Introduction
Chronic obstructive pulmonary disease (COPD) is a
common and frequently occurring disease of the respiratory system,
with high morbidity and mortality rates (1). The exact cause of COPD is not
currently clear, although COPD is widely considered to be
associated with an abnormal inflammatory response of the lungs to
noxious gases or particles, mainly due to cigarette smoke (CS)
(2). In the current definition of
COPD, there appears to be an immune basis for the abnormal pattern
of inflammation (3). An increasing
number of studies have indicated that in COPD, the numbers of
macrophages, T lymphocytes and neutrophils are increased in the
various parts of the lung. Antigen-presenting cells (APCs) usually
aid the organization of the recruited lymphocytes into lymphoid
follicles, and the presence of oligoclonal lymphocytes recognize,
process and present the processed antigen to the naïve lymphocytes
(4).
Dendritic cells (DCs), a type of APC, may be
involved in the development of COPD, as DCs normally exert a
predominant role in the initiation and orchestration of immune
responses. DCs are recruited from the circulation and migrate
toward epithelial surfaces, where the cells capture antigens and
recognize danger signals. Following antigen uptake, DCs migrate to
regional draining lymph nodes for antigen presentation. During
migration, DCs ingest and process antigens, and upregulate the
expression of co-stimulatory molecules at the cell surface, a
process known as maturation using the cluster of differentiation
(CD)83+ cell surface marker (5). In the lymph nodes, DCs present the
processed antigen to naive T lymphocytes, resulting in the
initiation, suppression or termination of adaptive immune responses
if no longer required (6). The
airways and lungs contain a rich network of DCs, localized near the
epithelial surface. DCs thus normally control immunologic
homeostasis, but may function inappropriately in COPD (7).
During the organization of the recruited lymphocytes
into lymphoid follicles, chemokines and the corresponding receptors
are key determinants of lymphoid tissue organization (8). Among the chemokine receptors (CCRs),
CCR7 is an notable receptor with the potential to influence
responses in the peripheral tissues and lymph nodes. CCR7 is
crucial for the homing of immature T cells and mature DCs to the
lymph nodes via dedicated ligands, that is, the lymphoid chemokine
(C-C) motif ligand (CCL)19 and CCL21 (9). A previous study observed increased
pulmonary CCL19 expression levels in a model of murine COPD, with
typical pulmonary lymphoid neogenesis upon CS exposure (10). The determining role of
CCR7+ in the recirculation of lymphocytes from mucosal
tissues is indicated by the finding that CCR7+
deficiency results in the marked appearance of ectopic lymphoid
follicles in the lung and other mucosal sites (11).
In the present study, the hypothesis that small
airways have fewer mature DCs in patients with COPD was analyzed.
The study additionally investigated the role of human pulmonary DCs
in the pathogenesis of COPD. DC infiltration in the peripheral
airways was compared among non-smokers, smokers without airway
obstruction and patients with COPD.
Materials and methods
Patient tissue sample collection
This study was approved by the medical ethical
committee of the Affiliated Hospital of Zunyi Medical College
(Zunyi, China). COPD was diagnosed and classified by the Global
Initiative for Chronic Obstructive Lung Disease (GOLD) criteria
(12). Tissues were obtained from
surgical lung resection specimens of the following patients
diagnosed with solitary pulmonary lesions: Eight smokers with COPD,
eight non-smokers with COPD, eight smokers without COPD and eight
non-smokers without COPD (which served as a control group). Lung
tissues at the maximum distance from the pulmonary lesion, and
without signs of retro-obstructive pneumonia or tumor invasion,
were collected by a pathologist. None of the patients who underwent
surgery for malignancy were treated with neo-adjuvant chemotherapy.
All patients signed informed consent prior to the surgery and were
interviewed with regard to smoking habits and medication use. COPD
diagnosis and severity were defined using pre-operative spirometry
according to the GOLD classification.
Determination of CD83+,
CD1a+ and CCR7+ gene expression levels in
COPD and control tissues
The lung tissue samples were ground using liquid
nitrogen. Total RNA was extracted from the samples using a human
tissue RNA purification kit (Norgen Biotek, Thorold, ON, USA)
according to the manufacturer’s instructions. The RNA samples were
analyzed for OD260, OD280 and OD230 using an ultraviolet
spectrometer (Applied Biosystems, Foster City, CA, USA) to
determine RNA purity (OD260/280>1.8 and OD260/230>1.5).
Formaldehyde denaturing agarose gel electrophoresis was employed to
determine RNA integrity through examination of the 28S to 18S ratio
in the RNA samples.
Reverse transcription quantitative polymerase chain
reaction (RT-qPCR) analysis was performed to determine the relative
expression levels of CCR7+ gene transcripts in the COPD
and control tissues. For qPCR analysis, the 7900HT Real-Time PCR
detection system (Applied Biosystems) was utilized. To generate
cDNA for qPCR analysis, SuperScript® III Reverse
Transcriptase (Invitrogen, Carlsbad, CA, USA) was employed. To
quantify the final cDNA PCR products, SYBR® Green PCR
Master Mix (Life Technologies, Grand Island, NY, USA) was used. The
conditions for the PCR reactions were as follows: 50°C for 2 min,
95°C for 2 min, followed by 40 cycles of 95°C for 15 sec, 60°C for
30 sec and 72°C for 30 sec. During the 72°C stage, analysis of the
SYBR fluorophore for quantification was conducted. The relative
expression levels of CCR7 mRNA were calculated by normalization of
these levels to GAPDH mRNA levels using the comparative threshold
cycle (ct) method, in which fold difference = 2-(Δct of target
gene-Δct of reference). The mRNA amplification primers were as
follows: CCR7+, 5′-GGTGGTGGCTCTCCTTGT CATT-3′ and
5′-GCTTTAAAGTTCCGCACGTCCTT-3′; CD83+,
5′-GCTGGAAATGCTGGGCTGA-3′ and 5′-CAT GCAACAGCCTTGTGGTTTAC-3′;
CD1a+, 5′-CAG GGACATGGGAGCATTG-3′ and 5′-AACAAGTCTGAT
GTGGCATTGAA-3′; GAPDH, 5′-CGGTATTTGGTC TATTGGGC-3′ and
5′-TGGAAGATGGTGATGGGATTTC-3′.
Immunohistochemical detection of mature
DCs in COPD tissues
A two-step indirect immunohistochemical method
involving unlabeled primary antibodies and labeled secondary
antibodies was used to determine the presence of the
CD83+, CD1a+ and CCR7+ cell
markers in all paraffin-embedded lung tissue samples. The
antibodies used were as follows: FITC-conjugated mouse monoclonal
antibodies to human to CD3, CD20 and CD14 (#340546; 1:100; BD
Biosciences, Franklin Lanes, NJ, USA), FITC mouse monoclonal
antibodies to human CD80 (#555683; 1:100; BD Biosciences), FITC
mouse monoclonal antibodies to human CD86 (#555657; 1:100; BD
Biosciences), FITC mouse monoclonal antibodies to human monoclonal
antibodies to HLA-DR (#555811; 1:100; BD Biosciences),
FITC-conjugated mouse monoclonal antibody to human CD83 (#MAB1774;
1:200; R&D Systems Inc., Minneapolis, MN, USA), FITC-conjugated
mouse monoclonal antibody to human CCR7 (#MAB197-100; 1:50; R&D
Systems Inc.) and FITC-conjugated mouse monoclonal antibody to
human CD1a (#MAB7076; 1:50; R&D Systems Inc.).
Positive-staining cells were assessed using a minimum of five
images from each slide. All images were captured using an IPWIN32
catch system (Life Technologies).
Myeloid DC isolation from lung
tissue
BDCA+ cells were purified from peripheral
blood monocytes (PMCs) using a commercially available isolation kit
(Miltenyi Biotec, Bergisch Gladbach, Germany). Briefly, PMCs were
first depleted of T cells, monocytes/macrophages and natural killer
(NK) cells using anti-CD3, -CD11b and -CD16 beads. Subsequently,
this depleted population was incubated with anti-CD4 beads, thus
only the CD4+ cells were retained. Since all
BDCA+ cells express CD4, this method allows the
pre-enrichment of BDCA+ cells and prevents contamination
with lymphocytes, monocytes/macrophages and NK cells.
Flow-cytometric analysis of DCs
For cell surface staining, 200 μl aliquots of
bronchial tissue brushings were added to the labeled
fluorescence-activated cell sorting tubes. To decrease nonspecific
binding, 20 μl normal human immunoglobulin (60 g/l;
Intragam®; Commonwealth Serum Laboratories, Sydney,
Australia) was added to each tube for 20 min at room temperature.
After additional incubation for 20 min in the dark, with directly
conjugated monoclonal antibodies to surface markers of interest,
cells were washed with 0.5% bovine serum albumin in Isoton II
(Beckman Coulter, Hialeah, FL, USA; hereafter referred to as wash
buffer), centrifuged at 1,500g for 90 sec and the supernatant was
discarded. A total of 20 ml wash buffer was added and events were
acquired immediately with a FACSCalibur flow cytometer (BD
Biosciences) and analyzed with FlowJo software (FlowJo LLC,
Ashland, OR, USA). A total of 10,000 events were collected from
bronchial brushings.
Statistical analysis
Statistical analysis was conducted using SPSS 17.0
(SPSS, Inc., Chicago, IL, USA). When evaluating differences in
continuous variables among multiple independent groups, the one-way
analysis of variance test was used. P<0.05 was considered to
indicate a statistically significant difference.
Results
Subject characteristics
Evident chronic bronchitis and emphysema
pathological changes were observed in the lung tissues of patients
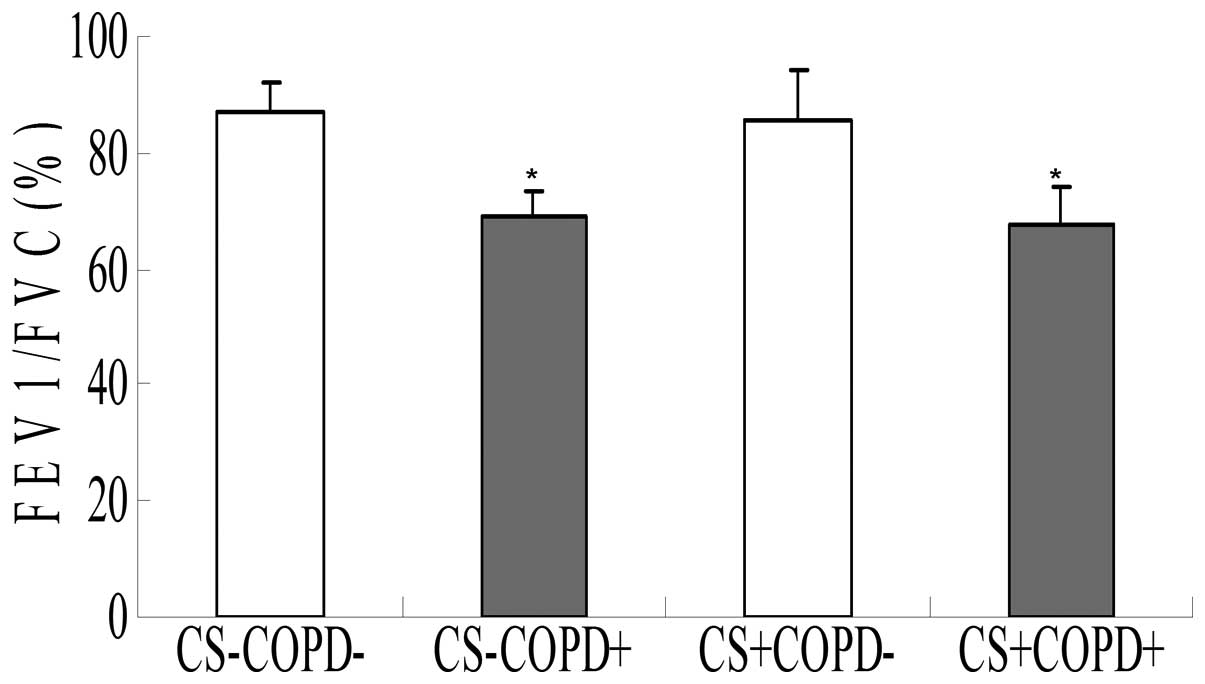
with COPD (Fig. 1). Clinical lung
function data from the patients are presented in Fig. 2. As expected from the selection
criteria, the forced expiratory volume 1/forced vital capacity
ratio was significantly lower in the patients with COPD, as
compared with the non-smokers and the asymptomatic smokers
(P<0.05). All patients, with the exception of the non-smokers
group, were current smokers. No significant differences in age,
weight and height among the patients were identified. Furthermore,
no substantial differences in smoking history pack-years between
the asymptomatic smokers without COPD and those with COPD were
detected (P>0.05). The non-smokers group contained approximately
equal numbers of males and females but the asymptomatic smokers and
the patients with COPD were predominantly males.
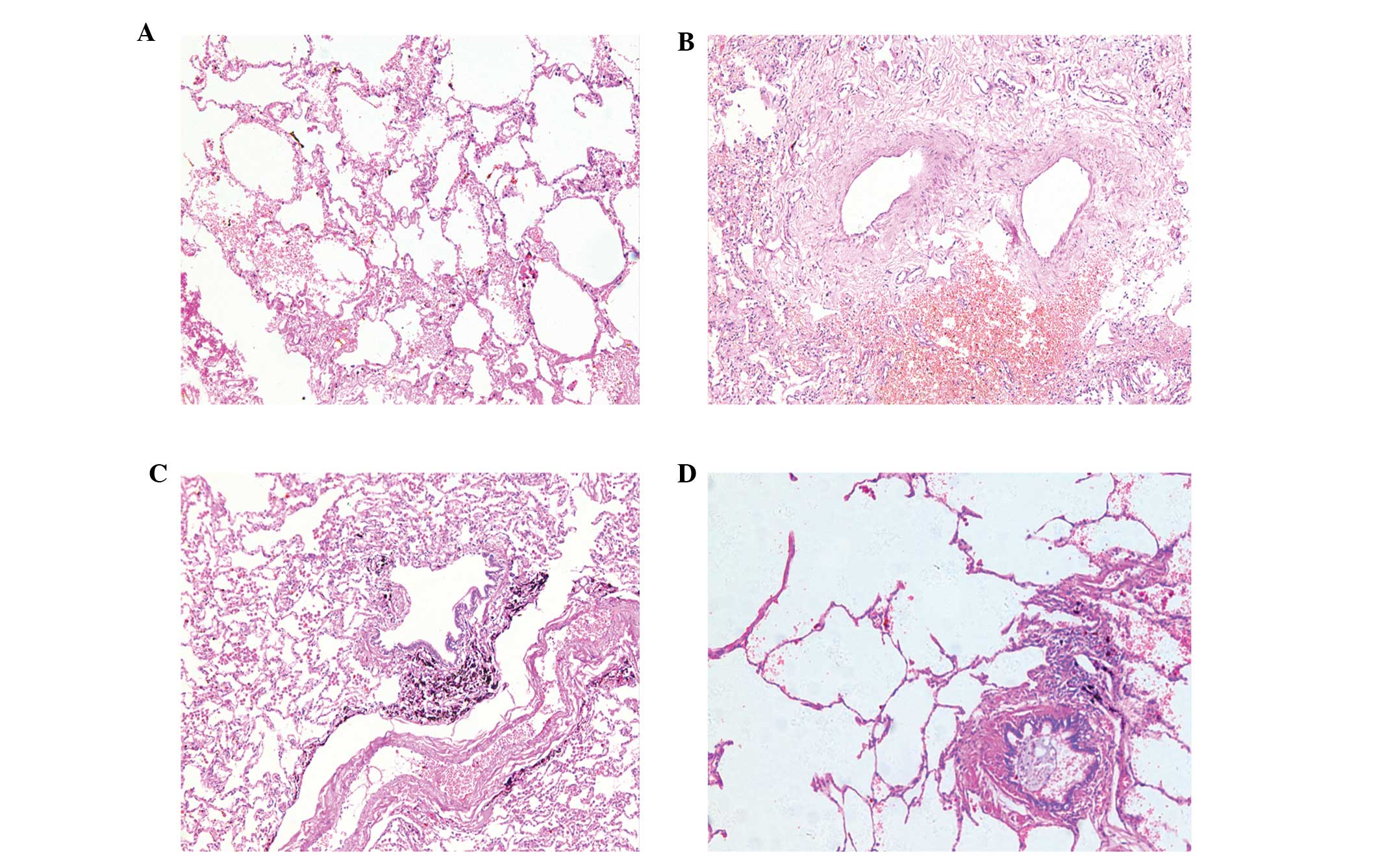 | Figure 1Lung tissue hematoxylin and eosin
staining of different groups of patients (magnification, ×200). (A)
CS−COPD−, a transverse section of a small
airway of normal appearance with a patent lumen and a relatively
thin airway wall with numerous surrounding alveolar attachments.
(B) CS−COPD+, chronic obstructive
bronchiolitis with thickening of the airway wall and infiltration
with lymphocytes, macrophages and neutrophils. (C)
CS+COPD−, black dust accumulated in the small
airway, heavily infiltration with lymphocytes but without narrowing
of the airway wall. (D) CS+COPD+, patients
with peribronchiolar destruction of alveolar walls, resulting in
the loss of alveolar attachments, airway collapse and enlargement
of the air spaces distal to the terminal bronchioles. CS, cigarette
smoker; COPD, chronic obstructive pulmonary disease;
CS−COPD−, non-smoker without COPD;
CS−COPD+, non-smoker with COPD;
CS+COPD−, cigarette smoker without COPD;
CS+COPD+, cigarette smoker with COPD. |
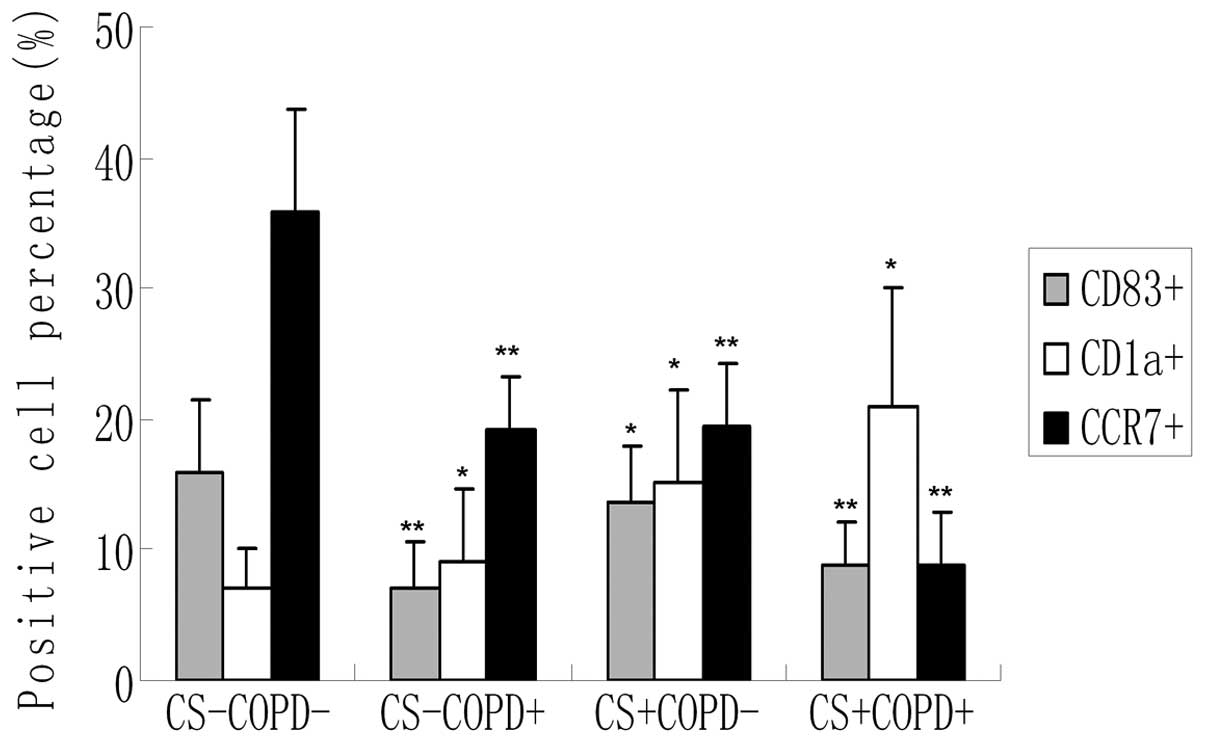
Reduced numbers of mature
CD83+ and CCR7+ DCs, and increased numbers of
immature CD1a+ DCs in COPD patients
Immunohistochemical analysis of CD83+,
CD1a+ and CCR7+ DCs revealed that a greater
number of CD1a+ DCs were detected in virtually all
smoker and patients with COPD, whereas CD83+ and
CCR7+ were specific for the control, asymptomatic
non-smoker group (Fig. 3). The
results of the cell number count (Fig.
4) demonstrated that the numbers of CD83+ and
CCR7+ DCs were significantly reduced, but the numbers of
CD1a+ DCs were significantly increased in the COPD and
smoker groups as compared with the control group (P<0.05).
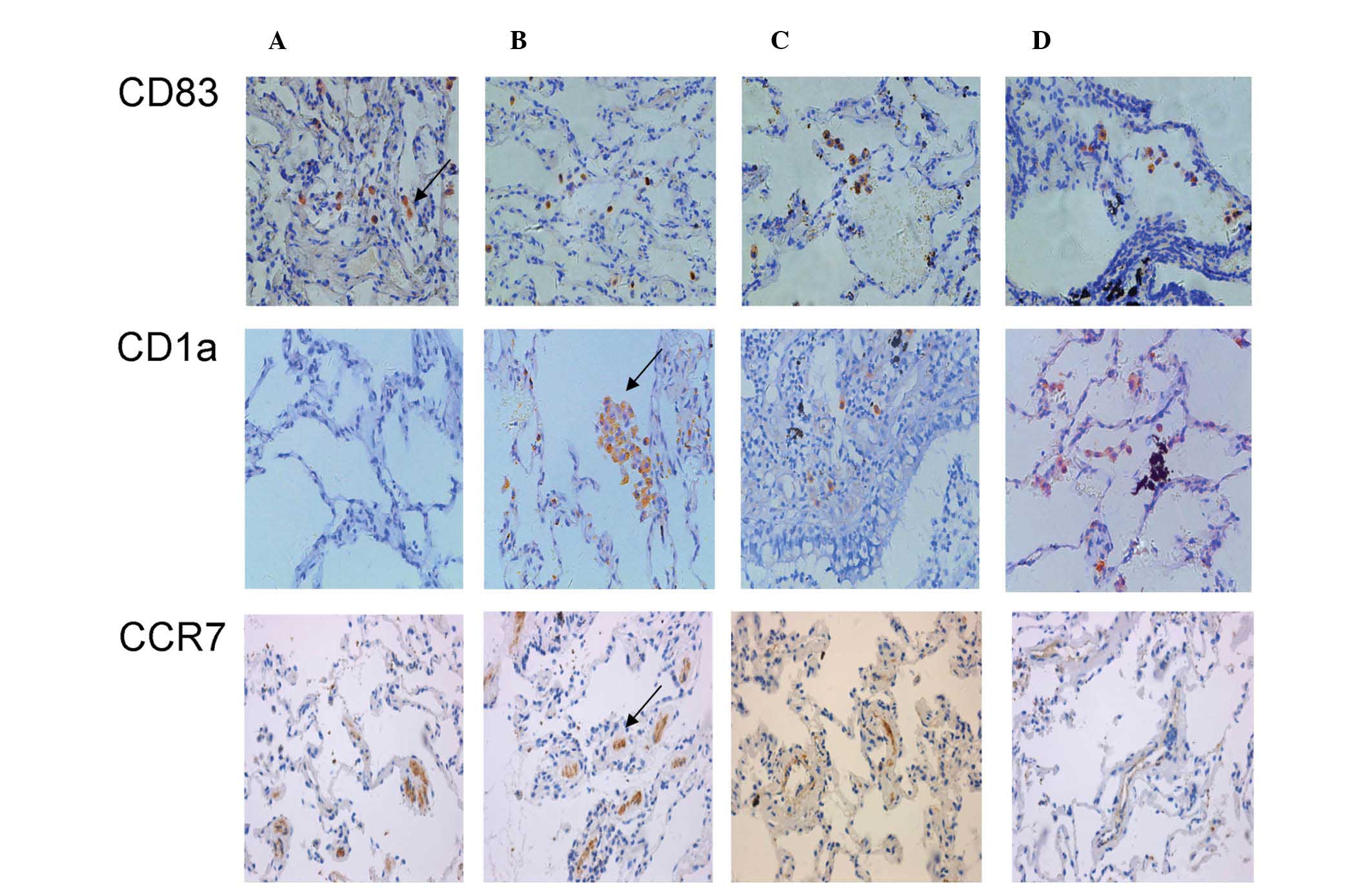 | Figure 3Immunohistochemical analysis of
CD83+, CD1a+ and CCR7+ cells in
the following groups: (A) CS−COPD−; (B)
CS−COPD+; (C) CS+COPD−
and (D) CS+COPD+. Magnification, ×400. Arrows
indicate CD83+, CD1A+ and CCR7+
stained cells. CD, cluster of differentiation; CCR, chemokine
receptor; CS, cigarette smoker; COPD, chronic obstructive pulmonary
disease; CS−COPD−, non-smoker without COPD;
CS−COPD+, non-smoker with COPD;
CS+COPD−, cigarette smoker without COPD;
CS+COPD+, cigarette smoker with COPD. |
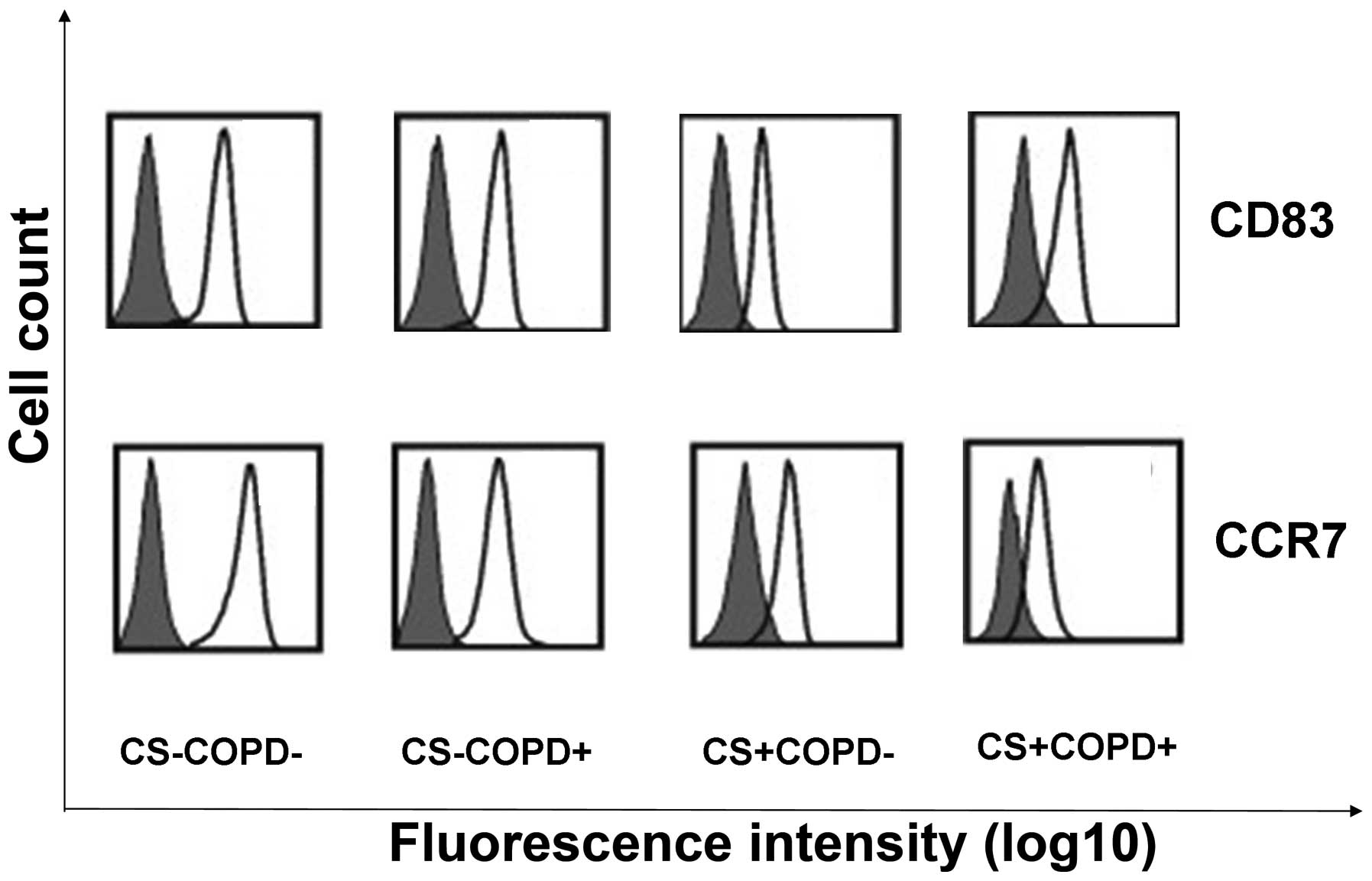
Reduced CD83+ and
CCR7+ expression during lung myeloid DC maturation in
COPD
The levels of cell surface CCR7 and CD83 expression
during myeloid DCs maturation in the four groups was analyzed.
Representative histograms for each group are shown in Fig. 5. The CD83 and CCR7 myeloid DC
expression levels were reduced as compared with the control group,
as detected by flow cytometric analysis.
Reduced CD83+ and
CCR7+, and increased CD1a+ expression levels
in lung tissues from COPD patients
The expression levels of CD83+ and
CCR7+ mRNA transcripts were significantly lower, and
CD1a+ expression levels were significantly higher
(P<0.05) in COPD lung tissues as compared with lung tissues from
non-smokers and smokers without COPD (Fig. 6).
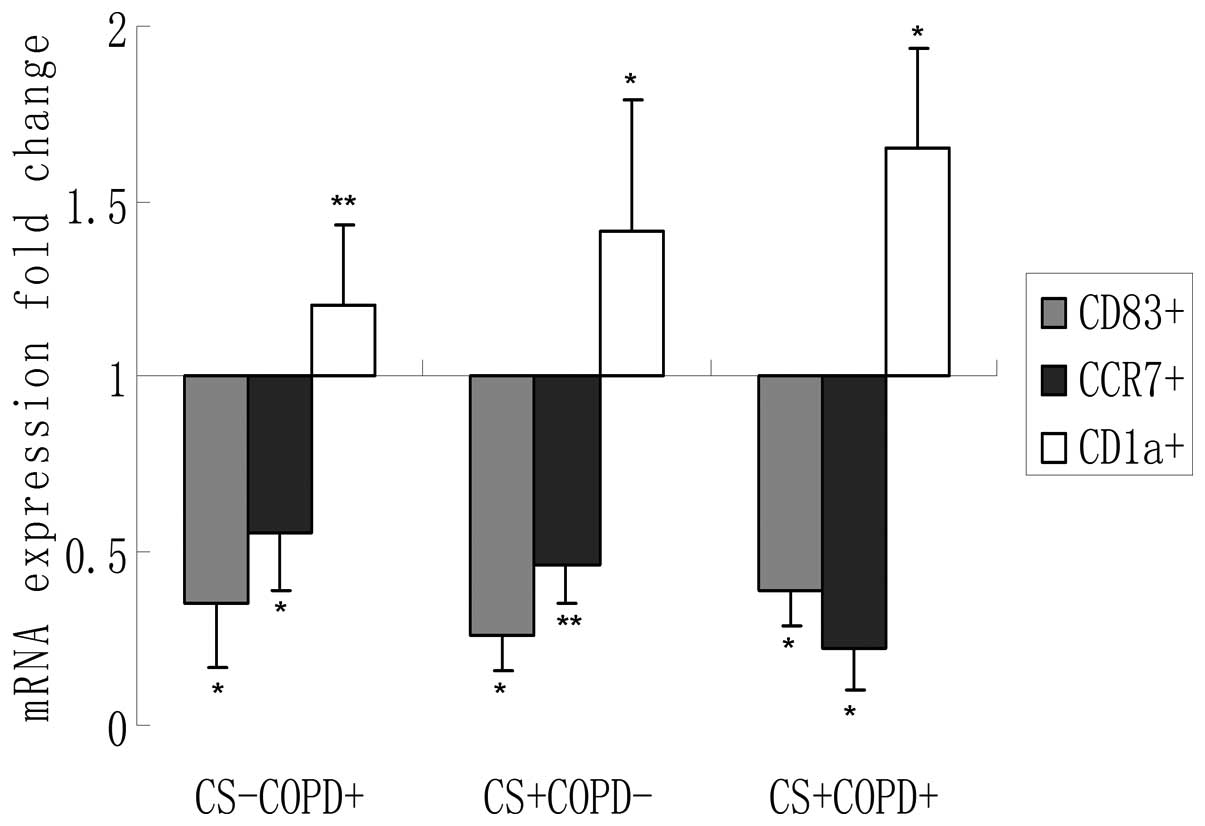 | Figure 6CD83+, CD1a+
and CCR7+ expression levels in lung tissues from the
different groups. The results are presented as the mean ± SD, n=8,
*P<0.05, **P<0.01, as compared with the
control for each marker at a baseline of 1. CD, cluster of
differentiation; CCR, chemokine receptor; CS, cigarette smoker;
COPD, chronic obstructive pulmonary disease;
CS−COPD−, non-smoker without COPD;
CS−COPD+, non-smoker with COPD;
CS+COPD−, cigarette smoker without COPD;
CS+COPD+, cigarette smoker with COPD. |
Discussion
In the present study, the results verified the
hypothesis that chronic exposure to CS impairs the normal DC
maturation process, and subsequently alters or suppresses normal DC
function and interaction with naive lymphocytes, resulting in an
imbalance of immunity that may increase the susceptibility of
patients with COPD to respiratory infections. A number of markers
that have been associated with the maturation of DCs were analyzed.
Since active smoking may reduce the numbers of DCs, the data from
patients with COPD, from smokers without COPD and from non-smokers
were compared.
Traditionally, DCs have been described as key cells
in linking the innate and adaptive immune responses, which are both
involved in chronic inflammation in COPD (13). The hypothesis that DCs are involved
in the development of COPD in smokers has been determined by
previous studies. For example, evidence in humans demonstrates that
CS induces the recruitment of a large numbers of immature DCs into
the small airways of patients with COPD (14). DCs also are ideally localized to
initiate an inflammatory reaction in response to inhaled CS.
However, few studies concerning the potential of DC involvement in
the pathogenesis of COPD have been published. In patients with
COPD, significantly increased numbers of small airway
langerin-expressing DCs in the bronchoalveolar lavage fluid (BALF)
of smokers have been observed (15). An immunohistochemical study
demonstrated an increase in the numbers of Langerhans cells in the
airways of smokers with COPD, as compared with smokers without COPD
and non-smokers (16). Further
studies revealed that the numbers of mature DCs were reduced in
patients with COPD (17,18). Active smoking in patients with COPD
is associated with reduced numbers of bronchial mucosal DCs that
express the CD83+ maturation marker (19).
The present study quantified airway DC cells in
groups of smokers and non-smokers, with and without COPD. The
numbers of CD83+ DCs were reduced but the numbers of
CD1a+ DCs were increased in individuals with COPD and
those who smoked cigarettes. An early study indicated that the
numbers of CD1a+ immature DCs are increased in the
alveoli and in the BALF of smokers (20). However, normally relatively few
CD1a+ DCs are detected in human alveoli and these cells
comprise <1% BAL cells. Soler et al (21) reported no differences in the
numbers of CD1a+ DCs in the bronchial epithelium between
smokers and non-smokers. In concurrence with this finding, another
study observed no difference in the numbers of pulmonary
langerin-positive immature DCs in small airways between healthy
smokers and non-smokers, or between smokers with COPD and
ex-smokers (22). By contrast,
during analysis of cells in the large airways, a recent study
identified mucosal DCs by their ultra-structure in endobronchial
biopsies of smokers and ex-smokers with COPD, and demonstrated
markedly reduced numbers in those who continued to smoke (16). Furthermore, sputum data have
indicated that the numbers of mature CD83+ and
DC-lysosome-associated membrane glycoprotein 1 (LAMP1) DCs, and the
ratios of mature CD83++ and mature DC-LAMP1 DCs to total
DCs are reduced in current smokers as compared with healthy
subjects (23). The reduction in
the numbers of mature DCs appears to be associated with smoking
status, as a comparable reduction in the number of
immunohistologically detected CD83++ mature bronchial
mucosal DCs has recently been reported in large airways of smokers
with asthma, as compared with non-smokers with asthma (24).
In the present study, to further investigate whether
the increase in the number of mature DCs in the airways of patients
with COPD may be explained by an increase in the numbers of
CCR7+ cells, CCR7+ expression in the human
lung at the mRNA level was determined, and the CCR7+
expression levels among non-smokers, smokers without COPD and
patients with COPD were compared. The data suggest that CS may
stimulate these local immune responses by impairing airway DC
homing to the lymph nodes, thus promoting local antigen
presentation within the airway wall. Pulmonary DC migration to the
draining lymph nodes is induced by antigen capture and is
characterized by the downregulation of DC antigen capture capacity
and the upregulation of DC lymph node homing receptors,
predominantly CCR7+ (25,26).
A consistent and specific association has been detected between
reduced CCR7+ expression levels in myeloid DCs, and
airflow limitation and pulmonary hyperinflation in smokers
(27). The possible underlying
mechanism that links reduced myeloid DC CCR7+ expression
levels and airway obstruction may be that impaired homing of
myeloid DCs to the lymph nodes results in the accumulation of
myeloid DCs in the airways. This accumulation may stimulate local
adaptive immune responses, which induce airway remodeling and
obstruction. Notably, in the presence of pathogen- and
damage-associated molecular patterns, DC migration is accompanied
by full DC maturation, a differentiation process characterized by
an increase in various cell surface and intracellular molecule
expression levels (28,29). Therefore, excessive local adaptive
immune responses are key elements in the pathogenesis of COPD. In
addition, a previous study reported that CS extracts suppress
maturation-associated CCR7+ expression in human myeloid
DCs in vitro (30).
Therefore, due to the essential role of CCR7+ in the
migration of myeloid DCs to draining lymph nodes, CS may reduce the
migratory potential of myeloid DCs.
The predominant concern is the definition of which
cells detected in the lungs constitute DCs. In mice, pulmonary DCs
include CD11c+ major histocompatibility complex II (MHC
II)+ conventional DCs and CD11c+
plasmatocytoid DCs (31). Human
lung DCs comprise three subsets: Myeloid DC type 1
(BDCA1+/MHC II+), myeloid DC type 2
(BDCA3+/MHC II+) and plasmacytoid DC
(BDCA2+/CD123+) (32). The expression of CCR7 or CD83 does
not define a cell as a DC. For example, CCR7 may also be expressed
by T cells and certain lung cancer cells. In the present study, to
validate the results, myeloid DCs were collected from the lung
tissue, and CCR7+ and CD83+ cells were
detected through flow cytometry.
Apparently conflicting results are presented in the
sparse human literature concerning methodological issues of DC
detection, and this may be due to assessment of distinct
DC-specific markers, sampling of different anatomic sites, and
distinguishing whether the findings are due to smoking status or
the disease process itself (33).
As an example of the first issue, langerin and CD1a+ are
considered to be markers of an immature myeloid DC phenotype,
whereas CD83+ and DC-LAMP are considered markers of
mature DCs. In the present study, DC-specific markers in lung
tissues samples and the common corresponding CCR7+
receptors were selected to improve the study.
As mentioned above, analysis of numerous cell
surface markers increased the validity of the present study.
However, certain limitations to the present study require further
consideration. Although reduced myeloid DC numbers were observed in
the patients with COPD, as well as lower CCR7+ levels,
demonstrating the causal role of DCs in the pathogenesis of COPD is
not possible with this approach. For this purpose, in vivo
animal experiments are required to investigate the effect of
overexpression or knockout of DC function on CS-induced
inflammation. Such animal models are also required to elucidate the
underlying mechanism responsible for the accumulation of DCs in the
airways.
In addition, the presence of cancer may have
influenced DC infiltration in the small airways, resulting in an
enhancement of inflammatory cell recruitment (34). However, primary bronchus carcinoma
was the main reason for surgery in all groups, which minimized the
risk of confounding factors among groups. Furthermore, recent data
have revealed a suppression of DC accumulation in lung cancer,
rather than an increase (35).
Most importantly, in the present study, the tissue samples removed
for analysis were obtained at a distance from the primary
pathological lung tissues.
In conclusion, these data indicate that smoking
affects the expression profile of function-associated surface
molecules on airway myeloid DCs. The involvement of DCs in the
pathogenesis of COPD is becoming recognized. The present study
provides evidence that reduced CCR7+ expression levels
on airway myeloid DCs may be associated with airflow limitation in
smokers. Future advances in the understanding of pulmonary DCs,
combined with recent advances in the pharmaceutical manipulation of
DC function, may aid in the identification of novel therapeutic
methods with which to prevent or treat COPD more effectively.
Acknowledgements
The authors would like to thank Dr Gang Xu from the
Department of Cardiothoracic Surgery, Dr Shuang-ming Xu, Yong Zhao,
Xiao-ming Zhao, You-jing Cheng and Li-na Wang from the Department
of Respiration for sample collection and Dr Shang-fu Xu from the
Department of Pharmacology for suggesting employing RT-PCR
analysis. Support was provided by Guizhou Province Programs for
Science and Technology Development.
References
|
1
|
Kim V and Criner GJ: Chronic bronchitis
and chronic obstructive pulmonary disease. Am J Respir Crit Care
Med. 187:228–237. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baraldo S, Turato G and Saetta M:
Pathophysiology of the small airways in chronic obstructive
pulmonary disease. Respiration. 84:89–97. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vassallo R, Walters PR, Lamont J, et al:
Cigarette smoke promotes dendritic cell accumulation in COPD; a
Lung Tissue Research Consortium study. Respir Res. 11:452010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mortaz E, Kraneveld AD, Smit JJ, Kool M,
Lambrecht BN, et al: Effect of cigarette smoke extract on dendritic
cells and their impact on T-cell proliferation. PLoS One.
3:e49462009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Idoyaga J and Steinman RM: SnapShot:
Dendritic Cells. Cell. 146:660–660.e2. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu K, Victora GD, Schwickert TA,
Guermonprez P, Meredith MM, Yao K, et al: In vivo analysis of
dendritic cell development and homeostasis. Science. 324:392–397.
2009.PubMed/NCBI
|
|
7
|
Steinman RM: Decisions about Dendritic
Cells: Past, Present, and Future. Ann Rev Immunol. 30:1–22. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jang MH, Sougawa N, Tanaka T, Hirata T,
Hiroi T, Tohya K, et al: CCR7+ is critically important for
migration of dendritic cells in intestinal lamina propria to
mesenteric lymph nodes. J Immunol. 176:803–810. 2006. View Article : Google Scholar
|
|
9
|
Bouchon A, Hernández-Munain C, Cella M and
Colonna M: A DAP12-mediated pathway regulates expression of CC
chemokine receptor 7 and maturation of human dendritic cells. J Exp
Med. 194:1111–1122. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Demoor T, Bracke KR, Vermaelen KY, Dupont
L, Joos GF and Brusselle GG: CCR7+ modulates pulmonary and lymph
node inflammatory responses in cigarette smoke-exposed mice. J
Immunol. 183:8186–8194. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Demedts IK, Bracke KR, Van Pottelberge G,
Testelmans D, Verleden GM, Vermassen FE, et al: Accumulation of
dendritic cells and increased CCL20 levels in the airways of
patients with chronic obstructive pulmonary disease. Am J Respir
Crit Care Med. 175:998–1005. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vestbo J, Hurd SS, Agustí AG, Jones PW,
Vogelmeier C, Anzueto A, et al: Global strategy for the diagnosis,
management, and prevention of chronic obstructive pulmonary
disease: GOLD executive summary. Am J Respir Crit Care Med.
187:347–365. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Condon TV, Sawyer RT, Fenton MJ and Riches
DW: Lung dendritic cells at the innate-adaptive immune interface. J
Leukoc Biol. 90:883–895. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tsoumakidou M, Demedts IK, Brusselle GG
and Jeffery PK: Dendritic Cells in Chronic Obstructive Pulmonary
Disease: new players in an old game. Am J Respir Crit Care Med.
177:1180–1186. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lommatzsch M, Bratke K, Knappe T, Bier A,
Dreschler K, Kuepper M, et al: Acute effects of tobacco smoke on
human airway dendritic cells in vivo. Eur Respir J. 35:1130–1136.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rogers AV, Ädelroth E, Hattotuwa K, Dewar
A and Jeffery PK: Bronchial mucosal dendritic cells in smokers and
ex-smokers with COPD: an electron microscopic study. Thorax.
63:108–114. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Galgani M, Fabozzi I, Perna F, Bruzzese D,
Bellofiore B, Calabrese C, et al: Imbalance of circulating
dendritic cell subsets in chronic obstructive pulmonary disease.
Clin Immunol. 137:102–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Su YW, Xu YJ and Liu XS: Quantitative
differentiation of dendritic cells in lung tissues of smokers with
and without chronic obstructive pulmonary disease. Chin Med J
(Engl). 123:1500–1504. 2010.PubMed/NCBI
|
|
19
|
Tsoumakidou M, Koutsopoulos AV, Tzanakis
N, Dambaki K, Tzortzaki E, Zakynthinos S, et al: Decreased small
airway and alveolar CD83+ dendritic cells in COPD. Chest.
136:726–733. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Robbins CS, Dawe DE, Goncharova SI,
Pouladi MA, Drannik AG, Swirski FK, et al: Cigarette smoke
decreases pulmonary dendritic cells and impacts antiviral immune
responsiveness. Am J Respir Cell Mol Biol. 30:202–211. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Soler P, Moreau A, Basset F and Hance AJ:
Cigarette smoking-induced changes in the number and differentiated
state of pulmonary dendritic cells/Langerhans cells. Am Rev Respir
Dis. 139:1112–1117. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Souto GR, Segundo TK, Costa FO, Aguiar MC
and Mesquita RA: Effect of smoking on Langerhans and dendritic
cells in patients with chronic gingivitis. J Periodontol.
82:619–625. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tsoumakidou M, Bouloukaki I, Koutala H,
Kouvidi K, Mitrouska I, Zakynthinos S, et al: Decreased sputum
mature dendritic cells in healthy smokers and patients with chronic
obstructive pulmonary disease. Int Arch Allergy Immunol.
150:389–397. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tsoumakidou M, Elston W, Zhu J, Wang Z,
Gamble E, Siafakas NM, et al: Cigarette smoking alters bronchial
mucosal immunity in asthma. Am J Respir Crit Care Med. 175:919–925.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ohl L, Mohaupt M, Czeloth N, Hintzen G,
Kiafard Z, Zwirner J, Blankenstein T, Henning G and Förster R:
CCR7+ governs skin dendritic cell migration under inflammatory and
steady-state conditions. Immunity. 21:279–288. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nickel T, Pfeiler S, Summo C, Kopp R,
Meimarakis G, Sicic Z, Lambert M, Lackermair K, David R,
Beiras-Fernandez A, et al: oxLDL downregulates the dendritic cell
homing factors CCR7 and CCL21. Mediators Inflamm. 2012:3209532012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Demoor T, Bracke KR, Joos GF and Brusselle
GG: Increased T-regulatory cells in lungs and draining lymph nodes
in a murine model of COPD. Eur Respir J. 35:688–689. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Givi ME, Redegeld FA, Folkerts G and
Mortaz E: Dendritic cells in pathogenesis of COPD. Curr Pharm Des.
18:2329–2335. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Boislève F, Kerdine-Römer S,
Rougier-Larzat N and Pallardy M: Nickel and DNCB Induce CCR7
Expression on Human Dendritic Cells Through Different Signalling
Pathways: Role of TNF-alpha and MAPK. J Invest Dermatol.
123:494–502. 2004.PubMed/NCBI
|
|
30
|
Marcenaro E, Cantoni C, Pesce S, Prato C,
Pende D, Agaugué S, Moretta L and Moretta A: Uptake of CCR7+ and
acquisition of migratory properties by human KIR+ NK cells
interacting with monocyte-derived DC or EBV cell lines: regulation
by KIR/HLA-class I interaction. Blood. 114:4108–4116. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kirby AC, Raynes JG and Kaye PM: CD11b
regulates recruitment of alveolar macrophages but not pulmonary
dendritic cells after pneumococcal challenge. J Infect Dis.
193:205–213. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Demedts IK, Brusselle GG, Vermaelen KY and
Pauwels RA: Identification and characterization of human pulmonary
dendritic cells. Am J Respir Cell Mol Biol. 32:177–184. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bratke K, Klug M, Bier A, Julius P,
Kuepper M, Virchow JC and Lommatzsch M: Function-associated surface
molecules on airway dendritic cells in cigarette smokers. Am J
Respir Cell Mol Biol. 38:655–660. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kurabayashi A, Furihata M, Matsumoto M,
Hayashi H and Ohtsuki Y: Distribution of tumor-infiltrating
dendritic cells in human non-small cell lung carcinoma in relation
to apoptosis. Pathol Int. 54:302–311. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Almand B, Resser JR, Lindman B, Nadaf S,
Clark JI, Kwon ED, Carbone DP and Gabrilovich DI: Clinical
significance of defective dendritic cell differentiation in cancer.
Clin Cancer Res. 6:1755–1766. 2000.PubMed/NCBI
|