Introduction
Retinitis pigmentosa (RP) is the most common form of
progressive hereditary retinal degeneration, with a worldwide
prevalence of ~1 in 4,000 (1,2). To
date, mutations in at least 61 genes have been reported to cause RP
(RetNet, https://sph.uth.edu/Retnet/).
However, mutations in these genes contribute to only half of the
clinical cases (3). Therefore,
identification of additional genes responsible for RP is important
to determine the molecular basis of RP and aid in the development
of novel therapeutic strategies. Genes known to cause other forms
of hereditary retinal degeneration may be good candidates for genes
causing RP.
Leber congenital amaurosis (LCA) is the most severe
form of hereditary retinal degeneration, with ~20 causative genes
identified. Our previous study has shown that about half of the
variants were detected in nine frequently mutated exons (4). Mutations in eight of the 20
LCA-associated genes have been reported to cause RP as well
(4–7). However, systemic evaluation of
LCA-associated genes in patients with RP is limited (8–10),
particularly for those 12 of the 20 genes in which a mutation has
not been identified in patients with RP. The 12 genes known to
cause LCA but not RP are as follows: Aryl hydrocarbon interacting
protein-like 1 (AIPL1) (11), calcium-binding protein 4
(CABP4) (12), centrosomal
protein 290 kDa (CEP290) (13), death domain containing 1
(DTHD1) (14), guanylate
cyclase 2D, membrane (retina-specific; GUCY2D) (15), IQ motif-containing protein B1
(IQCB1) (16), potassium
inwardly-rectifying channel, subfamily J, member 13 (KCNJ13)
(17), Leber congenital amaurosis
5 (LCA5) (18),
nicotinamide nucleotide adenylyltransferase 1 (NMNAT1)
(19), orthodenticle homeobox 2
(OTX2) (20), retinal
degeneration 3 (RD3) (21)
and retinitis pigmentosa GTPase regulator interacting protein 1
(RPGRIP1) (22).
In the present study, variations in LCA-associated
genes were evaluated in a cohort of patients with RP using two
methods: i) The most commonly mutated nine exons were analyzed by
Sanger sequencing in 293 patients with RP; and ii) for the 12 genes
known to associate with LCA but not RP, variants that resulted from
exome sequencing in 157 of the 293 patients with RP were selected
and then further confirmed by Sanger sequencing. Mutations in four
patients with RP were identified in LCA-associated genes.
Subjects and methods
Subjects
Probands with a clinical diagnosis of RP from 293
unrelated families were recruited from the Pediatric and Genetic
Eye Clinic, Zhongshan Ophthalmic Center (Guangzhou, China) since
1996. The diagnosis of RP was based on phenotypes described in a
previous study (23). Written
informed consent from each participant or their guardians was
obtained prior to collection of their clinical data and venous
blood samples. Genomic DNA was prepared from leukocytes in venous
blood as previously described (24). The present study was approved by
the Institutional Review Board of the Zhongshan Ophthalmic
Center.
Analysis of the nine frequently mutated
exons
The most frequently mutated nine exons were selected
based on our previous study (4),
as listed in Table I. The genomic
fragments of the nine exons were amplified by polymerase chain
reaction, using primers encompassing each of the nine exons and the
adjacent intronic regions (Table
II). Touchdown PCR amplifications of the genomic fragments,
sequencing and result analysis was processed as previously
described (4).
 | Table IMost frequently mutated nine exons
analyzed. |
Table I
Most frequently mutated nine exons
analyzed.
| Gene | OMIM | Map location | Number of coding
exons | Targeted
exon(s) | Frequency for
mutant alleles in Chinese LCA patients (4), % |
|---|
| GUCY2D | 600179 | 17p13.1 | 18 | 2 | 9.00 |
| | | | 11 | 3.85 |
| | | | 12 | 3.85 |
| CRB1 | 604210 | 1q31-q32.1 | 12 | 6 | 7.70 |
| | | | 9 | 5.14 |
| | | | 11 | 6.41 |
| RPE65 | 180069 | 1p31 | 14 | 4 | 5.14 |
| RPGRIP1 | 605446 | 14q11 | 24 | 3 | 5.14 |
| CEP290 | 610142 | 12q21.32 | 53 | 6 | 3.85 |
| Table IIPrimers used for the most frequently
mutated nine exons. |
Table II
Primers used for the most frequently
mutated nine exons.
| Primer name | Primer sequence,
5′-3′ | Size of amplicon,
bp |
|---|
| GUCY2D-E2a-FW |
ccttggccccagttagtctt | 552 |
| GUCY2D-E2a-RV |
gttcaccggacccacgag | |
| GUCY2D-E2b-FW |
gtccccgcttcgaggtag | 552 |
| GUCY2D-E2b-RV |
accgagtgcatcaccatga | |
| GUCY2D-E11-FW |
gatagttgcagggctggtct | 397 |
| GUCY2D-E11-RV |
gtttcatcactgggctttgc | |
| GUCY2D-E12-FW |
tgaacctctgatgtaaagaaacc | 398 |
| GUCY2D-E12-RV |
gtagcctggaaggccagag | |
| CRB1-E6a-FW |
ttcatgcacttctgcaagatt | 598 |
| CRB1-E6a-RV |
tgaacagaagcacctttgactg | |
| CRB1-E6b-FW |
cgaagcaacagggatgtgtt | 648 |
| CRB1-E6b-RV |
tttcatagcaggcagaagca | |
| CRB1-E9a-FW |
atgtatcaaatagtcaatatgcaatgt | 598 |
| CRB1-E9a-RV |
gagataaatgcctccgatttc | |
| CRB1-E9b-FW |
tgtgggagacagagctattga | 594 |
| CRB1-E9b-RV |
cttgaggagagagctttccaa | |
| CRB1-E11-FW |
agactgtgctgttccagagaga | 344 |
| CRB1-E11-RV |
ctgttcaccccactcaacaa | |
| RPE65-E4-FW |
ccctttattcttcatgttgtgc | 380 |
| RPE65-E4-RV |
gtcagtaacctctactcctcgaaa | |
| RPGRIP1-E3-FW |
tgtggttaatagatcacggtagatg | 530 |
| RPGRIP1-E3-RV |
gcagaaaggagggagtgaga | |
| CEP290-E6-FW |
gcttgttgttgactcatttgaa | 375 |
| CEP290-E6-RV |
ttggtgatgacaaaatgaaca | |
Variants in 12 genes as determined by
exome sequencing
Whole exome sequencing was performed on 157 of the
293 unrelated patients with RP, using a commercial service from BGI
Shenzhen (Shenzhen, China; http://www.genomics.cn/index) as previously described
(25,26). Mutations in 60 genes responsible
for RP were identified in approximately half of these patients
(27). The variants in the 12
genes known to be associated with LCA but not RP, resulted from
exome sequencing of the 157 patients with RP, were collected for
further analysis. Heterozygous variants for dominant genes and
homozygous or compound heterozygous variants for recessive genes
were selected and verified by Sanger sequencing, using primers to
amplify the individual fragments harboring variants (Table III). The mutation hot spot,
c.2991+1655A>G in CEP290 (13), is at the position beyond the scope
of exome sequencing, thus genomic fragments of CEP290
encompassing c.2991+1655A>G were amplified and analyzed by
Sanger sequencing in all 157 RP patients.
| Table IIIPrimers used for validating variants
from exome sequencing. |
Table III
Primers used for validating variants
from exome sequencing.
| Primer name | Primer sequence,
5′-3′ | Target
variant/sequenced sample |
|---|
| CEP290-E7-FW |
tttgaaaattttggcctattatttatg |
CEP290:c.442-10_11insT/RP397 |
| CEP290-E7-RV |
tccctgagacaaagtcatacca | |
|
CEP290-IVS26+1655-FW |
ggttcaggccgttctcct |
CEP290:c.2991+1655A>G/All
patients |
|
CEP290-IVS26+1655-RV |
agtttttaaggcggggagtc | |
| CEP290-E28-FW |
tccaggtctgatggaattcag |
CEP290:c.3104-2delA/RP276 |
| CEP290-E28-RV |
ttcagagatccagacaaaccac | |
| CEP290-E32-FW |
tttgtcatgtagtttgacaaaagat |
CEP290:c.4040G>A/RP276 |
| CEP290-E32-RV |
cggatcatgaggtcaggaga | |
| CEP290-E49-FW |
agcatttagagccccaggtt |
CEP290:c.6736A>G/RP397 |
| CEP290-E49-RV |
ctgttcatcaggaagaaacca | |
| LCA5-E4-FW |
caagagaaagaacgggcaac |
LCA5:c.634G>T/RP374 |
| LCA5-E4-RV |
atgcccaatgagaaacatcc | |
| LCA5-E9-FW |
ccagagagaagccccaaaac |
LCA5:c.1642C>T/RP374 |
| LCA5-E9-RV |
tggatttgacctctctgatgtt | |
Bioinformatics analysis
In total, two online computational prediction
algorithms, PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT
(http://sift.jcvi.org/), were used to predict the
functional impact of missense mutations identified (28). The PolyPhen-2 website can predict
the functional effect of variants and classify them into ‘probably
damaging’, ‘possibly damaging’, ‘benign’, and ‘unknown’ (29). The SIFT algorithm shows a
normalized probability score of a missense variant: When the
normalized probability is larger than 0.05, the variant is
predicted to be ‘tolerated’, otherwise, the variant is predicted to
be ‘damaging’ (30). The impact of
variants on the splice site was predicted by NNSPLICE version 0.9
(http://fruitfly.org/seq_tools/splice.html) (7). The description of variants referred
to the nomenclature of the Human Genomic Variation Society
(http://www.hgvs.org/mutnomen/). Novel
variants were further evaluated in 192 normal controls.
Results
Sanger sequencing
Analysis of the nine frequently mutated exons
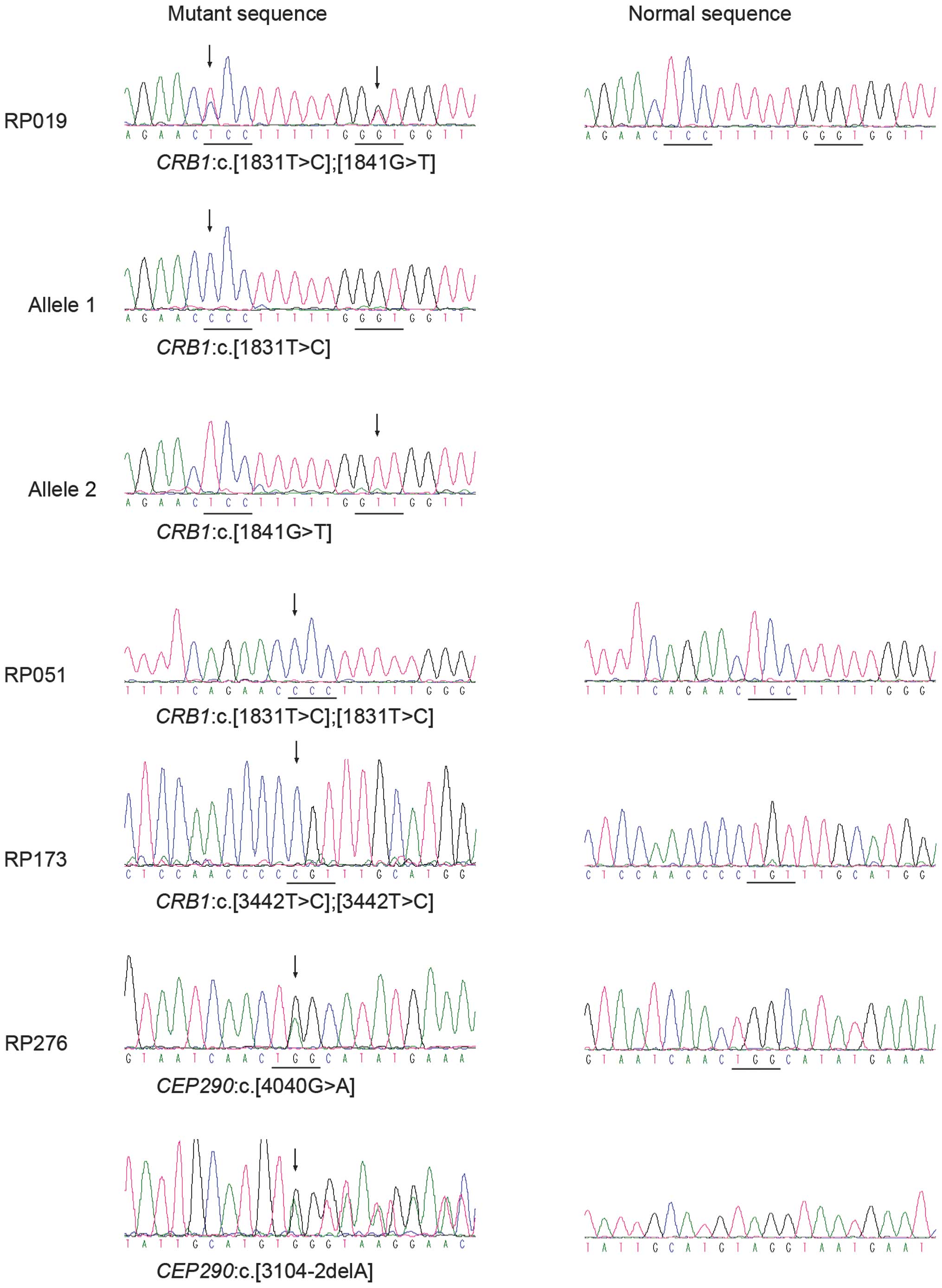
identified potential pathogenic mutations in the CRB1 gene
in three patients, including one known and two novel mutations,
i.e. c.1831T>C (p.S611P), c.1841G>T (p.G614V) and
c.3442T>C (p.C1148R) (Table
IV; Fig. 1). These variants
were not present in the 192 unaffected controls. One patient had
compound heterozygous mutations and the other two had homozygous
mutations (Table V).
| Table IVPotential pathogenic mutations
detected in patients with RP. |
Table IV
Potential pathogenic mutations
detected in patients with RP.
| Gene | Method | Nucleotide
change | Amino acid
change | Bioinformatics | Status/patient
ID | Allele
frequency | Reported |
|---|
|
|
|---|
| P/SS | SIFT | Patients | Controls |
|---|
| CRB1 | Sanger | c.1831T>C | p.S611P | PrD | D | Het/RP019;
Hom/RP051 | 4/586 | 0/384 | Li et al
(4) |
| CRB1 | Sanger | c.1841G>T | p.G614V | PrD | D | Het/RP019 | 1/586 | 0/384 | Novel |
| CRB1 | Sanger | c.3442T>C | p.C1148R | PrD | D | Hom/RP173 | 1/586 | 0/384 | Novel |
| CEP290 | Exome | c.3104-2delA | Splicing
defect | SSA | NA | Het/RP276 | 1/314 | 0/192 | Novel |
| CEP290 | Exome | c.4040G>A | p.W1347* | NA | NA | Het/RP276 | 1/314 | 0/192 | Novel |
| Table VClinical data of the four patients
with mutations. |
Table V
Clinical data of the four patients
with mutations.
| Patient ID | Gene | Variations | Inheritance | Gender | Age (years) | First symptom | Visual acuity (OD;
OS) | Fundus changes | Electroretinography
response |
|---|
|
|
|---|
| Exam | Onset | Rod | Cone |
|---|
| RP019 | CRB1 |
c.[1831T>C];[1841G>T] | AR | Male | 17 | EC | PV, NB | FC; FC | AV, WPD, PD,
MD | NA | NA |
| RP051 | CRB1 |
c.[1831T>C];[1831T>C] | Isolated | Female | 5 | 3 | NB, CB | 0.1; 0.3 | PD, MD | Severely
reduced | Severely
reduced |
| RP173 | CRB1 |
c.[3442T>C];[3442T>C] | Isolated/cons | Female | 29 | EC | PV | HM; HM | AV, PD | NA | NA |
| RP276 | CEP290 |
c.[4040G>A];[3104-2delA] | Isolated | Male | 6 | EC | NB | ND | AV, CRD | Extinguished | Extinguished |
Exome sequencing
Exome sequencing identified six variants in two of
the 12 genes, including four variants in CEP290 and two
variants in LCA5, i.e. c.[442-10_11insT];[6736A>G] in
CEP290 of RP397, c.[4040G>A];[3104-2delA] in
CEP290 of RP276, and c.[1642C>T];[634G>T] in
LCA5 of RP374. The compound heterozygous
c.[4040G>A];[3104-2delA] mutations in CEP290 of RP276
were considered to be potential pathogenic mutations (Tables IV and V, Fig.
1). The other two compound heterozygous variants in
CEP290 and LCA5, respectively, were unlikely
pathogenic since the c.6736A>G (p.K2246E) in CEP290 and
the c.1642C>T (p.P548S) in LCA5 were predicted to be
benign or tolerated by PolyPhen-2 and SIFT, while mutations in
these two genes are associated with recessive retinal diseases. No
potentially pathogenic variants were detected in the remaining 10
of the 12 genes, including AIPL1, CABP4,
DTHD1, GUCY2D, IQCB1, KCNJ13,
NMNAT1, OTX2, RD3 and RPGRIP1.
Clinical data of patients with
LCA-associated gene mutations
Clinical data of the four RP patients with mutations
in the LCA-associated genes were summarized in Table V. The patients examined were
between 5 and 29 years old. Although they presented with poor
vision or night blindness, none of thee patients exhibited
nystagmus or oculardigital sign. These patients were likely to have
early onset severe RP.
Discussion
In the present study, potentially pathogenic
mutations in LCA-associated genes were identified in four of the
293 patients with RP. By screening the most frequently mutated nine
exons, homozygous or compound heterozygous mutations were
identified in two exons of the CRB1 gene in 1.0% (3/293) of
RP probands. No such variant was detected in the rest of the six
exons of the other four genes. This indicates that the mutation
rate of these nine exons in RP patients is markedly lower compared
with that in the LCA patients (4).
After analyzing the variants in 12 genes, associated with LCA but
not RP, resulting from exome sequencing of 157 patients, it was
confirmed that one patient harbored compound heterozygous mutations
in CEP290. Previously, the CEP290 mutation in
patients with RP has rarely been reported, except for the compound
heterozygous mutations, c.[4705-1G>T];[3559delC], in an
autosomal recessive RP patient (32).
All four patients with RP demonstrated early onset
severe retinal degeneration, but also, they were marginally
different from LCA due to the absence of nystagmus and oculodigital
signs. According to previous studies, among RP patients caused by
CRB1 mutations, patients with null mutations (i.e.,
nonsense, frameshift and splice-site mutations) on the two alleles
are likely to result in a more severe form of retinal degeneration
(e.g. LCA), while a missense mutation on at least one allele may
suffer from a milder phenotype (e.g. RP) (33,34).
Previous studies have revealed that CRB1 can cause autosomal
recessive RP, and it was recently reported that CRB1
mutations are a relatively frequent cause of autosomal recessive
early onset retinal degeneration in Israeli, Palestinian and
Spanish populations (34,35).
Hereditary retinal degeneration is a complicated
group of diseases causing blindness. For each form, including RP,
LCA or cone-rod dystrophies, a number of causative genes have been
identified. Sometimes, atypical phenotypes or phenotypic
progression may hinder proper classification of the diseases and as
a result analysis of pertinent candidate genes may not be
available. Conversely, a number of genes responsible for one form
of retinal degeneration may also lead to other forms of the
disease. Those genes that are considered to cause a certain form of
retinal degeneration may remain potential candidate genes for other
forms of retinal dystrophy. Extensive analysis of all the potential
candidate genes, not just a subset of well-defined known causative
genes, may lead to the identification of the genetic defects in
more patients with retinal degeneration. This may be increasingly
significant in the era of exome sequencing.
Acknowledgements
The authors would like to thank all of the patients
and controls for their participation. The present study was
supported by the National Natural Science Foundation of China
(grant nos. 81170881 and U1201221) and the Guangdong Department of
Science & Technology Translational Medicine Center (grant no.
2011A080300002).
References
|
1
|
Parmeggiani F: Clinics, epidemiology and
genetics of retinitis pigmentosa. Curr Genomics. 12:236–237. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hu DN: Prevalence and mode of inheritance
of major genetic eye diseases in China. J Med Genet. 24:584–588.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hartong DT, Berson EL and Dryja TP:
Retinitis pigmentosa. Lancet. 368:1795–1809. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li L, Xiao X, Li S, et al: Detection of
variants in 15 genes in 87 unrelated Chinese patients with Leber
congenital amaurosis. PLoS One. 6:e194582011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
den Hollander AI, Heckenlively JR, van den
Born LI, et al: Leber congenital amaurosis and retinitis pigmentosa
with Coats-like exudative vasculopathy are associated with
mutations in the crumbs homologue 1 (CRB1) gene. Am J Hum Genet.
69:198–203. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zernant J, Külm M, Dharmaraj S, et al:
Genotyping microarray (disease chip) for Leber congenital
amaurosis: detection of modifier alleles. Invest Ophthalmol Vis
Sci. 46:3052–3059. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Reese MG, Eeckman FH, Kulp D and Haussler
D: Improved splice site detection in Genie. J Comput Biol.
4:311–323. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Morimura H, Fishman GA, Grover SA, et al:
Mutations in the RPE65 gene in patients with autosomal recessive
retinitis pigmentosa or leber congenital amaurosis. Proc Natl Acad
Sci USA. 95:3088–3093. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rivolta C, Sharon D, DeAngelis MM and
Dryja TP: Retinitis pigmentosa and allied diseases: numerous
diseases, genes, and inheritance patterns. Hum Mol Genet.
11:1219–1227. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Booij JC, Florijn RJ, ten Brink JB, et al:
Identification of mutations in the AIPL1, CRB1, GUCY2D, RPE65, and
RPGRIP1 genes in patients with juvenile retinitis pigmentosa. J Med
Genet. 42:e672005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sohocki MM, Bowne SJ, Sullivan LS, et al:
Mutations in a new photoreceptor-pineal gene on 17p cause Leber
congenital amaurosis. Nat Genet. 24:79–83. 2000. View Article : Google Scholar
|
|
12
|
Aldahmesh MA, Al-Owain M, Alqahtani F,
Hazzaa S and Alkuraya FS: A null mutation in CABP4 causes Leber’s
congenital amaurosis-like phenotype. Mol Vis. 16:207–212.
2010.PubMed/NCBI
|
|
13
|
den Hollander AI, Koenekoop RK, Yzer S, et
al: Mutations in the CEP290 (NPHP6) gene are a frequent cause of
Leber congenital amaurosis. Am J Hum Genet. 79:556–561. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Abu-Safieh L, Alrashed M, Anazi S, et al:
Autozygome-guided exome sequencing in retinal dystrophy patients
reveals pathogenetic mutations and novel candidate disease genes.
Genome Res. 23:236–247. 2013. View Article : Google Scholar :
|
|
15
|
Perrault I, Rozet JM, Calvas P, et al:
Retinal-specific guanylate cyclase gene mutations in Leber’s
congenital amaurosis. Nat Genet. 14:461–464. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Estrada-Cuzcano A, Koenekoop RK,
Coppieters F, et al: IQCB1 mutations in patients with leber
congenital amaurosis. Invest Ophthalmol Vis Sci. 52:834–839. 2011.
View Article : Google Scholar
|
|
17
|
Sergouniotis PI, Davidson AE, Mackay DS,
et al: Recessive mutations in KCNJ13, encoding an inwardly
rectifying potassium channel subunit, cause leber congenital
amaurosis. Am J Hum Genet. 89:183–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
den Hollander AI, Koenekoop RK, Mohamed
MD, et al: Mutations in LCA5, encoding the ciliary protein
lebercilin, cause Leber congenital amaurosis. Nat Genet.
39:889–895. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Koenekoop RK, Wang H, Majewski J, et al:
Mutations in NMNAT1 cause Leber congenital amaurosis and identify a
new disease pathway for retinal degeneration. Nat Genet.
44:1035–1039. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Henderson RH, Williamson KA, Kennedy JS,
et al: A rare de novo nonsense mutation in OTX2 causes early onset
retinal dystrophy and pituitary dysfunction. Mol Vis. 15:2442–2447.
2009.PubMed/NCBI
|
|
21
|
Friedman JS, Chang B, Kannabiran C, et al:
Premature truncation of a novel protein, RD3, exhibiting subnuclear
localization is associated with retinal degeneration. Am J Hum
Genet. 79:1059–1070. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dryja TP, Adams SM, Grimsby JL, et al:
Null RPGRIP1 alleles in patients with Leber congenital amaurosis.
Am J Hum Genet. 68:1295–1298. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ji Y, Wang J, Xiao X, Li S, Guo X and
Zhang Q: Mutations in RPGR and RP2 of Chinese patients with
X-linked retinitis pigmentosa. Curr Eye Res. 35:73–79. 2010.
View Article : Google Scholar
|
|
24
|
Wang Q, Wang P, Li S, et al: Mitochondrial
DNA haplogroup distribution in Chaoshanese with and without myopia.
Mol Vis. 16:303–309. 2010.PubMed/NCBI
|
|
25
|
Chen Y, Zhang Q, Shen T, et al:
Comprehensive mutation analysis by whole-exome sequencing in 41
Chinese families with Leber congenital amaurosis. Invest Ophthalmol
Vis Sci. 54:4351–4357. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li Y, Vinckenbosch N, Tian G, et al:
Resequencing of 200 human exomes identifies an excess of
low-frequency non-synonymous coding variants. Nat Genet.
42:969–972. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu Y, Guan L, Shen T, et al: Mutations of
60 known causative genes in 157 families with retinitis pigmentosa
based on exome sequencing. Hum Genet. 133:1255–1271. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Flanagan SE, Patch AM and Ellard S: Using
SIFT and PolyPhen to predict loss-of-function and gain-of-function
mutations. Genet Test Mol Biomarkers. 14:533–537. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ramensky V, Bork P and Sunyaev S: Human
non-synonymous SNPs: server and survey. Nucleic Acids Res.
30:3894–3900. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kumar P, Henikoff S and Ng PC: Predicting
the effects of coding non-synonymous variants on protein function
using the SIFT algorithm. Nat Protoc. 4:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang J, Liu J and Zhang Q: FOXL2 mutations
in Chinese patients with blepharophimosis-ptosis-epicanthus
inversus syndrome. Mol Vis. 13:108–113. 2007.PubMed/NCBI
|
|
32
|
Neveling K, Collin RW, Gilissen C, et al:
Next-generation genetic testing for retinitis pigmentosa. Hum
Mutat. 33:963–972. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Beryozkin A, Zelinger L, Bandah-Rozenfeld
D, et al: Mutations in CRB1 are a relatively common cause of
autosomal recessive early-onset retinal degeneration in the Israeli
and Palestinian populations. Invest Ophthalmol Vis Sci.
54:2068–2075. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bujakowska K, Audo I, Mohand-Saïd S, et
al: CRB1 mutations in inherited retinal dystrophies. Hum Mutat.
33:306–315. 2012. View Article : Google Scholar :
|
|
35
|
Corton M, Tatu SD, Avila-Fernandez A, et
al: High frequency of CRB1 mutations as cause of Early-Onset
Retinal Dystrophies in the Spanish population. Orphanet J Rare Dis.
8:202013. View Article : Google Scholar : PubMed/NCBI
|