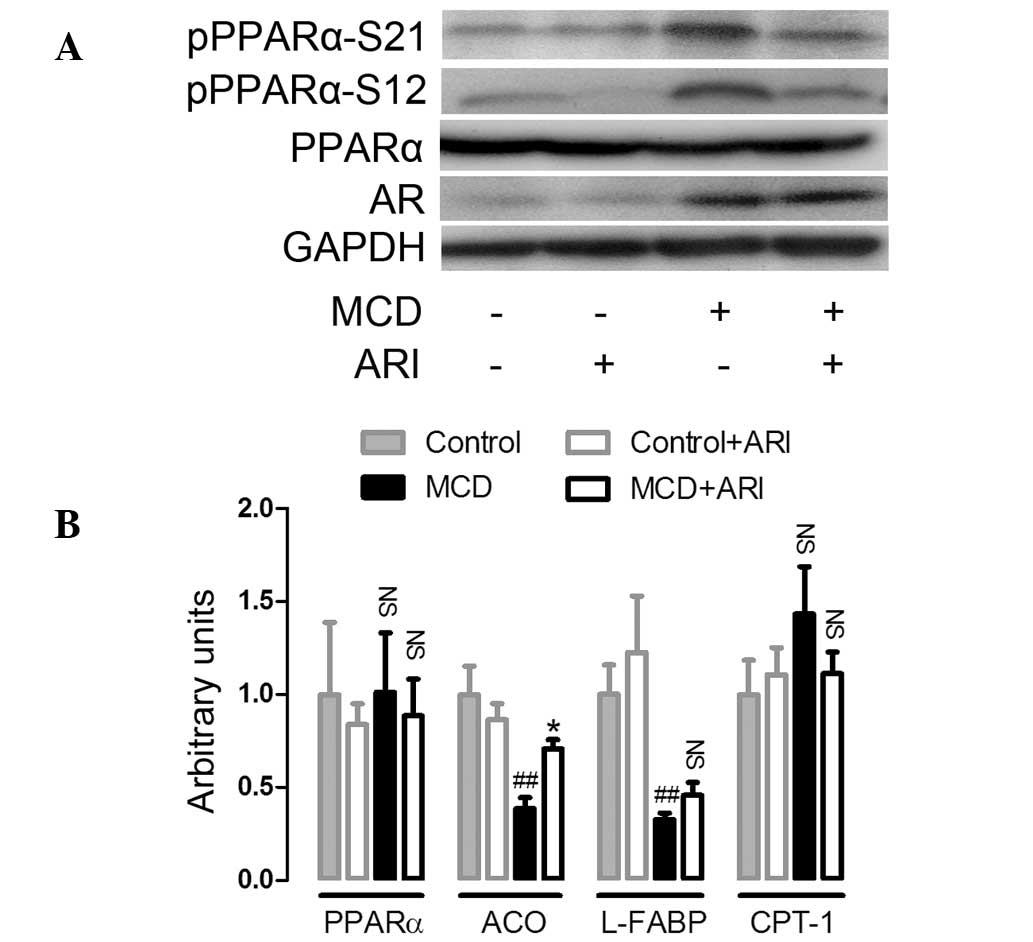 | Figure 4AR inhibition increased hepatic PPARα
transcriptional activity in MCD diet-fed C57BL/6 mice. (A) The
effects of AR inhibition on PPARα expression and phosphorylation in
mice fed the MCD diet for 4 weeks. Western blotting was performed
as described in the Materials and methods section (n=3). (B)
Hepatic mRNA expression of PPARα, ACO, L-FABP and CPT-1 analyzed by
quantitative polymerase chain reaction, standardized against an
internal control (β-actin) and are expressed as fold differences
compared with values obtained in mice fed the control diet (n=4).
Values are expressed as the mean ± standard error of the mean.
##P<0.01, *P<0.05, compared with mice
fed the control diet. NS, no significant difference; PPAR-α,
peroxisome proliferator-activated receptor-α; pPPARα S12,
phosphoserine-12 PPARα; pPPARα S21, phosphoserine-21 PPARα; MCD,
methionine-choline-deficient; AR, aldose reductase; ACO; acetyl
coenzymeA oxidase; L-FABP, liver fatty acid binding protein; CPT-1,
carnitine palmitoyl transferase-1. |
Discussion
AR induction has been observed in a number of liver
diseases, including alcoholic liver disease, chronic hepatitis B
and C, and hepatocellular carcinoma in humans, and in hereditary
hepatitis in rats (8–10). We previously demonstrated that
hepatic AR induction in db/db mice with diet-induced
steatohepatitis contributed to the development of liver
inflammation and fibrosis (15).
However, the importance of AR in the development of hepatic
steatosis in db/db mice with steatohepatitis was not established.
The present study demonstrated that hepatic AR protein levels were
also increased in MCD diet-induced steatohepatitis in C57BL/6 mice.
It also showed that the induction of AR protein resulted in an
increase in PPARα phosphorylation and thus led to the accumulation
of lipid in the liver. Furthermore, AR inhibition ameliorated MCD
diet-induced hepatic steatosis and inflammation, suppressed the
phosphorylation of PPARα and increased its transcriptional
activity. The results therefore indicate that AR may affect the
development of hepatic steatosis by modulating the phosphorylation
of PPARα and suppressing the activity of this protein.
Notably, the current study demonstrated the
beneficial effect of pharmacological inhibition of AR on hepatic
steatosis whereas a previous study did not report a significantly
beneficial effect of genetic ablation of AR, which results in
complete loss of activity, on hepatic steatosis in the same rodent
model (15). One possible
explanation for the discrepancy between complete AR deficiency and
ARI treatment is the possibility that any of a number of AR-related
enzymes, such as mouse vas deferens protein, mouse fibroblast
growth factor regulated protein and AR-like-1, may be upregulated
to compensate for the AR deficiency. These AR-like enzymes have a
number of the same functions as AR. However, little data is
available regarding the significance of these enzymes in lipid
disorders. Further studies are required using genetic ablation of
AR-like enzymes in order to investigate their involvement in
hepatic steatosis.
It is well established that PPARα is a central
regulator of hepatic lipid catabolism. The ablation or inhibition
of PPARα causes the development of a number of lipid disorders
including hepatic steatosis and non-alcoholic fatty liver disease
(16,17). Post-translational modification by
phosphorylation is one of the most important mechanisms whereby
PPARα transcriptional activity is modulated (18,19).
Multiple phosphorylation sites have been identified on different
domains of mouse PPARα, which include the A/B domain, the
DNA-binding domain and the ligand-binding domain. Further, PPARα
phosphorylation was shown to be catalyzed by a diverse group of
kinases, including protein kinase A, protein kinase C,
extracellular signal-regulated kinases and glycogen synthase
kinase. Depending on the types of cells and stimuli involved,
phosphorylation can either lead to activation or inactivation of
PPARα (19–21). In the current study, it was
demonstrated that MCD diet-induced PPARα phosphorylation at S12 and
S21 contributed to suppression of PPARα transcriptional activity in
the mouse liver and that AR inhibition attenuated this PPARα
phosphorylation, which may contribute to the amelioration of
diet-induced steatosis. However, further studies are required to
determine the signaling pathways involved in the MCD diet-induced
phosphorylation of PPARα.
The molecular mechanisms involved in progression
from liver steatosis to NASH remain unclear. However, oxidative
stress is a possible candidate (22,23).
4-Hydroxynonenal (4HNE) is a cytotoxic byproduct of lipid
peroxidation that is hypothesized to participate in the
pathogenesis of a number of diseases (24). In addition to glucose, AR can
catalyze the reduction of a number of aldehydes and carbonyls,
including 4HNE (25). Thus, AR has
been postulated to serve a cytoprotective function by rapidly
detoxifying aldehydes. In vitro studies have shown that AR
expression is induced by 4HNE in rat vascular smooth muscle cells
and that inhibition of AR sensitizes cells to 4HNE cytotoxicity
(26). Furthermore, an in
vivo study showed that inhibition of AR was associated with
increased numbers of apoptotic cells and increased 4HNE content in
the arterial wall of a murine model of giant cell arteritis
(27). However, AR inhibitors have
also been reported to exert beneficial effects on injuries in a
number of rodent models, including allergic airway inflammation,
ischemic myocardial injury, arterial balloon injury and uveitis
(28–31). The current study demonstrated that
zopol, an AR inhibitor, attenuated MCD diet-induced oxidative
stress and improved liver inflammation. This provides further
evidence of the beneficial effect of AR inhibitors on
inflammation.
In conclusion, the present study demonstrated the
protective effect of an AR inhibitor against MCD diet-induced
hepatic steatosis and liver damage. This effect was mediated, at
least in part, through modulation of the phosphorylation of PPARα
and its transcriptional activity.
Acknowledgements
This study was supported by Educational Commission
of Fujian Province, China (grant no. JB12198) and the Natural
Science Foundation of Fujian Province, China (grant no.
2009J01180).
References
|
1
|
Ludwig J, Viggiano TR, McGill DB and Oh
BJ: Nonalcoholic steatohepatitis: Mayo Clinic experiences with a
hitherto unnamed disease. Mayo Clin Proc. 55:434–438.
1980.PubMed/NCBI
|
|
2
|
Day CP and James OF: Steatohepatitis: a
tale of two ‘hits’? Gastroenterology. 114:842–845. 1998.
|
|
3
|
Kota BP, Huang TH and Roufogalis BD: An
overview on biological mechanisms of PPARs. Pharmacol Res.
51:85–94. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Abdelmegeed MA, Yoo SH, Henderson LE, et
al: PPARalpha expression protects male mice from high fat-induced
nonalcoholic fatty liver. J Nutr. 141:603–610. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ip E, Farrell G, Hall P, et al:
Administration of the potent PPARalpha agonist, Wy-14,643, reverses
nutritional fibrosis and steatohepatitis in mice. Hepatology.
39:1286–1296. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ip E, Farrell GC, Robertson G, et al:
Central role of PPARalpha-dependent hepatic lipid turnover in
dietary steatohepatitis in mice. Hepatology. 38:123–132. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Clements RS Jr: The polyol pathway. A
historical review. Drugs. 32(Suppl 2): 3–5. 1986. View Article : Google Scholar
|
|
8
|
Brown KE, Broadhurst KA, Mathahs MM, et
al: Immunodetection of aldose reductase in normal and diseased
human liver. Histol Histopathol. 20:429–436. 2005.PubMed/NCBI
|
|
9
|
O’Connor T, Ireland LS, Harrison DJ and
Hayes JD: Major differences exist in the function and
tissue-specific expression of human aflatoxin B1 aldehyde reductase
and the principal human aldo-keto reductase AKR1 family members.
Biochem J. 343:487–504. 1999.PubMed/NCBI
|
|
10
|
Takahashi M, Hoshi A, Fujii J, et al:
Induction of aldose reductase gene expression in LEC rats during
the development of the hereditary hepatitis and hepatoma. Jpn J
Cancer Res. 87:337–341. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qiu L, Lin J, Xu F, et al: Inhibition of
aldose reductase activates hepatic peroxisome
proliferator-activated receptor-α and ameliorates hepatosteatosis
in diabetic db/db mice. Exp Diabetes Res.
2012:7897302012.PubMed/NCBI
|
|
12
|
Qiu L, Wu X, Chau JF, et al: Aldose
reductase regulates hepatic peroxisome proliferator-activated
receptor alpha phosphorylation and activity to impact lipid
homeostasis. J Biol Chem. 283:17175–17183. 2008. View Article : Google Scholar
|
|
13
|
Brunt EM: Nonalcoholic steatohepatitis:
definition and pathology. Semin Liver Dis. 21:3–16. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sahai A, Malladi P, Pan X, et al: Obese
and diabetic db/db mice develop marked liver fibrosis in a model of
nonalcoholic steatohepatitis: role of short-form leptin receptors
and osteopontin. Am J Physiol Gastrointest Liver Physiol.
287:G1035–G1043. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qiu L, Lin J, Ying M, et al: Aldose
reductase is involved in the development of murine diet-induced
nonalcoholic steatohepatitis. PLoS One. 8:e735912013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lefebvre P, Chinetti G, Fruchart JC and
Staels B: Sorting out the roles of PPAR alpha in energy metabolism
and vascular homeostasis. J Clin Invest. 116:571–580. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Djouadi F, Weinheimer CJ, Saffitz JE, et
al: A gender-related defect in lipid metabolism and glucose
homeostasis in peroxisome proliferator- activated receptor alpha-
deficient mice. J Clin Invest. 102:1083–1091. 1998. View Article : Google Scholar
|
|
18
|
Burns KA and Vanden Heuvel JP: Modulation
of PPAR activity via phosphorylation. Biochim Biophys Acta.
1771:952–960. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gelman L, Michalik L, Desvergne B and
Wahli W: Kinase signaling cascades that modulate peroxisome
proliferator-activated receptors. Curr Opin Cell Biol. 17:216–222.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Barger PM, Brandt JM, Leone TC, et al:
Deactivation of peroxisome proliferator-activated receptor-alpha
during cardiac hypertrophic growth. J Clin Invest. 105:1723–1730.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tamasi V, Miller KK, Ripp SL, et al:
Modulation of receptor phosphorylation contributes to activation of
peroxisome proliferator activated receptor alpha by
dehydroepiandrosterone and other peroxisome proliferators. Mol
Pharmacol. 73:968–976. 2008. View Article : Google Scholar
|
|
22
|
Koek GH, Liedorp PR and Bast A: The role
of oxidative stress in non-alcoholic steatohepatitis. Clin Chim
Acta. 412:1297–1305. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Voican CS and Perlemuter G: Insulin
resistance and oxidative stress: two therapeutic targets in
non-alcoholic steatohepatitis. J Hepatol. 54:388–391. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Esterbauer H, Schaur RJ and Zollner H:
Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and
related aldehydes. Free Radic Biol Med. 11:81–128. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vander Jagt DL, Kolb NS, Vander Jagt TJ,
et al: Substrate specificity of human aldose reductase:
identification of 4-hydroxynonenal as an endogenous substrate.
Biochim Biophys Acta. 1249:117–126. 1995.PubMed/NCBI
|
|
26
|
Spycher S, Tabataba-Vakili S, O’Donnell
VB, et al: 4-hydroxy-2,3-trans-nonenal induces transcription and
expression of aldose reductase. Biochem Biophys Res Commun.
226:512–516. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rittner HL, Hafner V, Klimiuk PA, et al:
Aldose reductase functions as a detoxification system for lipid
peroxidation products in vasculitis. J Clin Invest. 103:1007–1013.
1999. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ruef J, Liu SQ, Bode C, et al: Involvement
of aldose reductase in vascular smooth muscle cell growth and
lesion formation after arterial injury. Arterioscler Thromb Vasc
Biol. 20:1745–1752. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tracey WR, Magee WP, Ellery CA, et al:
Aldose reductase inhibition alone or combined with an adenosine
A(3) agonist reduces ischemic myocardial injury. Am J Physiol Heart
Circ Physiol. 279:H1447–H1452. 2000.PubMed/NCBI
|
|
30
|
Yadav UC, Ramana KV, Aguilera-Aguirre L,
et al: Inhibition of aldose reductase prevents experimental
allergic airway inflammation in mice. PLoS One. 4:e65352009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yadav UC, Srivastava SK and Ramana KV:
Aldose reductase inhibition prevents endotoxin-induced uveitis in
rats. Invest Ophthalmol Vis Sci. 48:4634–4642. 2007. View Article : Google Scholar : PubMed/NCBI
|