Introduction
Corneal avascularity, the absence of blood vessels
in the cornea, is required for corneal transparency and the
maintenance of vision. Corneal neovascularization, which is induced
by a wide range of inflammatory, infectious and traumatic
disorders, can lead to visual impairment and even blindness
(1,2). It has been reported that 4.14% of
ophthalmic patients in the USA have corneal neovascularization
(3). Corneal angiogenesis is a
common histopathological feature of corneal diseases, leading to
corneal transplantation (4).
Although several medical and surgical options are currently
available for the management of corneal neovascularization,
treatment remains challenging and problematic (5).
Under normal physiological conditions, corneal
avascularity is maintained by a balance between high levels of
antiangiogenic factors and low levels of angiogenic factors
(1,5). The presence of neovascularization
indicates the activation of several angiogenic factors promoting
angiogenesis, which is likely to be associated with a
downregulation of antiangiogenic factors (6,7).
Several angiogenic molecules have been identified, including
vascular endothelial growth factors (VEGFs), fibroblast growth
factors and matrix metalloproteinases (MMPs) (1). VEGFs have been identified as being
important in the angiogenic process, which involves the
proliferation, migration and differentiation of endothelial cells
(ECs) and the degradation of the surrounding extracellular matrix
(ECM) (1,8,9).
VEGF-A, also termed VEGF 165, is a member of the
VEGF family that is important in angiogenesis. VEGF-A binds to two
tyrosine kinase receptors, VEGFR1 and VEGFR2. The latter receptor,
VEGFR2, is the direct signal transducer for angiogenesis. By
comparison, VEGFR-1 has weaker kinase activity and negatively
modulates VEGFR-2-mediated angiogenesis (10). The binding of VEGF-A to VEGFR2
triggers several signaling pathways, including the activation of
phosphoinositide 3-kinase (PI3K) and Akt, the recruitment of
protein lipase C (PLC) and subsequent activation of the
mitogen-activated protein kinase (MAPK) cascade through protein
kinase C (PKC) and the activation of focal adhesion kinase (FAK)
and its substrate paxillin (9,11).
In addition, the activation of VEGFR2 eventually leads to the
differentiation, proliferation, migration and capillary formation
of ECs (9,11,12).
Paxillin is a signal transduction adaptor protein,
which is associated with focal adhesion and is one of the major
substrates of FAK, a nonreceptor protein tyrosine kinase (13). Paxillin can be phosphorylated on
Tyr 31 (PY31) and Tyr 118 (PY118) by FAK or Src kinases (14). VEGF-A has been reported to recruit
FAK, which phosphorylates paxillin in ECs (15,16).
This phosphorylation promotes the formation of the
paxillin-Crk-Dock180 molecular complex (17,18),
regulates the activity of Rho guanine triphosphatase (19,20)
and activates the Rac and extracellular signal-regulated kinase
(ERK) signaling pathways (17,21,22),
which leads to increases in cell migration and adhesion (14). However, the role of paxillin in the
VEGF-A-induced proliferation, migration, adhesion and capillary
formation of ECs remains to be elucidated.
The purpose of the present study was to investigate
the role of paxillin in the VEGF-A-induced proliferation,
migration, adhesion and capillary formation of human umbilical vein
ECs (HUVECs) through the small interfering (si)RNA-based knockdown
of paxillin.
Materials and methods
The present study was performed in accordance with
the Declaration of Helsinki and the use of human cells/tissues was
approved by the Medical Ethical Committee of Wuhan University
(Wuhan, China). HUVECs were isolated from two female patients (24
and 30 years old). Written informed consent was obtained prior to
cell/tissue collection
HUVEC cell culture
The HUVECs, which were isolated from human umbilical
cord veins, as previously reported by Jaffe et al (23), were cryopreserved following primary
culture and stored at the Department of Ophthalmology of Wuhan
University. The HUVECs were seeded into poly-L-lysine-coated flasks
and maintained in endothelial complete medium supplemented with 5%
fetal bovine serum, 1% penicillin/streptomycin and 1% EC growth
supplement (ScienCell Research Laboratories, San Diego, CA, USA).
The cells were maintained at 37°C in a humidified incubator under
5% CO2, with the medium replaced every 2–3 days until
the cells reached confluency. The cells were harvested with 0.05%
trypsin-ethylene glycol tetraacetic acid solution (Wuhan Boster Bio
Engineering Co., Ltd., Wuhan, China) and were further cultured in
the poly-L-lysine-coated flasks for use in the subsequent
experiments, which used cells starting at passage five when they
exhibited a cobblestone appearance.
Von Willebrand factor immunofluorescence
staining
The HUVECs were grown on glass coverslips in sterile
six-well plates until they reached confluency. The cells were
rinsed with phosphate-buffered saline (PBS; Wuhan Boster Bio
Engineering Co., Ltd.) three times and fixed with 4%
paraformaldehyde for 30 min at room temperature (RT). The cells
were permeabilized with 0.1% Triton X-100 for 15 min and then
incubated in a 3% H2O2/ethanol solution to
inhibit the endogenous peroxidase. The cells were then washed with
PBS three times and were incubated with the primary antibody,
polyclonal rabbit anti-human von Willebrand factor (1:100; Wuhan
Boster Bio-Engineering Co., Ltd., Wuhan China), at 4°C overnight.
PBS without primary antibodies was used as a negative control.
After 24 h, the primary antibody was removed by washing the cells
with PBS and the immunoreactivity was detected by incubating the
cells with the fluorescein isothiocyanate-coupled secondary
antibody, goat anti-rabbit immunoglobulin (Ig)G (1:10; Wuhan Boster
Bio-Engineering Co., Ltd.), at RT for 45 min. Cell nuclei were
counter-stained with 4,6-diamino-2-phenylindole (Wuhan Boster Bio
Engineering Co., Ltd.). The coverslips were then washed with PBS,
the cells were examined with a fluorescence microscope (Olympus,
Tokyo, Japan) and images were captured with a DP70 digital camera
(Olympus).
Immunoprecipitation
The HUVECs were grown to confluence and stimulated
with 20 ng/ml VEGF-A (Cell Signaling Technology, Inc., Beverly, MA,
USA) at 37°C for 0, 20, 40 and 60 min. The cells were then washed
with ice-cold PBS and solubilized on ice with lysis buffer
containing 150 mM NaCl, 10 mM Tris-HCl, (pH 7.5) and 1% Triton
X-100 supplemented with a cocktail of phosphatase and proteinase
inhibitors containing 1 mM vanadate, 10 mg/ml leupeptin, 10 mg/ml
aprotinin, 1 mM phenylmethylsulfonyl fluoride and 0.36 mM
phenanthroline. The lysates were centrifuged at 10,000 xg for 15
min at 4°C and the supernatants were incubated with polyclonal
mouse anti-human paxillin (Abcam, Cambridge, MA, USA), anti-mouse
IgG and protein A-agarose at 4°C overnight. The immunoprecipitates
were then collected by centrifugation and the agarose pellet was
suspended in 2X SDS-PAGE buffer. The expression of total paxillin
and phosphorylated paxillin was determined by western blot
analysis.
Knockdown of paxillin in the HUVECs
The HUVECs were seeded into six-well plates at a
density of 5×106 cells/ml. The cells were maintained
overnight at 37°C in a humidified incubator supplemented with 5%
CO2 followed by transfection with a duplex of
oligonucleotides targeting paxillin mRNA using
LipofectamineTM 2000 (Invitrogen Life Technologies,
Carlsbad, CA, USA). All the siRNA constructs were obtained from
Guangzhou RiboBio Co., Ltd. (Guangzhou, China). The following three
pairs of paxillin siRNAs were used: siRNA1 forward,
5′GCUGGAACUGAACGCUGUAdTdT3′ and reverse,
5′UACAGCGUUCAGUUCCAGCdTdT3′ (target sequence: GCTGGAACTGAACGCTGTA);
siRNA2 forward, 5′GUGUGGAGCCUUCUUUGGUdTdT3′ and reverse,
5′ACCAAAGAAGGCUCCACACTdTd3′ (target sequence: GTGTGGAGCCTTCTTTGGT);
and siRNA3 forward, 5′GCAGCAACCUUUCUGAACUdTdT3′ and reverse,
5′AGUUCAGAAAGGUUGCUGCTdTd3′ (target sequence: GCAGCAACCTTTCTGAACT).
A scramble siRNA construct was used as a negative control. The
efficiency of the siRNA-mediated paxillin-knockdown was examined by
reverse transcription quantitative polymerase chain reaction
(RT-qPCR) and western blot analysis.
RT-qPCR
Total RNA was isolated from the HUVECs using TRIzol
reagent (Invitrogen Life Technologies) according to the
manufacturer’s instructions. The RNA was reverse-transcribed into
complementary DNA using a Plexor™ qPCR System (Promega Corporation,
Madison, WI, USA). The RT-qPCR was performed with a final volume of
20 μl containing 2 μl cDNA, 0.5 μl of each primer and 10 μl SYBR
green. The primers used for the amplification of paxillin are shown
in Table I. The β-actin gene was
used as a control housekeeping gene. The cycling parameters were as
follows: 95°C for 20 sec, followed by 40 cycles of 95°C for 10 sec,
60°C for 20 sec and 70°C for 1 sec, with a final extension at 65°C
for 15 sec. Melting curve analyses were performed to verify the
amplification specificity. The mRNA expression ΔCt values of
paxillin from each sample were calculated by normalizing against
the internal control β-actin and the relative expression of
paxillin was calculated using the 2−ΔΔCT method
(24).
 | Table IPrimers used for amplification of the
paxillin gene, reverse transcribed from human umbilical vein
endothelial cell-derived mRNA. |
Table I
Primers used for amplification of the
paxillin gene, reverse transcribed from human umbilical vein
endothelial cell-derived mRNA.
| Primer | Sequences
(5′-3′) | Length (bp) | Tm (°C) | GC (%) | Size |
|---|
| Primer 1 | Forward,
AGTGCTTTGTGTGCCGGGAA | 20 | 57.01 | 55.00 | 195 bp |
| Reverse,
AGGCACAGACGAAGTGCTCG | 20 | 57.03 | 60.00 | |
| Primer 2 | Forward,
TCATGGCCCAGGGGAAGACA | 20 | 56.97 | 60.00 | 141 bp |
| Reverse,
CGCAGACTCCTTTGGCGACT | 20 | 57.01 | 60.00 | |
| Primer 3 | Forward,
TTCTGAACTCGACCGCCTGC | 20 | 57.02 | 60.00 | 176 bp |
| Reverse,
TCTCTTTCGTCAGGGGCCCA | 20 | 57.02 | 60.00 | |
Western blot analysis
The HUVECs were homogenized on ice in lysis buffer
after 48 h siRNA treatment. The proteins were resolved by SDS-PAGE
and transferred onto polyvinylidene difluoride membranes by
electroblotting. The membranes were inhibited with 10% non-fat dry
milk in Tris-buffered saline with Tween-20 (pH 8.0) and incubated
with the polyclonal primary antibody rabbit anti-human paxillin
(1:1,000; Epitomics, Burlingame, CA, USA). For immunoprecipitation,
a primary antibody against phosphorylated paxillin at Y118 and Y31
(polyclonal rabbit anti-human p-paxillin; 1:1,000; Epitomics) was
also used. The membranes were then incubated with the antibodies at
4°C overnight. β-actin was used as a loading control. The membranes
were then incubated with a polyclonal horseradish peroxidase-linked
goat anti-rabbit secondary antibody (1:5,000; KPL, Gaithersburg,
MD) at RT for 2 h. The bands were visualized using a
chemiluminescence detection system with Novex® ECL
chemiluminescent substrate reagent kit (Pierce, Rockford, IL,
USA)
Cell adhesion assay
Following siRNA and VEGF-A treatment, the HUVECs in
the experimental and control groups were harvested and counted.
Equal quantities of cells (1×105 cells/ml) were seeded
into 96-well plates coated with Matrigel (Wuhan Boster Bio
Engineering Co., Ltd.) and cultured for 2 h at 37°C in a humidified
incubator with 5% CO2. Following incubation, the cells
were washed twice with Hank’s solution to remove the non-adherent
cells. Images of the adherent cells were captured followed by
further incubation with 20 μl MTT (5 mg/ml, Wuhan Boster Bio
Engineering Co., Ltd.) for 4 h at RT. The optical density of the
plates was measured at a wavelength of 490 nm using a microplate
reader (Model 550; Bio-Rad, Tokyo, Japan).
Cell proliferation assay
The cell proliferation was evaluated using an
5-ethynyl-2′-deoxyuridine (EdU) Cell Proliferation assay kit
(Molecular Probes, Invitrogen Life Technologies) according to the
manufacturer’s instructions. Briefly, the HUVECs, grown in
multiwell plates, were incubated in 100 μl endothelial complete
medium containing 50 μM EdU (Wuhan Boster Bio Engineering Co.,
Ltd.) for 2 h. The cells were washed twice with PBS and fixed with
4% paraformaldehyde in PBS for 30 min at RT. The cells were then
washed with glycine (2 mg/ml) for 5 min in a shaker and
permeabilized with 0.5% Triton X-100 for 10 min. The cells were
again washed twice with PBS and incubated in 1X Apollo®
reaction buffer, containing 100 mM Tris-HCl (pH 8.5), 1 mM
CuSO4, 100 μm Apollo 550 fluorescent azide and 100 mM
ascorbic acid, for 30 min at RT, protected from light. The cells
were then permeabilized three times with 0.5% Triton X-100,
followed by 1–2 washes with 100 μl methanol. The cells were
subsequently stained with Hoechst 33342 for 30 min at RT, protected
from light, and washed with PBS.
Images were captured and analyzed using a BD Pathway
855 High Content Bioimager (BD Biosciences, San Jose, CA, USA). A
total of 16 random fields were selected in each well. The number of
EdU-positive cells and Hoechst-stained cells were calculated and
the EdU-positive cells were expressed as the ratio of the total
cell number. These ratios were normalized to the control
ratios.
Cell cycle analysis by flow
cytometry
The HUVECs were harvested, washed twice in ice-cold
PBS and centrifuged at 160 × g for 5 min. They were then
resuspended in chilled ethanol (250 ml/l) and incubated at 4°C
overnight. Subsequently, the cells were washed with PBS, stained
with propidium iodide for 30 min and analyzed by flow cytometry.
Data were acquired using a flow cytometer (Beckman Coulter, Miami,
FL, USA), and analyzed with Multicycle software (Version 2.0,
Phoenix Flow Systems, San Diego, CA, USA), with 10,000 events
analyzed in each sample. The cell proliferation index (PI) was
calculated as follows: PI = (S + G2/M)/(G0/G1 + S + G2/M) ×100%
(25).
Boyden chamber assay of HUVEC
migration
Following siRNA and VEGF-A treatment, the HUVECs in
the experimental and control groups were harvested, suspended with
ECM and seeded into six-well plates at a density of
1×106 cells/well. Following culture for 24 h,
2×105 cells were seeded into Transwell inserts (Corning
Costar, Costar, NY, USA). Inserts containing the HUVECs were placed
into a six-well plate containing endothelial complete medium
supplemented with 20 ng/ml VEGF-A in the lower well of a Boyden
chamber. The cells were cultured for 12 to 18 h and the upper
surface of the insert was then swabbed to remove unmigrated cells.
The inserts were fixed with 4% paraformaldehyde for 30 min and
stained with crystal violet for 20 min. Cell migration was
quantified by counting the number of migrated cells, expressed as
the percentage of migrated cells in the control.
Tube formation in vitro
At 48 h after siRNA treatment, the HUVECs
(4×104 cells/well) in the experimental and control
groups were resuspended in endothelial complete medium without
serum and seeded onto 96-well plates coated with Matrigel (70 μl).
Photomicrographs of the center of each well were obtained following
incubation of the cells at 37°C for 48 h. The tubes were stained
using a Cellomics Cytoskeletal Rearrangement kit and were analyzed
with Cellomics ArrayScan (Cellomics, Pittsburgh, PA, USA). Cell
images were acquired with the ArrayScan® HCS Reader
(Cellomics, Pittsburgh, PA, USA). Tube formation was assessed by
measuring the tube length by using the Image-Pro Plus 6.0 image
processing system (Media Cybernetics, Inc., Rockville, MD, USA).
Data are expressed as mm/mm2.
Statistical analysis
Analyses were performed using SPSS 13.0 (SPSS, Inc.,
Chicago, IL, USA). All values are expressed as the mean ± standard
deviation. One-way analysis of variance was performed to determine
the statistical significance. P<0.05 was considered to indicate
a statistically significant difference.
Results
Von Willebrand factor expression by
HUVECs
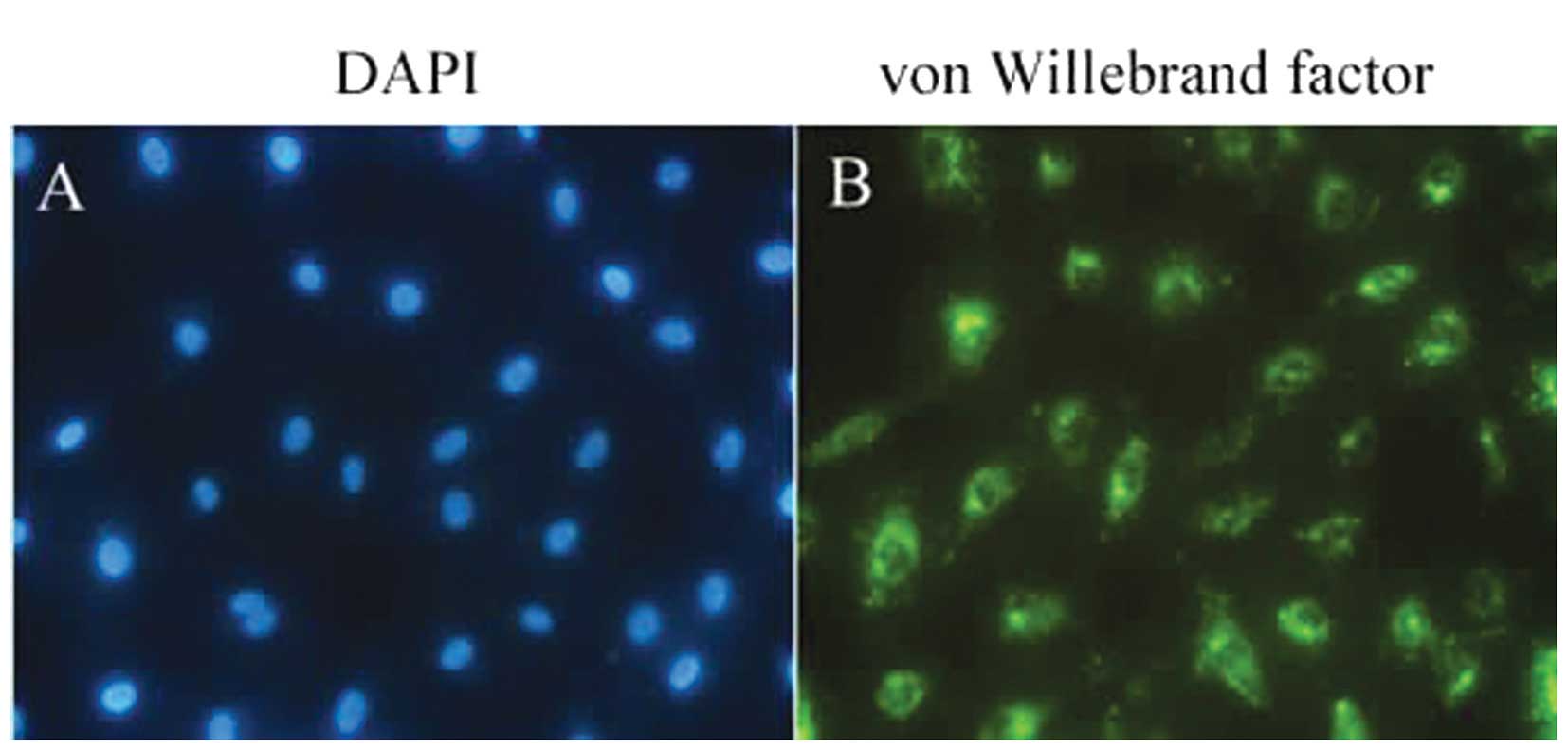
The von Willebrand factor is a glycoprotein
synthesized by ECs and is commonly used as a marker for HUVECs
(26). In the present study, the
HUVECs exhibited a round or spindle-shaped morphology under a
bright-field microscope. Under a fluorescence microscope, the
majority of the HUVECs stained positive for von Willebrand factor.
The cells had a cobblestone-like appearance and were homogeneous
with green fluorescence in the cytoplasm (Fig. 1). No fluorescence was detected in
the control cells.
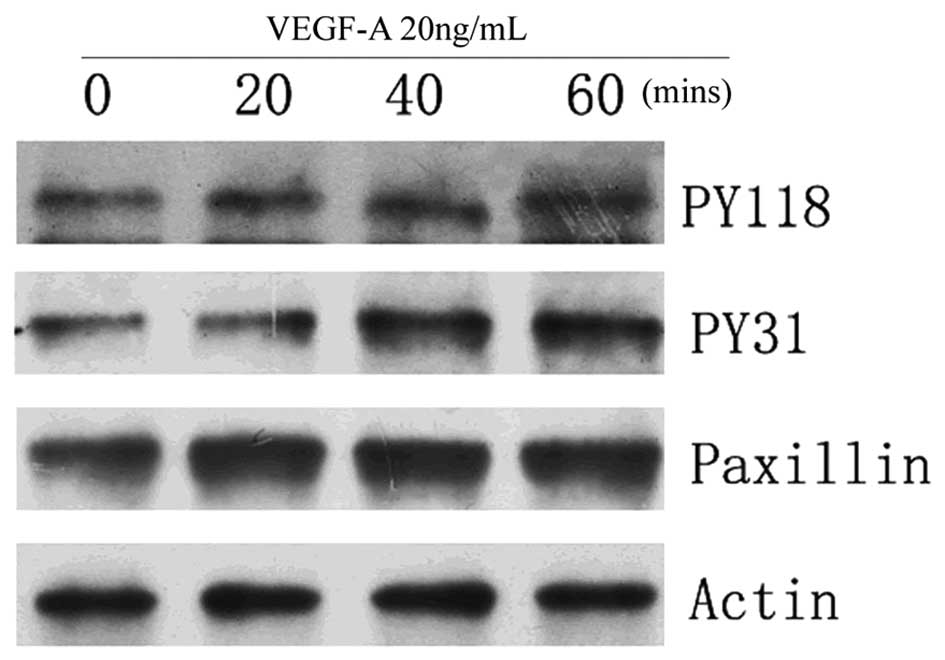
VEGF-A upregulates the expression of
phosphorylated paxillin in HUVECs
The present study subsequently investigated the
effects of VEGF-A on the phosphorylation of paxillin in HUVECs by
immunoprecipitation. The HUVECs were treated with 20 ng/ml VEGF-A
for 0, 20, 40 and 60 min. Immunoblots using antibodies specific to
paxillin phosphorylated at PY118 or PY31 revealed that the
phosphorylation of paxillin at PY118 and PY31 increased in an
incubation time-dependent manner (Fig.
2).
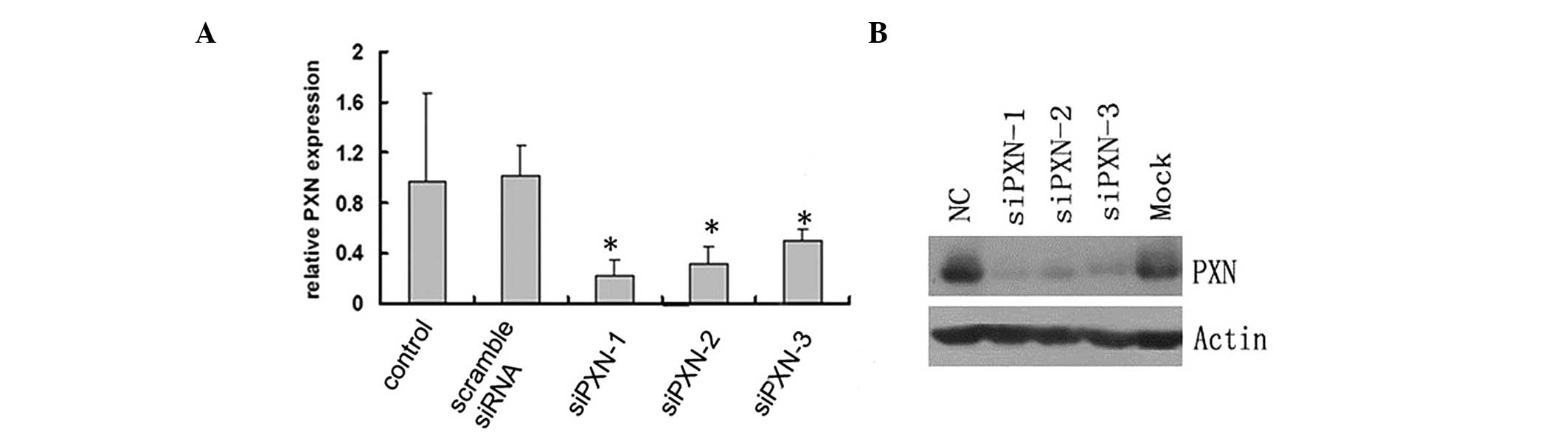
siRNA-mediated knockdown decreases the
mRNA and protein expression levels of paxillin in HUVECs
The role of paxillin in the HUVECs was examined by
knocking down the protein expression of paxillin by siRNA and
investigating the mRNA expression by RT-qPCR. Three pairs of siRNA
constructs, siPXN-1, siPXN-2 and siPXN-3, were assessed. The
control scramble siRNA did not alter the relative mRNA expression
of paxillin in the HUVECs; however, siPXN-1, siPXN-2 and siPXN-3
significantly decreased the mRNA expression of paxillin in the
HUVECs (Fig. 3A). Consistent with
the mRNA expression results, the protein expression of paxillin in
the HUVECS was also significantly inhibited by the three siRNAs
(Fig. 3B). The siPXN-1 construct
demonstrated the most marked inhibition of paxillin among the three
siRNAs (Fig. 3). Therefore,
siPXN-1 was selected for use in the subsequent experiments.
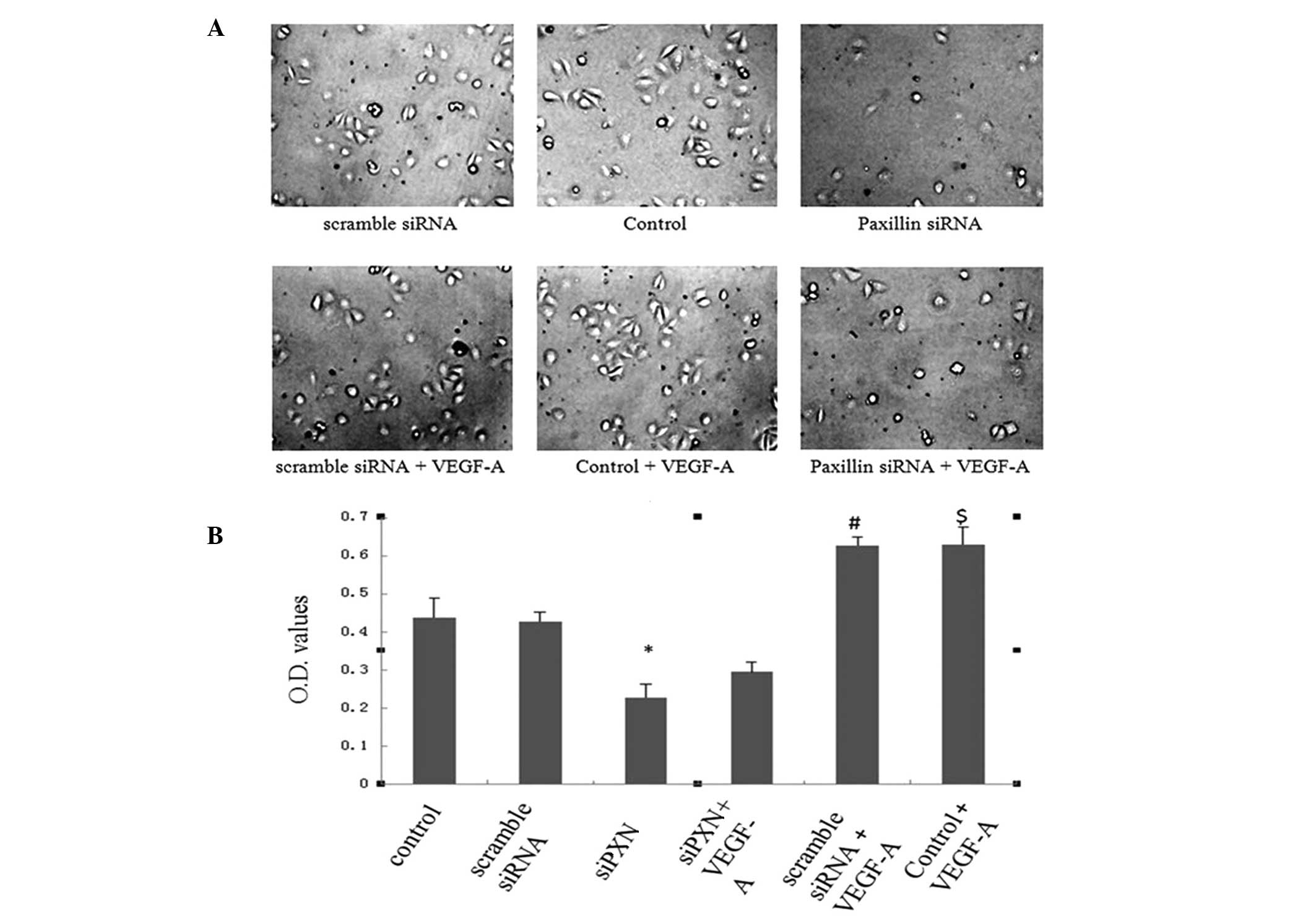
Knockdown of paxillin inhibits the
VEGF-A-induced adhesion of HUVECs
The expression of paxillin was then knocked down to
assess the role of the protein in the VEGF-A-induced adhesion of
HUVECs. Scramble siRNA did not alter the adhesive properties of
HUVECs compared with those of the control cells. Knockdown of
paxillin with siRNA significantly inhibited the adhesion of HUVECs
compared with that of the control and scramble siRNA cells
(Fig. 4A and B). VEGF-A treatment
significantly increased adhesion of the HUVECs in the control and
scramble siRNA groups; however, knockdown of paxillin with siRNA
inhibited the VEGF-A-induced adhesion of HUVECs (Fig. 4A and B).
Knockdown of paxillin inhibits the
VEGF-A-induced proliferation of HUVECs
The effects of paxillin on the VEGF-A-induced
proliferation of HUVECs were investigated using an EdU assay and
cell cycle analysis by flow cytometry. Scramble siRNA did not alter
HUVEC proliferation compared with that of the control; however, the
knockdown of paxillin with siRNA inhibited the proliferation of the
HUVECs compared with that of the control cells and cells treated
with scramble siRNA (P<0.05; Fig.
5A–C). VEGF-A treatment significantly increased the
proliferation of the HUVECs in the control and scramble siRNA
groups; however, knockdown of paxillin inhibited the VEGF-A-induced
proliferation of the HUVECs (Fig.
5A–C).
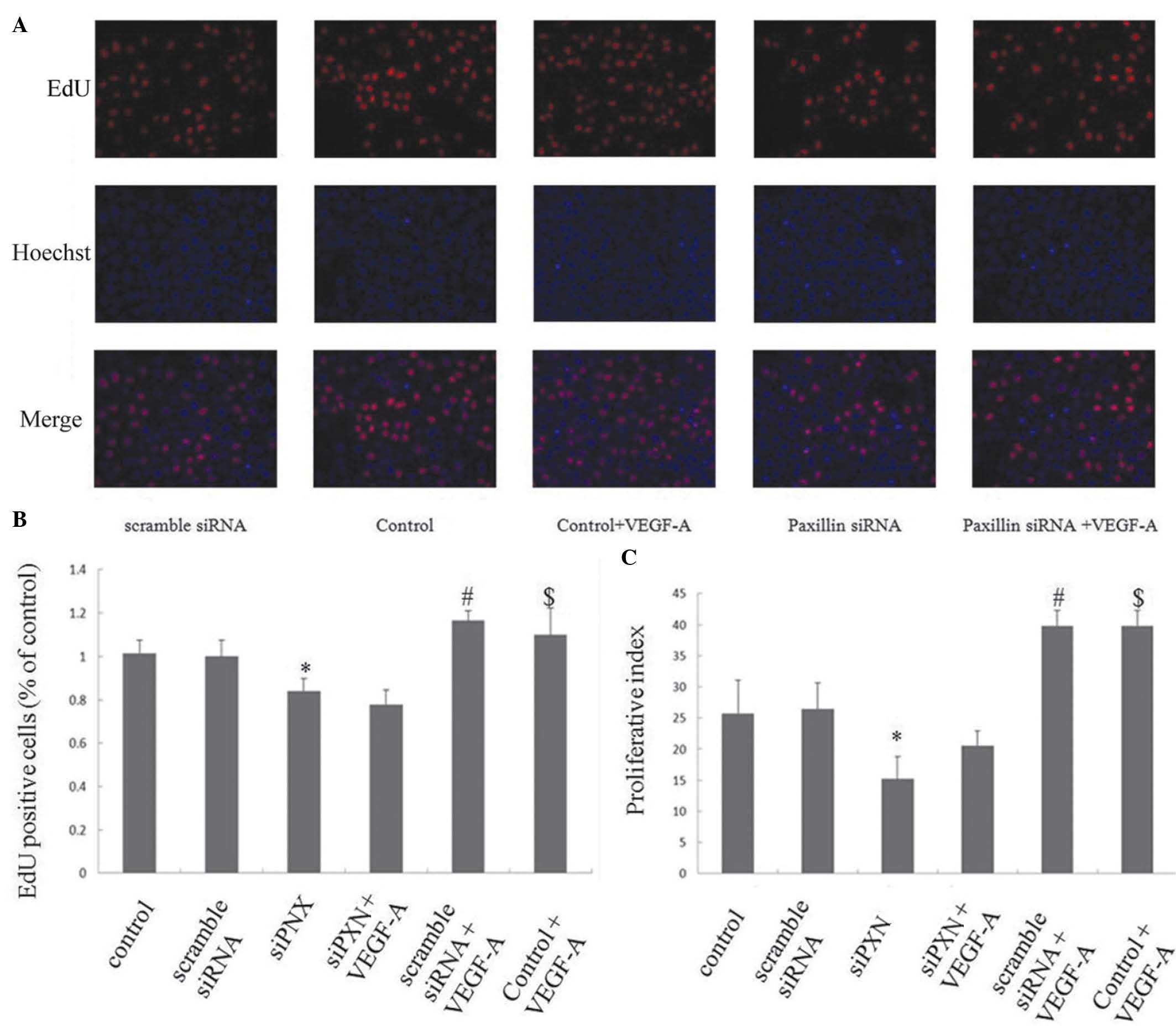 | Figure 5Knock down of paxillin inhibits the
VEGF-A-induced proliferation of HUVECs. The HUVECs were cultured on
plates following either mock-transfected or transfected with
scramble siRNA or siPXN-1, respectively. VEGF-A (20 ng/ml) was
added to the medium. (A) Representative photomicrographs of the
HUVECs stained with EdU (red) and Hoechst 33342 (blue).
Magnification ×200. (B) Percentage of EdU-positive cells.
*P<0.05, vs. control and scramble siRNA;
#P<0.05, vs. control; $P<0.05, vs.
scramble siRNA. (C) Cell proliferation index (PI) determined with
cell cycle analysis by flow cytometry. PI = (S + G2/M)/(G0/G1 + S +
G2/M) × 100%. Three independent experiments were performed.
*P<0.05, vs. control and scramble siRNA;
#P<0.05, vs. control; $P<0.05, vs.
scramble siRNA. VEGF-A, vascular endothelial growth factor A;
HUVECs, human umbilical vein endothelial cells; siRNA, small
interfering RNA. Data are presented as the mean ± standard
deviation. |
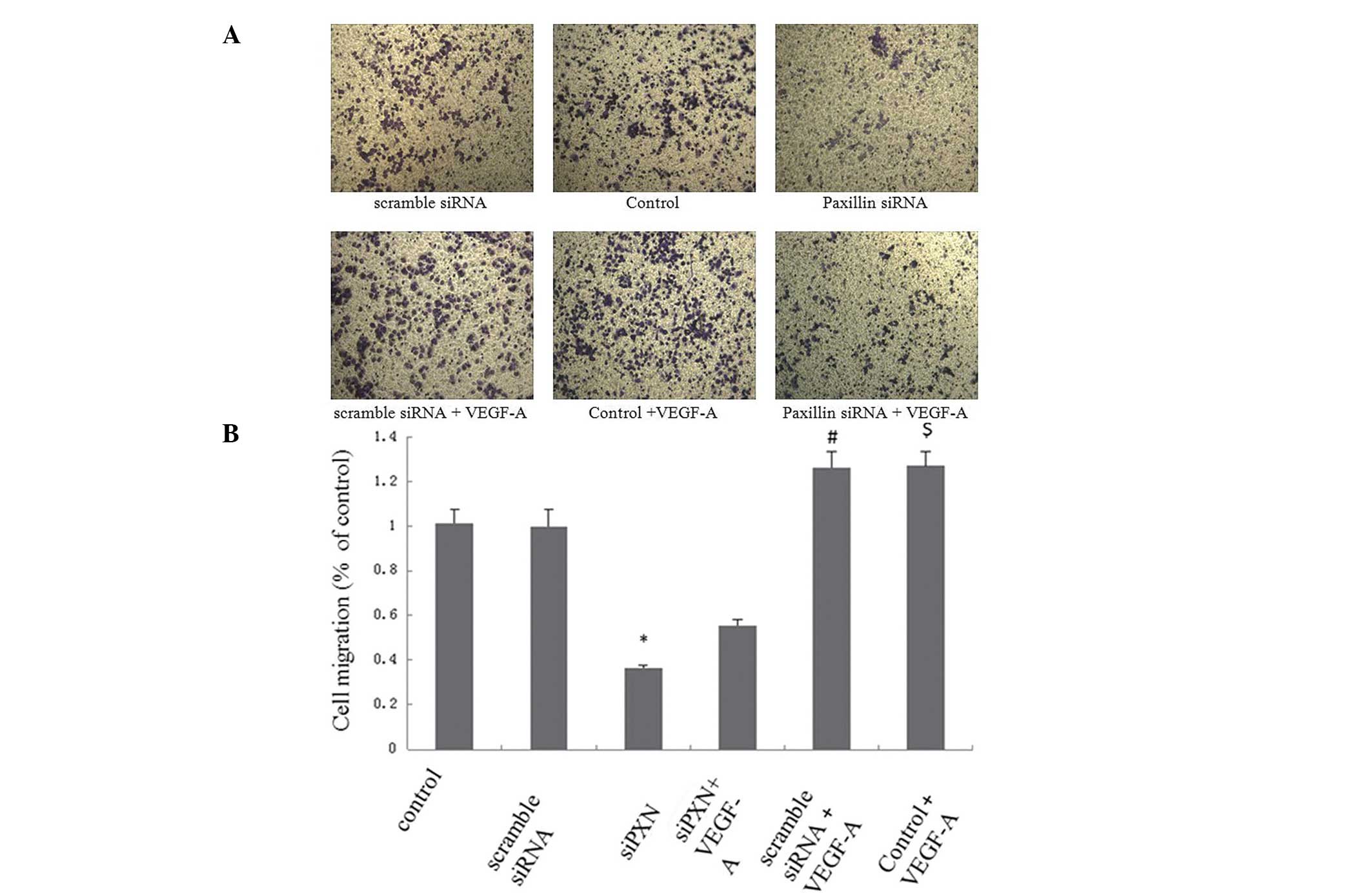
Knockdown of paxillin inhibits the
VEGF-A-induced migration of HUVECs
HUVEC migration was analyzed in a Boyden chamber
using VEGF-A as a chemoattractant. The siRNA-mediated knockdown of
paxillin was used to investigate the role of paxillin in the
VEGF-A-induced migration of the HUVECs. No significant differences
were observed in the number of migrating cells between the control
cells and the cells treated with scramble siRNA; however, knockdown
of paxillin significantly decreased the cell migration (P<0.01;
Fig. 6A and B). VEGF-A treatment
increased the number of migrated cells in the control and scramble
siRNA groups (Fig. 6A and B). Of
note, scramble siRNA did not affect VEGF-A-induced cell migration
compared with the control; however, the knockdown of paxillin by
siRNA inhibited the VEGF-A-induced migration of the HUVECs
(Fig. 6A and B).
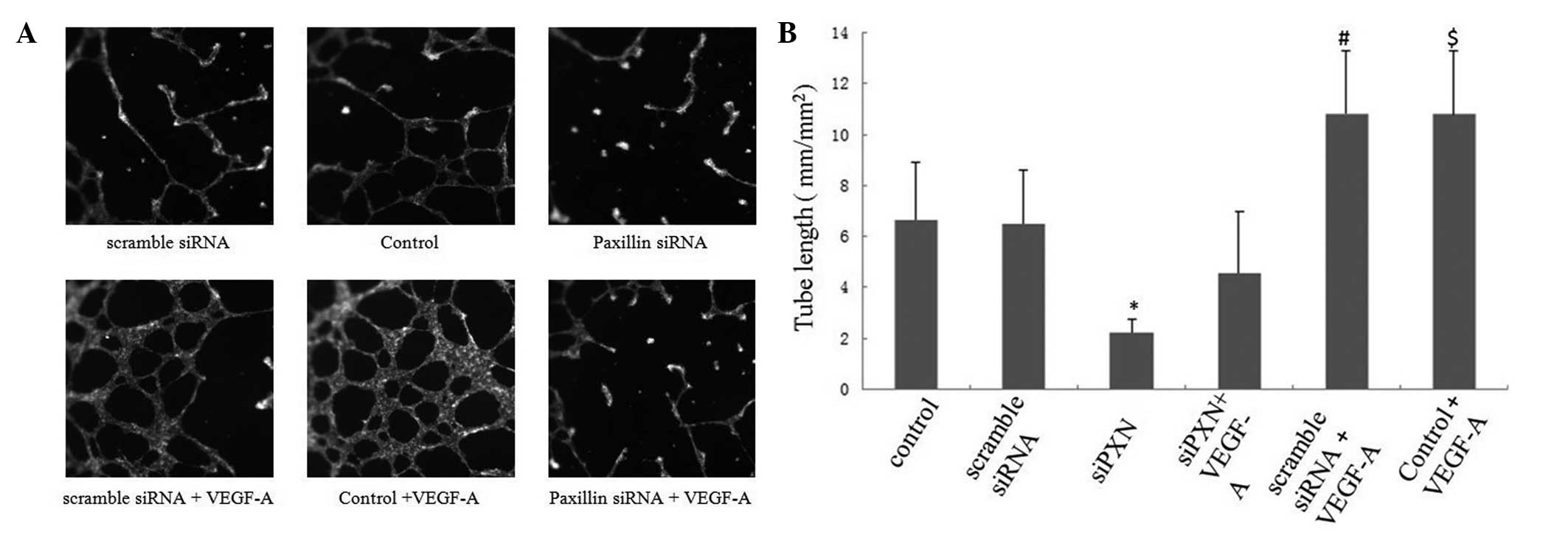
Knock down of paxillin inhibits the
VEGF-A-induced tube formation of HUVECs
The effects of paxillin on VEGF-A-induced tube
formation were evaluated using an in vitro Matrigel assay.
No significant difference was observed in the number of tubes
between the control group and the scramble siRNA group. Knockdown
of paxillin alone significantly decreased HUVEC tube formation
(Fig. 7A and B). VEGF-A treatment
increased the number of tubes that formed in the control and
scramble siRNA groups (Fig. 7A and
B). Scramble siRNA did not affect the VEGF-A-induced tube
formation compared with that of the control; however, knockdown of
paxillin with siRNA inhibited the VEGF-A-induced tube formation of
the HUVECs (Fig. 7A and B).
Discussion
Angiogenesis is a natural process that occurs during
healthy growth, development and wound healing. It also contributes
to numerous malignant, ischemic and inflammatory diseases (9). During angiogenesis, the ECs receive
angiogenic signals from growth factors to initiate proliferation,
migration and the formation of a tubular structure (9). VEGF-A and its receptors are important
in the promotion of the permeability, proliferation, migration and
capillary tube formation of ECs (9,11,27,28)
and contribute to several pathological processes, including
diabetic retinopathy, retinopathy of prematurity, age-associated
macular degeneration and corneal neovascularity (1,29–31).
The downstream effectors and targets of VEGF receptors that mediate
the diverse biological functions of VEGF-A include PKC, PLC, PI3K
and FAK (9,11). However, the key targets that
mediate VEGF-A-induced angiogenesis in the ECs remain to be fully
elucidated. It has been reported that VEGF-A increases the
phosphorylation of FAK and its substrate paxillin in the ECs
(15,16). Previous studies have demonstrated
that the expression of FAK, paxillin and MMPs are associated with
the angiogenic activities of the ECs (32), particularly in the
neovascularization observed in corneal diseases (33,34),
and our previous findings suggested that FAK mediates the
TNF-α-induced increase in MMPs in herpes simplex keratitis
(33). In vivo studies have
been conducted, confirming the hypothesis of the present study with
regard to corneal neovascularization. However, few studies have
been performed to investigate the role of paxillin in
VEGF-A-mediated angiogenesis. In the present study, the siRNA-based
knockdown of paxillin was used to investigate the role of the
paxillin protein in VEGF-A-induced effects on the HUVECs. The
results demonstrated that VEGF-A promoted the adhesion,
proliferation, migration and capillary formation of the HUVECs and
that the effects of VEGF-A were inhibited by paxillin knockdown.
These findings suggested that paxillin is essential for
VEGF-A-mediated angiogenesis in ECs.
During angiogenesis, the adhesion of ECs to each
other and to the ECM is required for the growth, differentiation
and survival of the cells (35).
Integrins, which are heterodimeric adhesion molecules, are
important in cell-cell and cell-matrix interactions (36). FAK mediates integrin signaling to
recruit cytoskeletal and signaling proteins, including paxillin,
which regulates the neovascularization of ECs (37). In addition, FAK is the downstream
signaling molecule of VEGF and its receptors, which promote EC
adhesion (15,16,37).
As a downstream signaling molecule of FAK, paxillin localizes to
focal adhesions and transduces adhesion and growth factor signals
to regulate cell functions (14,38).
In the present study, knockdown of paxillin reduced the adhesion of
HUVECs in the presence and absence of VEGF-A treatment, suggesting
that paxillin may mediate integrin and VEGF-A signaling in the
regulation of EC adhesion, possibly though FAK.
ECs receive cues from soluble proteins and growth
factors to elongate, migrate and proliferate (9) and VEGF-A, a proangiogenic factor,
induces the proliferation, migration and tube-like formation of ECs
(9,27,28).
Consistent with previous findings, the present study found that
VEGF-A treatment enhanced the proliferation, migration and
tube-like formation of the ECs. VEGF-A has also been demonstrated
to activate several downstream proteins. The activation of VEGFR2
by VEGF-A results in the activation of PI3K and Akt, leading to EC
survival (11) and activation of
the MAPK pathway, which leads to EC proliferation (39). It also leads to the activation of
FAK, which induces EC migration (9,11).
In the present study, the knockdown of paxillin inhibited the
VEGF-A-induced proliferation, migration and tube-like structure
formation of the HUVECs, suggesting that paxillin may mediate
multiple signaling pathways of VEGF-A in the regulation of EC
angiogenesis.
As an adapter protein within focal adhesions,
paxillin provides multiple docking sites for various signaling and
cytoskeletal proteins (40).
Studies investigating the effects of paxillin gene disruption have
demonstrated that paxillin-null cells exhibit abnormal focal
adhesions, a disrupted cytoskeleton, decreased phosphorylation of
FAK, decreased MAPK activation and reduced cell migration (41). Loss of Src and paxillin contributes
to EC defects, including decreased proliferation, migration and
angiogenesis of the ECs (42). The
tyrosine kinases FAK and Src, which are activated by adhesion and
growth factors, can phosphorylate paxillin and are, therefore,
important in the paxillin-mediated angiogenesis of the ECs
(9,11). In agreement with these previous
studies, the present study found that VEGF-A treatment increased
the phosphorylation of paxillin at Tyr 118 and Tyr 31. It has been
reported that the phosphorylation of paxillin at Tyr 118 by Src
activates the ERK signaling pathway, which leads to increased cell
survival (22). In response to
integrin activation, FAK activates PI3K and Akt indirectly through
Src, thereby leading to increased cell survival, elongation and
migration (43,44). Therefore, paxillin and FAK may
recruit and activate several signaling proteins to promote these
processes in the ECs.
In conclusion, the present study demonstrated that
paxillin knockdown inhibited the VEGF-A-induced proliferation,
migration, adhesion and capillary formation of HUVECs, suggesting
that these two factors are important in angiogenesis. Therefore,
paxillin may be a potential target for antiangiogenic therapies.
However, how paxillin is involved in cell migration, how it
regulates focal adhesions and how it interacts with VEGF and other
angiogenic factors remains to be elucidated. Therefore, further
understanding of the role of paxillin in angiogenesis is required
to facilitate the development of antiangiogenic therapies.
Acknowledgements
The authors would like to thank Mr. Hong Xia for his
administrative support for this study. Grant support was provided
by the National Natural Science Foundation of China (no.
81070708).
References
|
1
|
Chang JH, Gabison EE, Kato T and Azar DT:
Corneal neovascularization. Curr Opin Ophthalmol. 12:242–249. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Regenfuss B, Bock F, Parthasarathy A and
Cursiefen C: Corneal (lymph) angiogenesis - from bedside to bench
and back: a tribute to Judah Folkman. Lymphat Res Biol. 6:191–201.
2008. View Article : Google Scholar
|
|
3
|
Chevez-Barrios P: Are we getting closer to
prevention and treatment of corneal neovascularization? Clin
Experiment Ophthalmol. 35:689–690. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cursiefen C, Kuchle M and Naumann GO:
Angiogenesis in corneal diseases: histopathologic evaluation of 254
human corneal buttons with neovascularization. Cornea. 17:611–613.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gupta D and Illingworth C: Treatments for
corneal neovascularization: a Review. Cornea. 30:927–938. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lim P, Fuchsluger TA and Jurkunas UV:
Limbal stem cell deficiency and corneal neovascularization. Semin
Ophthalmol. 24:139–148. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rundhaug JE: Matrix metalloproteinases and
angiogenesis. J Cell Mol Med. 9:267–285. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ebrahem Q, Chaurasia SS, Vasanji A, et al:
Cross-talk between vascular endothelial growth factor and matrix
metalloproteinases in the induction of neovascularization in vivo.
Am J Pathol. 176:496–503. 2010. View Article : Google Scholar :
|
|
9
|
Hoeben A, Landuyt B, Highley MS, et al:
Vascular endothelial growth factor and angiogenesis. Pharmacol Rev.
56:549–580. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shibuya M: Differential roles of vascular
endothelial growth factor receptor-1 and receptor-2 in
angiogenesis. J Biochem Mol Biol. 39:469–478. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Olsson AK, Dimberg A, Kreuger J and
Claesson-Welsh L: VEGF receptor signalling - in control of vascular
function. Nat Rev Mol Cell Biol. 7:359–371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stefanini MO, Wu FT, Mac Gabhann F and
Popel AS: The presence of VEGF receptors on the luminal surface of
endothelial cells affects VEGF distribution and VEGF signaling.
PLoS Comput Biol. 5:e10006222009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Turner CE: Paxillin. Int J Biochem Cell
Biol. 30:955–959. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Brown MC and Turner CE: Paxillin: adapting
to change. Physiol Rev. 84:1315–1339. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Abedi H and Zachary I: Vascular
endothelial growth factor stimulates tyrosine phosphorylation and
recruitment to new focal adhesions of focal adhesion kinase and
paxillin in endothelial cells. J Biol Chem. 272:15442–15451. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Birukova AA, Cokic I, Moldobaeva N and
Birukov KG: Paxillin is involved in the differential regulation of
endothelial barrier by HGF and VEGF. Am J Respir Cell Mol Biol.
40:99–107. 2009. View Article : Google Scholar :
|
|
17
|
Oh J, Diaz T, Wei B, et al: TIMP-2
upregulates RECK expression via dephosphorylation of paxillin
tyrosine residues 31 and 118. Oncogene. 25:4230–4234. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Petit V, Boyer B, Lentz D, et al:
Phosphorylation of tyrosine residues 31 and 118 on paxillin
regulates cell migration through an association with CRK in NBT-II
cells. J Cell Biol. 148:957–970. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rios A, Hernandez-Ramirez VI, Moguel M, et
al: Participation of Rho, ROCK-2, and GAP activities during actin
microfilament rearrangements in Entamoeba histolytica induced by
fibronectin signaling. Cell Biol Int. 32:984–1000. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tsubouchi A, Sakakura J, Yagi R, et al:
Localized suppression of RhoA activity by Tyr31/118-phosphorylated
paxillin in cell adhesion and migration. J Cell Biol. 159:673–683.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dobkin-Bekman M, Naidich M, Rahamim L, et
al: A preformed signaling complex mediates GnRH-activated ERK
phosphorylation of paxillin and FAK at focal adhesions in L beta T2
gonadotrope cells. Mol Endocrinol. 23:1850–1864. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sachdev S, Bu Y and Gelman IH:
Paxillin-Y118 phosphorylation contributes to the control of
Src-induced anchorage-independent growth by FAK and adhesion. BMC
Cancer. 9:122009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jaffe EA, Nachamann RL, Becker CG and
Minick CR: Culture of human endothelial cells derived from
umbilical cord veins. Identification by morphologic and immunologic
criteria. J Clin Invest. 52:2745–2756. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
25
|
Turner NA and Moake J: Assembly and
activation of alternative complement components on endothelial
cell-anchored ultra-large von Willebrand factor links complement
and hemostasis-thrombosis. Plos one. 8:e593722013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shan C, Xu F, Zhang S, et al: Hepatitis B
virus X protein promotes liver cell proliferation via a positive
cascade loop involving arachidonic acid metabolism and p-ERK1/2.
Cell Res. 20:563–575. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ng YS, Krilleke D and Shima DT: VEGF
function in vascular pathogenesis. Exp Cell Res. 312:527–537. 2006.
View Article : Google Scholar
|
|
28
|
Tammela T, Enholm B, Alitalo K and
Paavonen K: The biology of vascular endothelial growth factors.
Cardiovasc Res. 65:550–563. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Van Geest RJ, Lesnik-Oberstein SY, Tan HS,
et al: A shift in the balance of vascular endothelial growth factor
and connective tissue growth factor by bevacizumab causes the
angiofibrotic switch in proliferative diabetic retinopathy. Br J
Ophthalmol. 96:587–590. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Velez-Montoya R, Clapp C, Rivera JC, et
al: Intraocular and systemic levels of vascular endothelial growth
factor in advanced cases of retinopathy of prematurity. Clin
Ophthalmol. 4:947–953. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ferrara N: Vascular endothelial growth
factor and age-related macular degeneration: from basic science to
therapy. Nat Med. 16:1107–1111. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen JY, Tang YA, Huang SM, et al: A novel
sialyltransferase inhibitor suppresses FAK/paxillin signaling and
cancer angiogenesis and metastasis pathways. Cancer Res.
71:473–483. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang YN, Wang F, Zhou W, Wu ZQ and Xing
YQ: TNF-alpha stimulates MMP-2 and MMP-9 activities in human
corneal epithelial cells via the activation of FAK/ERK signaling.
Ophthalmic Res. 48:165–170. 2012. View Article : Google Scholar
|
|
34
|
Lee S, Zheng M, Kim B and Rouse BT: Role
of matrix metalloproteinase-9 in angiogenesis caused by ocular
infection with herpes simplex virus. J Clin Invest. 110:1105–1111.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bischoff J: Cell adhesion and
angiogenesis. J Clin Invest. 100:S37–S39. 1997.PubMed/NCBI
|
|
36
|
Hynes RO: Integrins: versatility,
modulation, and signaling in cell adhesion. Cell. 69:11–25. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wary KK, Kohler EE and Chatterjee I: Focal
adhesion kinase regulation of neovascularization. Microvasc Res.
83:64–70. 2012. View Article : Google Scholar
|
|
38
|
Teranishi S, Kimura K and Nishida T: Role
of formation of an ERK-FAK-paxillin complex in migration of human
corneal epithelial cells during wound closure in vitro. Invest
Ophthalmol Vis Sci. 50:5646–5652. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Takahashi T, Yamaguchi S, Chida K and
Shibuya M: A single autophosphorylation site on KDR/Flk-1 is
essential for VEGF-A-dependent activation of PLC-gamma and DNA
synthesis in vascular endothelial cells. EMBO J. 20:2768–2778.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Turner CE: Paxillin interactions. J Cell
Sci. 113:4139–4140. 2000.PubMed/NCBI
|
|
41
|
Hagel M, George EL, Kim A, et al: The
adaptor protein paxillin is essential for normal development in the
mouse and is a critical transducer of fibronectin signaling. Mol
Cell Biol. 22:901–915. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lai KM and Pawson T: The ShcA
phosphotyrosine docking protein sensitizes cardiovascular signaling
in the mouse embryo. Genes Dev. 14:1132–1145. 2000.PubMed/NCBI
|
|
43
|
Shaw LM, Rabinovitz I, Wang HH, Toker A
and Mercurio AM: Activation of phosphoinositide 3-OH kinase by the
alpha6beta4 integrin promotes carcinoma invasion. Cell. 91:949–960.
1997. View Article : Google Scholar
|
|
44
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|