Introduction
Hepatic fibrosis (HF) is recognized as one of the
most common types of liver disease, as well as one that is
resistant to the majority of current therapies, resulting in
significant global morbidity (1).
HF has been defined as a tissue-specific response to long-term
injury or illnesses, including chronic viral hepatitis, alcoholic
liver disease, cholestasis, circulatory disturbances, autoimmune
liver disease or one of a number of nutritional disorders (2,3).
Liver fibrosis is characterized by the excessive deposition of
extracellular matrix (ECM) proteins, consisting predominantly of
type I and type III collagen. These abnormal depositions disturb
the structure of the hepatic lobule, misdirecting blood flow in the
liver and thereby disturbing its healthy functioning. This leads to
liver cirrhosis and, ultimately, to liver carcinoma (4). Although numerous therapeutic options
are currently available for liver fibrosis, all have limited
degrees of success and none were capable of producing a complete
cure (5). Thus, there is an urgent
need to develop better preventative options as well as treatment
approaches, based on a more thorough understanding of the
pathogenesis of hepatic fibrosis.
Although the exact pathophysiological mechanisms
underlying the formation of hepatic fibrosis are elusive, there are
a number of potential processes that may be worthy of
investigation. Hepatic stellate cells (HSCs) are an important type
of fibrogenic liver cell. They are found during liver injury and
are known to be responsible for the progression of hepatic fibrosis
(6). These cells may be activated,
which induces their transdifferentiation into myofibroblasts
(MFBs). MFBs are characterized by a number of fibrotic functions,
including the induction of ECM deposition, α-smooth muscle actin
(α-SMA) expression, as well as the synthesis and secretion of type
I and type III collagen (7,8). A
growing body of evidence has documented that inhibition of the
transformation of HSCs may aid in the prevention and cure of liver
fibrosis (9). However, HSCs are
not the only mechanism through which fibrosis progresses. A number
of studies have indicated that this process is complicated and
involves numerous cytokines and signaling pathways (10,11).
Transforming growth factor (TGF)-β1 has been
identified as the most significant factor involved in the
activation and promotion of the transformation of HSCs (12). Previous studies have demonstrated
that TGF-β1 is highly expressed in numerous tissues, which exhibit
fibrosis. Furthermore, TGF-β1 has been shown to have a key role in
the development of fibrosis by promoting the proliferation of
tissue fibroblasts and the synthesis of collagen (13). The following stages of the
cell-signaling pathway of TGF-β1 have been elucidated: The initial
step is the transmission of a signal from the activated
transmembrane receptor, TβR. This signal is then transmitted to the
nucleus, predominantly through intracellular members of the Smad
family. Further evidence for this mechanism has been provided by
studies in which the TGF-β/Smad pathway was inhibited via RNA
interference. This resulted in a significant decrease of type I and
III collagen expression in fibrotic livers (14). Based on these findings, subsequent
studies have confirmed the potential use of members of the Smad
family as biomarkers with which to predict liver disease
progression (15). Thus, the
TGF-β/Smad pathway is hypothesized to be a target for the treatment
and cure of hepatic fibrosis.
Astragalus and Paeoniae Radix Rubra
extract (APE) is produced from a variety of herbs
(Astragali Radix, Paeoniae Radix Rubra,
Curcumae Rhizoma, Bupleuri Radix and Eupolyphaga)
with a standard ratio of 30:30:15:12:10 by crude herb weight and
contains the active components paeoniflorin, astragalosides and
curzenone (16). Astragali
Radix is thought to have restorative properties in Chinese
Medicine, and studies have demonstrated that it is a
hepatoprotective agent (17,18).
Extracts from Paeoniae Radix Rubra, Curcumae Rhizoma,
Bupleuri Radix and Eupolyphaga also have the potential to
suppress liver fibrosis and have been considered potent herbs with
which to treat liver disease (19). Previous in vivo experiments
have been conducted, which demonstrated that APE administered at a
ratio of 30:30:15:12:10 exhibited superior antifibrotic effects
compared to administration of individual herbs in a model of
CCl4-induced chronic liver injury (16). However, the molecular mechanisms
underlying the hepatoprotective effects of APE have remained
elusive. Therefore, the present study aimed to investigate the
effects of APE on proliferation, invasion and activation of HSCs.
In addition, the mechanisms underlying the effect of APE on the
TGF-β/Smad pathway were examined.
Materials and methods
Preparation of APE
The following herbs were purchased at Xi’an Chinese
Medicine Corporation (Xi’an, China): Astragali Radix,
Paeoniae Radix Rubra, Curcumae Rhizoma,
Bupleuri Radix and Eupolyphaga. Each herb was
identified by Dr Genquan Qiu, an author of the study and a
specialist in Traditional Chinese Herbal Medicine. Herb samples
were preserved in the specimen room of the Institute of Clinical
Pharmacology at the Xi’an Medical College (Xi’an, China). The
process of extracting and preparing the APE components from the
five herbs was as follows: A total amount 8.45 kg dried sliced
crude herbs (Astragali Radix, Paeoniae Radix Rubra,
Curcumae Rhizoma, Bupleuri Radix and
Eupolyphaga) in a standard ratio of 30:30:15:12:10 were
decocted in 80 l water three times at 95°C for 35 min. The decocted
solution was filtered though 150 μm gauze ( Sigma-Aldrich, St.
Louis, MO, USA) and the combined filtrates were then concentrated
to a mass of 4.22 kg in a vacuum desiccator at 70°C. The sediment
was dried into power using a spray drier at a temperature range of
80–160°C. This process yielded 2.11 kg of dry powder. It should be
noted that in all subsequent in vitro cell experiments, APE
powder was dissolved in Hank’s solution (Sigma-Aldrich).
Cell culture
Male Sprague-Dawley rats were obtained from the
Experimental Animal Center of Anhui Medical University (Anhui,
China) and had a weight range of 160–200g. HSCs were separated from
normal rat liver and maintained in Dulbecco’s modified Eagle’s
medium (DMEM; Sigma-Aldrich), supplemented with 10% fetal bovine
serum (FBS; Sigma-Aldrich) (20).
Cells were cultured in an environment of 95% air and 5%
CO2 at 37°C. All experiments were performed during the
exponential growth phase, once the cells had been plated for 24
h.
Cell proliferation assay
HSCs were seeded in 96-well plates at a density of
1×104 cells/well and cultured in 200 μl DMEM
supplemented with 10% FBS. Following overnight starvation in
serum-free medium, HSCs were incubated with 10% FBS with various
concentrations of APE (5, 10, 20, 40 or 80 μg/ml) for 16 h. Control
conditions consisted of cells which had an equal volume of
serum-free medium added without the addition of APE. Following
incubation, cell proliferation was assessed using an MTT assay
(Abcam, Cambridge, UK), as previously described (21). Absorbance was measured in an ELISA
microplate reader at 562 nm (FlexStation 3; Molecular Devices,
Sunnyvale, CA, USA). Data are expressed as the mean of a minimum of
three independent experiments.
Cell invasion assay
Transwell invasion chambers with 8-μm membrane pores
coated with Matrigel™ (BD Biosciences, San Jose, CA, USA) were
placed in 24-well plates. HSCs were starved overnight in serum-free
DMEM. Cells were then seeded in the upper compartment of the
chamber at a concentration of 1×105 cells/well and
incubated with or without the administration of APE at various
concentrations (10, 20, 40 or 80 μg/ml) for 24 h. Medium containing
TGF-β1 (40 pmol/l) was placed into the lower compartment of the
chamber. Serum-free medium was used for the control group.
Following incubation, cells on the upper side of the membrane were
completely removed using a cotton swab. Cells which had crossed the
Matrigel barrier and migrated to the lower side of the chamber were
fixed with 100% methanol for 1 min and stained with hematoxylin and
eosin (H&E; Sigma-Aldrich). The number of invading cells was
counted in five randomly selected fields at ×100 magnification
(CX31; Olympus Corp., Tokyo, Japan). The mean number of invading
cells in these fields was calculated and the experiments were run
in triplicate.
Collagen synthesis assay
Collagen synthesis was evaluated using a
3H-proline incorporation assay (22). In brief, HSC cells were seeded in
96-well plates at a density of 1×104 cells per well for
at least 24 h. Cells were synchronized by culturing in serum-free
medium overnight prior to incubation with 40 pmol/l TGF-β1 in the
presence of APE at various concentrations (10, 20, 40 or 80 μg/ml)
for 24 h. The control group received an equal volume of serum-free
medium without APE. The aforementioned cultures were subsequently
exposed to 3H-proline (37 kBq; Atom-Hitech, Beijing,
China) for 12 h. Following proline exposure, the cells were washed
twice with phosphate-buffered saline (PBS; Sigma-Aldrich), treated
with ice-cold 5% trichloroacetic acid (Sigma-Aldrich) for 1 h and
then washed twice more with distilled water. Finally, cells were
lysed with 0.25% trypsogen and counted in a liquid scintillation
counter (LS6000SE; Beckman Coulter, Fullerton, CA, USA).
Immunoblot analysis
HSCs were seeded at a density of 1×106
cells per dish. Once they reached sub-confluency, the cells were
cultured in serum-free medium for 24 h in the presence or absence
of APE at various concentrations (10, 20 or 40 μg/ml). Cells were
then treated with 40 pmol/l TGF-β1 for 30 min. Control cells were
treated with neither TGF-β1 nor APE. Following incubation, cells
were homogenized using a modified radioimmunoprecipitation assay
buffer (50 mM Tris-HCl, pH 7.4; 1% NP-40; 150 mM NaCl; and 1 mM
EDTA; Sigma-Aldrich) supplemented with protease and phosphatase
inhibitors (1 mM phenylmethyl sulfonyl fluoride, 0.1 mM
N-tosyl-L-phenylalanine chloromethyl ketone, 1 mg/ml
aprotinin, 1 mg/ml pepstatin, 0.5 mg/ml leupeptin, 1 mM NaF, 1 mM
Na4P2O4 and 2 mM
Na3VO4; Sigma-Aldrich). The extract was
centrifuged at 16,000 × g for 30 min at 4°C in order to remove cell
debris. The supernatant was collected and quantified using the
bicinchoninic acid protein assay (Pierce Biotechnology, Rockford,
IL, USA) and boiled for 5 min with SDS sample buffer (100 mM
Tris-HCl, pH 6.8; 4% SDS; 12% β-mercaptoethanol; 20% glycerol; and
0.01% bromophenol blue; Sigma-Aldrich) at the equivalent protein
level. Samples were subjected to SDS-PAGE and transferred to
polyvinylidene difluoride membranes (Bio-Rad Laboratories,
Hercules, CA, USA). The membranes were then blocked with 10%
skimmed milk powder (NanRong International Corp., Kaohsiung,
Taiwan) in PBS containing 0.1% Tween-20 (Sigma-Aldrich) and
incubated with the following primary antibodies: Mouse anti-human
monoclonal antibodies against PAI-1 (1:2,000 mouse monoclonal,
ab125687) and uPA (1:1,500, mouse monoclonal, ab82220), which were
purchased from Abcam; mouse anti-human monoclonal antibodies
against TGF-β1 (1:1,000, mouse monoclonal, sc-130348) Santa Cruz
Biotechnology, Inc., Dallas, TX, USA); rabbit anti-human
phospho-Smad2 antibody (1:500, mouse monoclonal, BS3725), rabbit
anti-human Smad2 antibody (1:500, mouse monoclonal, BS2993), rabbit
anti-human phospho-Smad3 antibody (1:500, mouse monoclonal,
BS4874), rabbit anti-human Smad3 antibody, (1:500, polyclonal,
AP0446), rabbit anti-Smad7 antibody (1:500, polyclonal, BS60366)
and rabbit anti-β-actin antibody (1:4,000, polyclonal, AP0060),
which were purchased from Bioworld Technology (St. Louis, MO, USA);
as well as rabbit anti-α-SMA antibody (1:2,000, polyclonal,
BS70000; Cell Signaling Technology, Danvers, MA, USA), overnight at
4°C. Following application of the primary antibodies, membranes
were incubated with the appropriate secondary antibodies against
mouse and rabbit (mouse IgG, 1:2,000, sc-2025; rabbit IgG 1:2,000,
sc-2027; Santa Cruz Biotechnology, Inc.) for 2 h at room
temperature. Finally, immunoreactivity was visualized using
enhanced chemiluminescnce (Amersham Pharmacia Biotech, Picastaway,
NJ, USA) and autoradiography. The resulting images were subjected
to densitometric analysis using Quantity One software (Bio-Rad
Laboratories).
Immunofluorescence
HSCs were seeded onto 24-well plates at
5×103 cells per well. Cells were then incubated in
serum-free medium for 24 h with or without treatment with APE at
various concentrations (10, 20 or 40 μg/ml) and stimulated with 60
pmol/l TGF-β1 for 1 h. Control cells were incubated in serum-free
medium with neither TGF-β1 nor APE. Following incubation, cells
were fixed with 4% paraformaldehyde for 30 min, permeabilized and
then blocked with 0.1% saponin and 0.5% bovine serum albumin
(Sigma-Aldrich) in PBS for 30 min at 4°C. Cells were incubated
overnight at 4°C with the primary antibodies described above, and
then incubated with fluorescein isothiocyanate-conjugated goat
anti-rabbit immunoglobulin G (1:100) for 2 h at room temperature.
Slides were then mounted with 80% phosphoglycerol, and visualized
under a fluorescence microscope (CX31; Olympus Corp.). In each
experiment, five randomly selected fields were analyzed from each
sample.
Reverse transcription-polymerase chain
reaction (RT-PCR)
HSCs were seeded at 1×106 cells per dish.
Cells were cultured in serum-free medium for 24 h with or without
treatment with APE at various concentrations (10, 20 or 40 μg/ml)
treatment and stimulated with 10 pmol/l TGF-β1 for 3 h prior to
harvesting. Control cells were cultured in an equal volume of
serum-free medium without TGF-β1 stimulation. The mRNA levels of
HSCs were analyzed using an RT-PCR assay. Total sample mRNA was
extracted using Trizol reagent (Invitrogen Life Technologies,
Carlsbad, CA, USA) according to the manufacturer’s instructions.
Revert Aid First Strand cDNA Synthesis kit (Fermentas, Vilnius,
Lithuania) was used to produce corresponding cDNA. The primers for
plasminogen activator inhibitor type 1 (PAI-1), urokinase-type
plasminogen activator (uPA) and β-actin (Sangon Biotech, Shanghai,
China) were as follows: Forward, 5′-CGGAGCACGGTCAAGCAAGTG-3′ and
reverse, 5′-GTTGAGGGCAGAGAGAGGCGC-3′ for PAI-1; forward,
5′-ACTACTACGGCTCTGAAGTCACCA-3′ and reverse,
5′-GAAGTGTGAGACTCTCGTGTAGAC-3′ for uPA; and forward,
5′-CTCCATCCTGGCCTCGCTGT-3′ and reverse, 5′-GCTGTCACCTTCACCGTTCC-3′
for β-actin. All target sequences were separately amplified for
30–35 cycles of 30 sec at 94°C, 30 sec at 55°C and 60 sec at 72°C.
Samples of each reaction product were separated by agarose gel
electrophoresis (Sigma-Aldrich), visualized by ethidium bromide
staining (Sigma-Aldrich) and visualized using 290 nm ultraviolet
illumination (E1617-T130; Bio-Rad Laboratories). The density of
each band was measured by densitometry using Quantity One 4.52
software (Bio-Rad Laboratories).
Statistical analyses
Statistical analysis was performed using SPSS
software (SPSS standard version 17.0; SPSS, Inc., Chicago, IL,
USA). Data are expressed as the mean ± standard deviation.
Comparisons between groups were made using Student’s t-test and the
Mann-Whitney rank sum test, the latter of which was used to compare
the degree of staining intensity. P<0.05 was considered to
indicate a statistically significant difference.
Results
APE suppresses HSC proliferation induced
by FBS
In order to detect the effect of APE on HSCs
proliferation, cells were stimulated with 10% FBS and treated with
varying concentrations of APE (5–80 μg/ml). As shown in Table I, 10% FBS increased the
proliferation of HSCs, while the administration of APE (5–80 μg/ml)
resulted in a suppression of HSC proliferation. The effect of
APE-induced HSC suppression occurred in a dose-dependent manner,
with an IC50 of 22.45 μg/ml.
 | Table IEffects of APE on viability of HSCs
treated with 10% FBS. |
Table I
Effects of APE on viability of HSCs
treated with 10% FBS.
| Group | APE dose
(μg/ml) | Absorbance
(A570) | Inhibition (%) |
|---|
| Control | - | 0.48±0.05 | - |
| FBS (12.5%) | - | 1.04±0.1c | - |
| APE | 5 | 0.91±0.07a | 18.03 |
| 10 | 0.83±0.05a | 29.46 |
| 20 | 0.73±0.04b | 43.68 |
| 40 | 0.51±0.04b | 78.42 |
| 80 | 0.48±0.04b | 90.63 |
APE inhibits cell invasion induced by
TGF-β1
The invasion capability of HSCs was investigated
using a Transwell invasion assay. As shown in Fig. 1A, HSCs moved to the lower
compartment of the chambers across the Matrigel-coated
polycarbonate membrane when stimulated by treatment with TGF-β1.
Treatment with APE (10, 20, 40, 80 μg/ml) significantly reduced the
number of TGF-β1-stimulated cells that invaded across the
polycarbonate membrane. In accordance with previous results, this
effect occurred in a dose-dependent manner (23). Thus, TGF-β1-induced HSC invasion
into the bottom chamber and across the collagen IV/collagen
I-coated polycarbonate membrane was inhibited by APE (Fig. 1A and B).
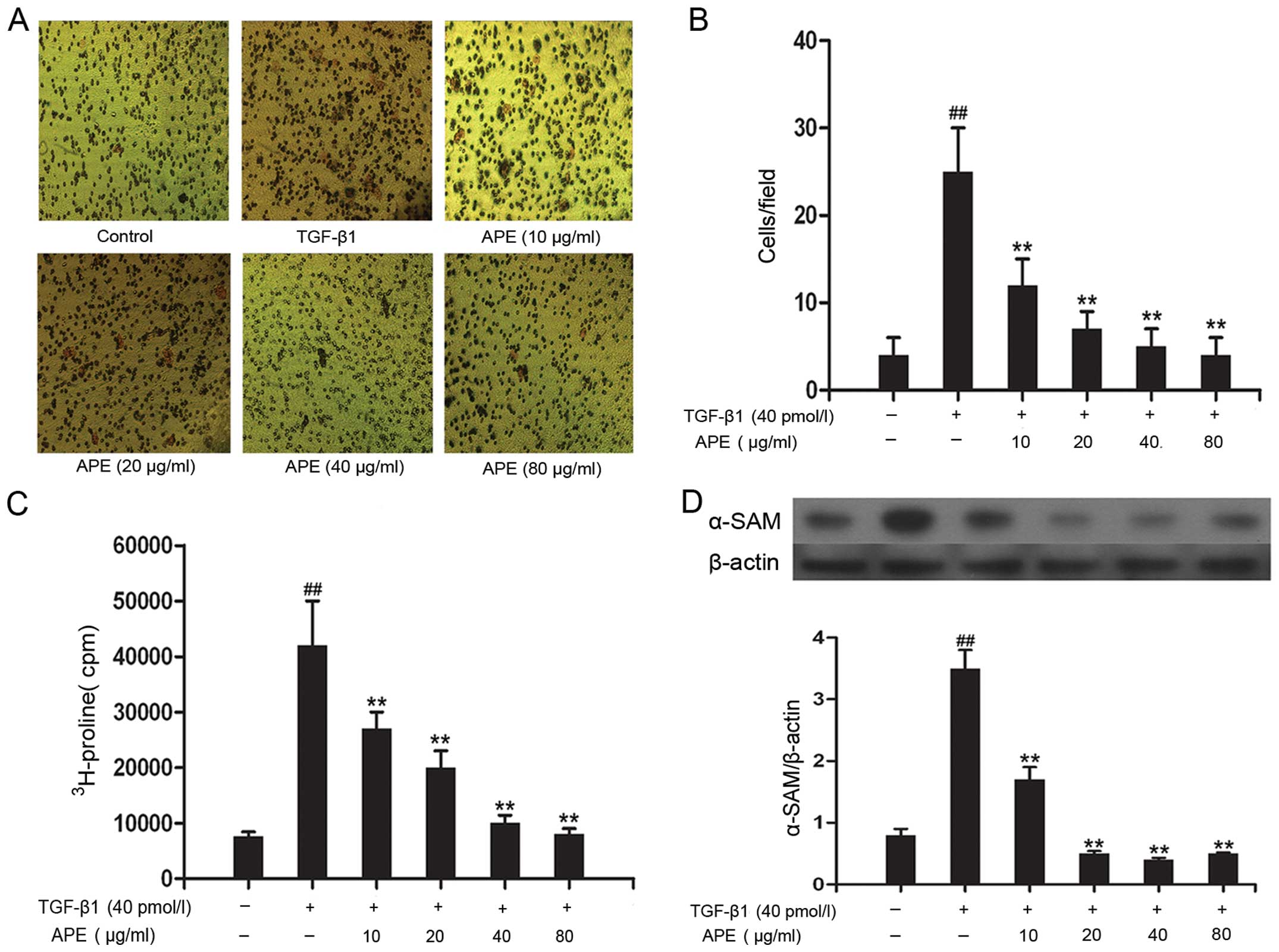 | Figure 1Effects of APE on TGF-β1-induced
invasion and activation of HSCs. (A and B) Number of cells
resulting from stimulation of HSCs by TGF-β1 treatment exhibited a
significant, dose-dependent suppression when treated with APE (10,
20, 40 or 80 μg/ml) (magnification, ×100). (C) TGF-β1-stimulated
HSC collagen synthesis was significantly and dose-dependently
decreased following treatment with APE (10, 20, 40 or 80 μg/ml).
(D) Expression of α-SAM was significantly decreased
following treatment with APE (10, 20, 40 or 80 μg/ml) compared with
that in the control. ##P<0.05 compared with control
group and **P<0.05 compared with group treated with
TGF-β1 alone. APE, Astragalus and Paeoniae Radix
Rubra extract; TGF-β1, transforming growth factor-β1; α-SAM,
α-sterile alpha motif; HSC, hepatic stellate cells. |
APE reduces TGF-β1-induced HSC
activation
Collagen synthesis was measured using a
3H-proline incorporation assay. As shown in Fig. 1C, collagen synthesis in
TGF-β1-stimulated HSCs was markedly increased compared with that of
control cells (P<0.01). Furthermore, treatment with APE (10, 20,
40, 80 μg/ml) significantly reduced TGF-β1-induced collagen
synthesis in a dose-dependent manner. Protein expression of α-SAM
was then used to measure HSC activation. As shown in Fig. 1D, the expression of the α-SAM
protein following treatment with TGF-β1 alone was 3.5-fold higher
than that in the control group. Furthermore, administration of APE
(10, 20, 40, 80 μg/ml) decreased α-SAM expression in a
dose-dependent manner. These results indicate that APE inhibits
liver fibrosis by suppressing HSC activation and collagen
synthesis.
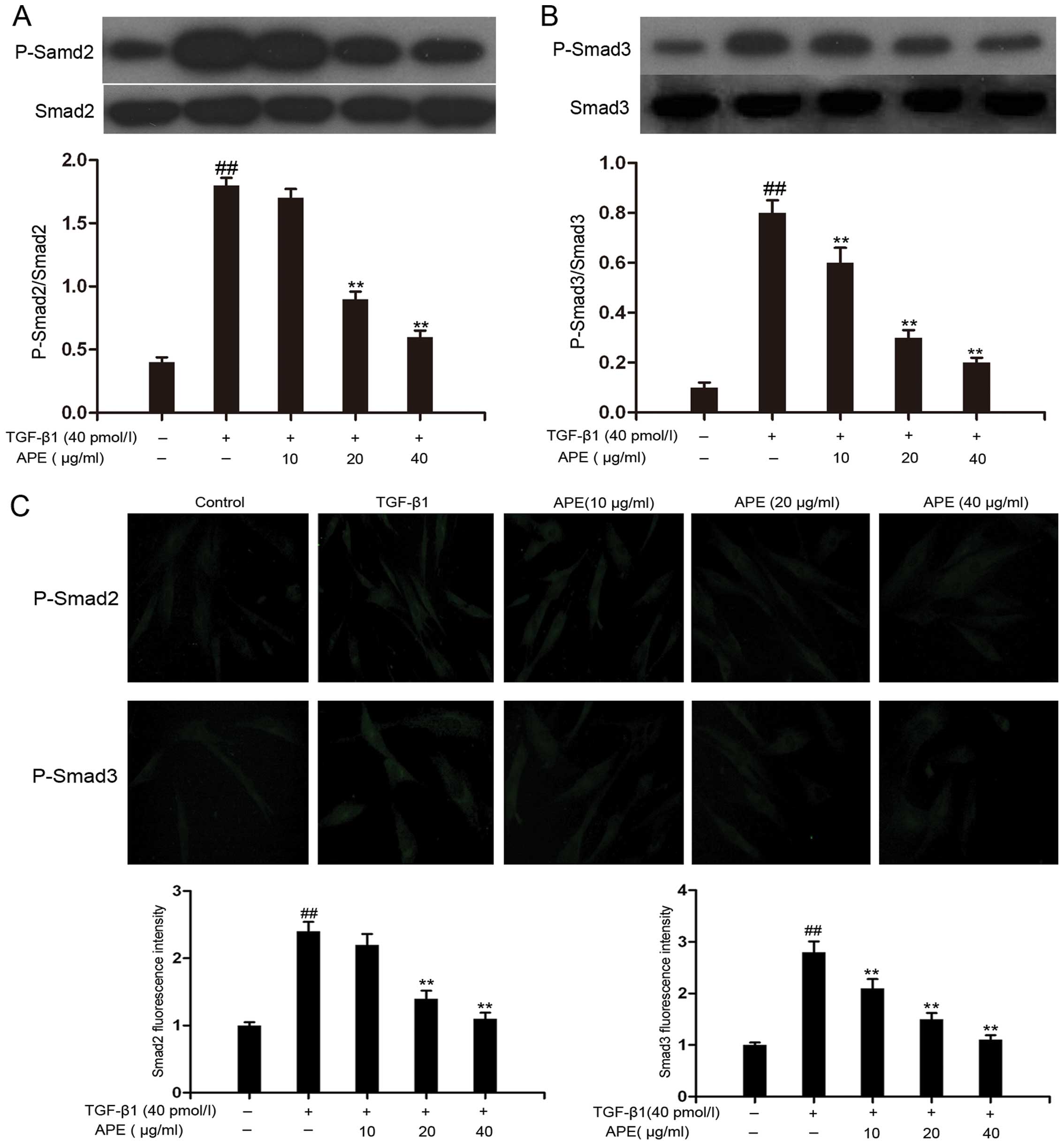
APE reduces TGF-β1-induced receptor
regulated (R)-Smad phosphorylation in HSCs
R-Smad phosphorylation was detected by western blot
and immunofluorescence analyses. As shown in Fig. 2A and B, western blotting results
demonstrated that TGF-β1 significantly increased the levels of
Smad-2 phosphorylation and moderately enhanced Smad-3
phosphorylation. This increase in R-Smad phosphorylation was
subsequently suppressed by the administration of APE (20, 40 μg/ml)
in a dose-dependent manner. Of note, a 10 μg/ml dosage of APE
significantly reduced Smad-3, but not Smad-2 phosphorylation.
Immunofluorescence also demonstrated that the expression of
phosphorylated R-Smad was elevated as a result of TGF-β1 treatment
(Fig. 2C). Treatment with APE (20,
40 μg/ml) significantly reduced the fluorescence intensity of
phosphorylated Smad2 and Smad3 in a dose-dependent manner. In
accordance with the results from the western blotting experiments,
a 10 μg/ml dosage of APE reduced the fluorescence intensity of
phosphorylated Smad3, but had no detectable effect on that of
phosphorylated Smad-2. These results indicated that APE decreased
TGF-β1-induced R-Smad phosphorylation.
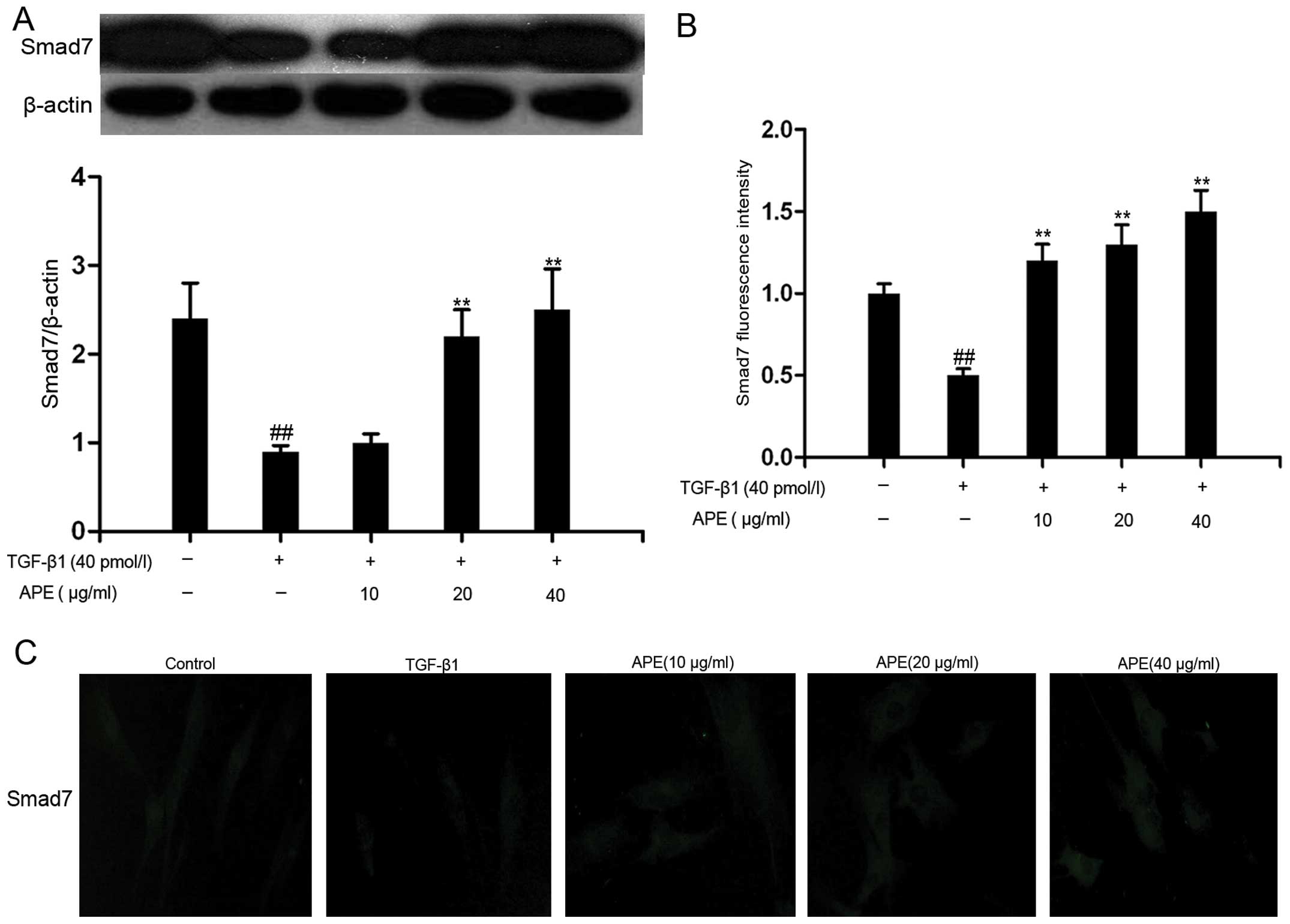
APE enhances Smad7 expression in HSCs,
reversing downregulation of Smad7 by TGF-β1
As shown in Fig.
3A, western blot analysis demonstrated that the protein
expression of Smad7 was significantly reduced following treatment
with TGF-β1 compared with that in the controls. However,
administration of high doses of APE (20 and 40 μg/ml) was able to
inverse this effect, leading to an increase in the expression of
Smad7. Immunofluorescence demonstrated that TGF-β1 reduced the
fluorescence intensity of Smad7, whereas treatment with APE (10, 20
or 40 μg/ml) increased Smad7 fluorescence intensity (Fig. 3B and C). These results indicated
that APE at high doses increased the expression of Smad-7.
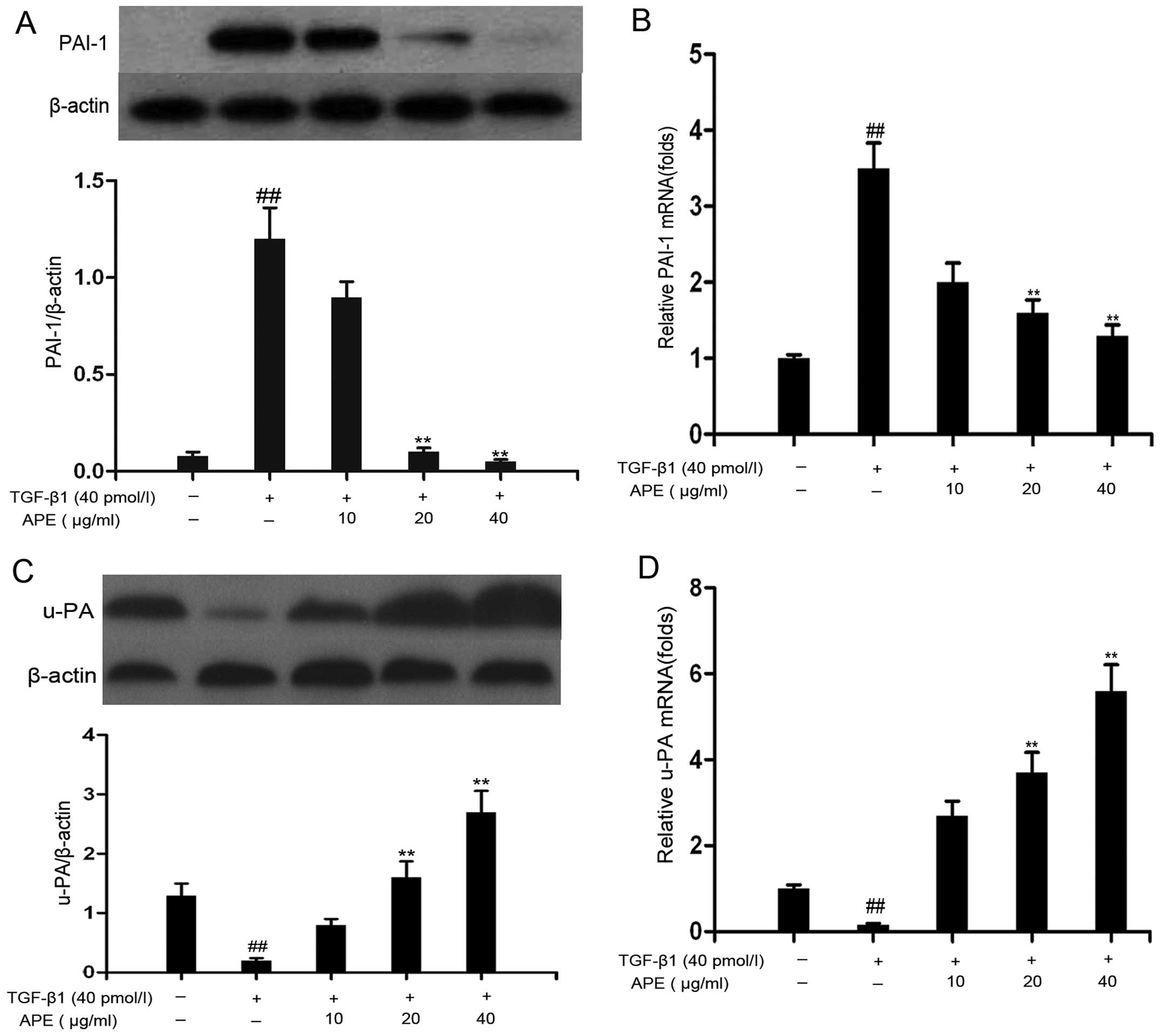
APE suppresses PAI-1 and increases uPA
expression, reversing the effects of TGF-β1 on HSCs
As shown in Fig.
4A, protein expression of PAI-1 was markedly increased in HSCs
treated with TGF-β1 compared with that in the control group.
However, treatment with APE (20 or 40 μg/ml) suppressed protein
expression in a dose-dependent manner. TGF-β1 induced an increase
of ~3.5-fold in PAI-1 mRNA expression compared with controls, while
APE suppressed the transcriptional activity of PAI-1 is a
dose-dependent manner. This inhibitory effect of APE was
statistically significant at higher concentrations (20 and 40
μg/ml; Fig. 4B). In accordance
with these results, TGF-β1 significantly reduced uPA protein levels
in HSCs, while APE, at concentrations of 20 or 40 μg/ml, completely
restored uPA protein expression levels in TGF-β1-treated cells
(Fig. 4C). The transcriptional
activity of uPA was also reduced following TGF-β1 treatment, while
administration of APE at doses of 20 or 40 μg/ml resulted in an
increase in uPA mRNA levels (Fig.
4D). These results demonstrated that APE treatment decreases
PAI-1 expression and restores that of uPA.
Discussion
Liver fibrosis is a chronic disease that is a
culmination of the insidious effects of liver degeneration. In
recent years, the clinical focus has concentrated predominantly on
the prophylaxis and treatment of primary liver disease, including
chronic hepatitis, or in trying to avoid contact with hepatotoxic
substances (24–27). It has been shown that certain
active components found in traditional herbal plants are effective
in suppressing liver fibrosis and collagen synthesis, which
indicated a potential therapeutic option for liver fibrosis
(28–30). One of these studies demonstrated
that APE significantly reduced levels of serum glutamic pyruvic
transaminase, glutamic oxaloacetic transaminase and hydroxyproline
in liver homogenates (31). Of
note, APE also attenuated the pathological changes characteristic
of liver fibrosis, which had been induced by CCl4
(16). In the present study, the
results showed that APE markedly suppressed hepatic stellate cell
proliferation and invasion, as well as their activation.
Furthermore, these inhibitory effects may be due to the effects of
APE on the TGF-β/Smad pathway.
HF results from the excessive secretion of matrix
proteins by HSCs, a process which is primarily triggered by TGF-β1.
A recent study suggested that garlic extract exhibits therapeutic
effects in liver fibrosis through inhibition of TGF-β1 in HSCs
(32). Other studies have
demonstrated that proanthocyanidin from grape seed extract exerts
protective hepatocellular effects, which resulted in the
amelioration of murine liver fibrosis, induced by TGF-β1 (33). In addition, using TGF-β1 small
interfering RNA to inhibit the expression of TGF-β1 attenuated rat
hepatic fibrosis induced by a high-fat diet and CCl4
(34). The results of the present
study confirmed that administration of TGF-β1 significantly
increased collagen synthesis in HSCs, which was hypothesized to be
involved in the formation of liver fibrosis. APE suppressed the
proliferation induced by FBS and the cell invasion induced by
TGF-β1 in HSCs in a dose-dependent manner. In addition, APE reduced
the collagen synthesis and the expression of α-SAM in HSCs, also in
a dose-dependent manner. These results indicated that TGF-β1 exerts
pro-fibrotic activity in HSCs, primarily through a dual action of
collagen synthesis promotion and the inhibition of collagen
degradation. These combined effects ultimately lead to excessive
collagen deposition.
TGF-β1 controls a diverse set of cellular process
and its canonical signaling is mediated via the TGF-β-induced
phosphorylation of receptor-activated Smad2 and Smad3 (35). It was recently shown that the
expression of the phosphorylated Smad2 and Smad3 proteins was
higher in samples of CCl4-induced liver fibrosis,
compared with that in the controls (36). Furthermore, overexpression of Smad
ubiquitin regulatory factor 2 suppressed TGF-β-mediated liver
fibrosis (37). Finally,
co-treatment with Boswellia serrata and Salvia
miltiorrhiza extracts reduced dimethylnotrosamine-induced
hepatic fibrosis in mice via downregulation of phosphorylated Smad3
(38). The present study showed
that APE inhibited the phosphorylation of Smad2 and Smad3 which was
induced by TGF-β1 in HSCs, in a dose-dependent manner. It was
therefore hypothesized that the reduction of phosphorylated Smad2
and -3 by APE may underlie its inhibitory effects on HSCs.
Smad7 serves as the negative feedback regulator for
TGF-β signaling, acting to antagonize the activity of the
receptor-regulated Smads, which leads to a termination of the TGF-β
signal (39). Suppression of Smad7
by DNA methyltransferase 1 promoted the phosphorylation of Smad2
and Smad3 that had been induced by HSC activation, or liver
fibrosis in general (40). Recent
studies have shown that hepatocytes are more sensitive to the
effects of TGF-β. This resulted in enhanced cell death in S7DeltaE1
and wild-type mice that had a deletion of exon I from the
endogenous Smad7 gene. A study demonstrated that hepatocytes are
more sensitive to the effects of TGF-β. In these mice, an increase
in oxidative stress and an increase in cell damage in response to
CCl4 was observed (41). Smad7-overexpression in common bile
duct ligation rats reduced the expression of collagen and α-SMA, as
well as the hydroxyproline content in the liver (42). The present study also showed that
TGF-β1-induced reduction in Smad7 expression was ameliorated by
treatment with APE in a dose-dependent manner. Smad7 has recently
been used successfully to abrogate TGF-β signaling in a number of
fibrotic diseases, resulting in decreased collagen or α-SMA
expression as a result of inhibition of the phosphorylation of
Smads (43). Therefore, it is
possible that the inhibition of Smad activation caused by the
application of APE may be partly due to an upregulation in Smad7
expression, with a resultant reduction in collagen production.
uPA and plasmin are involved in the cellular
proteolytic degradation of ECM proteins and in the maintenance of
tissue homeostasis. The activities of uPA/plasmin predominantly
rely on the functioning of a potent inhibitor of PA, PAI-1
(44). Under normal physiological
conditions, PAI-1 controls the activities of the uPA/plasmin system
and thus maintains tissue homeostasis. Recently, a number of
studies have demonstrated that co-expression of Smad7 and uPA
attenuates CCl4-induced liver fibrosis in rats (45,46).
Furthermore, the administration of Genistein was shown to modify
liver fibrosis and improve liver function by inducing uPA
expression in CCl4-treated rats (47). PAI-1 levels are known to be
significantly elevated in fibrotic tissues and a lack of PAI-1
protects certain organs from fibrosis in response to
injury-associated, pro-fibrotic signals (48). Liver fibrosis produces
intrinsically high levels of PAI-1 and low levels of urokinase-type
plasminogen activator. This altered ratio of activator and
inhibitor activities is an important factor contributing to altered
fibrin degradation and the subsequent ECM metabolism. The imbalance
ultimately aids in the formation of liver fibrosis (49). In the present study, TGF-β1 was
shown to increase mRNA and protein levels of PAI-1 and decrease
those of uPA, while APE significantly suppressed TGF-β1-induced
PAI-1 expression and increased uPA expression in the presence of
TGF-β1 in a dose-dependent manner, therefore reversing the effects
of TGF-β1. In view of growing evidence suggesting that elevated
PAI-1 expression and reduced uPA expression are essential in
collagen accumulation in HSCs, the APE-induced decrease in PAI-1
mRNA and protein expression may interfere with collagen deposition,
thus preventing keloid formation.
In conclusion, the present study demonstrated that
APE inhibited cell proliferation, invasion and collagen synthesis
in HSCs, and that the mechanisms underlying these effects may
involve the TGF-β/Smad signaling pathway. These results provided
evidence for the promising potential therapeutic use of APE in
liver fibrosis.
Acknowledgements
The authors would like to thank Miss Yin Zhou and
Mr. Xiaopeng Tian for their technical assistance, and the
Department of Pathology at Peking University, Shen Zhen Hospital
for providing and processing our samples. This study was supported
by research grants from the Nature Science Foundation of Xi’an
Medical College (grant no. L12C01).
References
|
1
|
Ray K: Liver: Hepatic stellate cells hold
the key to liver fibrosis. Nat Rev Gastroenterol Hepatol.
11:742014. View Article : Google Scholar
|
|
2
|
Kocabayoglu P and Friedman SL: Cellular
basis of hepatic fibrosis and its role in inflammation and cancer.
Front Biosci (Schol Ed). 5:217–230. 2013.
|
|
3
|
Mello T, Ceni E, Surrenti C and Galli A:
Alcohol induced hepatic fibrosis: role of acetaldehyde. Mol Aspects
Med. 29:17–21. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jian YC, Li W, He Y, Jiang M, Liu YB and
Xiong WJ: Effect of oxymatrine on hepatic gene expression profile
in experimental liver fibrosis of rats. Chin J Integr Med.
18:445–450. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Parise ER, de Oliveira AC, Conceicao RD,
Amaral AC and Leite K: Response to treatment with interferon-alpha
and ribavirin in patients with chronic Hepatitis C virus genotypes
2 and 3 depends on the degree of hepatic fibrosis. Braz J Infect
Dis. 10:78–81. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tsolaki E, Athanasiou E, Gounari E, Zogas
N, Siotou E, Yiangou M, Anagnostopoulos A and Yannaki E:
Hematopoietic stem cells and liver regeneration: differentially
acting hematopoietic stem cell mobilization agents reverse induced
chronic liver injury. Blood Cells Mol Dis. 53:124–132. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Trebicka J, Hennenberg M, Odenthal M, et
al: Atorvastatin attenuates hepatic fibrosis in rats after bile
duct ligation via decreased turnover of hepatic stellate cells. J
Hepatol. 53:702–712. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Elsharkawy AM, Oakley F and Mann DA: The
role and regulation of hepatic stellate cell apoptosis in reversal
of liver fibrosis. Apoptosis. 10:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moreira RK: Hepatic stellate cells and
liver fibrosis. Arch Pathol Lab Med. 131:1728–1734. 2007.PubMed/NCBI
|
|
10
|
Huang Q, Huang R, Zhang S, et al:
Protective effect of genistein isolated from Hydrocotyle
sibthorpioides on hepatic injury and fibrosis induced by chronic
alcohol in rats. Toxicol Lett. 217:102–110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Su LJ, Chang CC, Yang CH, et al:
Graptopetalum paraguayense ameliorates chemical-induced rat hepatic
fibrosis in vivo and inactivates stellate cells and Kupffer cells
in vitro. PLoS One. 8:e539882013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu MY, Hu JJ, Shen J, Wang ML, Zhang QQ,
Qu Y and Lu LG: Stat3 signaling activation crosslinking of TGF-β1
in hepatic stellate cell exacerbates liver injury and fibrosis.
Biochim Biophys Acta. 1842.2237–2245. 2014.
|
|
13
|
Lang Q, Liu Q, Xu N, et al: The
antifibrotic effects of TGF-β1 siRNA on hepatic fibrosis in rats.
Biochem Biophys Res Commun. 409:448–453. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen BL, Peng J, Li QF, Yang M, Wang Y and
Chen W: Exogenous bone morphogenetic protein-7 reduces hepatic
fibrosis in Schistosoma japonicum-infected mice via transforming
growth factor-beta/Smad signaling. World J Gastroenterol.
19:1405–1415. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee JH, Lee H, Joung YK, et al: The use of
low molecular weight heparin-pluronic nanogels to impede liver
fibrosis by inhibition the TGF-β/Smad signaling pathway.
Biomaterials. 32:1438–1445. 2011. View Article : Google Scholar
|
|
16
|
Hang W, Li L, Tian X, Yan J, Yang X, Wang
X, Liao G1 and Qiu G: Astragalus and Paeoniae radix rubra extract
inhibits liver fibrosis by modulating the transforming growth
factor-β/Smad pathway in rats. Mol Med Rep. Nov 5–2014.(Epub ahead
of print). View Article : Google Scholar
|
|
17
|
Yu KZ, Liu J, Guo BL, et al: Microscopic
research on a multi-source traditional Chinese medicine, Astragali
Radix. J Nat Med. 68:340–350. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yusufoglu HS, Alam A, Zaghloul AM,
Al-Salkini MA and Alam P: Comparative anti-inflammatory and
hepatoprotective activities of astragalus gummifer labill herb and
roots in rats. Afr J Tradit Complement Altern Med. 11:268–274.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liang J, Xu F, Zhang YZ, et al: The
profiling and identification of the absorbed constituents and
metabolites of Paeoniae Radix Rubra decoction in rat plasma and
urine by the HPLC-DAD-ESI-IT-TOF-MS(n) technique: a novel strategy
for the systematic screening and identification of absorbed
constituents and metabolites from traditional Chinese medicines. J
Pharm Biomed Anal. 83:108–121. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Knook DL, Seffelaar AM and de Leeuw AM:
Fat-storing cells of the rat liver. Their isolation and
purification. Exp Cell Res. 139:468–471. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Majumdar M, Ratho R, Chawla Y and Singh
MP: Evaluation of antigenicity and cell mediated immunity of
hepatitis E virus patients: using non radioactive MTT assay. Indian
J Med Microbiol. 31:64–68. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou G, Kandala JC, Tyagi SC, Katwa LC and
Weber KT: Effects of angiotensin II and aldosterone on collagen
gene expression and protein turnover in cardiac fibroblasts. Mol
Cell Biochem. 154:171–178. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hu X, Rui W, Wu C, He S, Jiang J, Zhang X
and Yang Y: Compound Astragalus and Salvia miltiorrhiza extracts
suppress hepatocarcinogenesis by modulating transforming growth
factor-β/Smad sigmaling. J Gastroenterol Hepatol. 29:1284–1291.
2014. View Article : Google Scholar
|
|
24
|
Hashiba M, Ono M, Hyogo H, et al: Glycemic
variability is an independent predictive factor for development of
hepatic fibrosis in nonalcoholic Fatty liver disease. PLoS One.
8:e761612013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hossain N, Afendy A, Stepanova M, et al:
Independent predictors of fibrosis in patients with nonalcoholic
fatty liver disease. Clin Gastroenterol Hepatol. 7:1224–1229. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Svegliati-Baroni G, Bugianesi E, Bouserhal
T, et al: Post-load insulin resistance is an independent predictor
of hepatic fibrosis in virus C chronic hepatitis and in
non-alcoholic fatty liver disease. Gut. 56:1296–1301. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kowdley KV, Belt P, Wilson LA, et al:
Serum ferritin is an independent predictor of histologic severity
and advanced fibrosis in patients with nonalcoholic fatty liver
disease. Hepatology. 55:77–85. 2012. View Article : Google Scholar
|
|
28
|
Chen HJ, Liang TM, Lee IJ, Huang YT and
Lin YL: Scutellariae radix suppresses LPS-induced liver endothelial
cell activation and inhibits hepatic stellate cell migration. J
Ethnopharmacol. 150:835–842. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Katz TM, Goldberg LH and Friedman PM:
Nonablative fractional photothermolysis for the treatment of striae
rubra. Dermatol Surg. 35:1430–1433. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhou YX, Chen J, Li JP, Wang YL and Jin
XD: Chinese medicinal herbs in treating model rats with hepatic
fibrosis. Afr J Tradit Complement Altern Med. 7:104–108. 2010.
|
|
31
|
Vitcheva V, Simeonova R, Krasteva I,
Nikolov S and Mitcheva M: Protective effects of a purified saponin
mixture from Astragalus corniculatus Bieb., in vivo hepatotoxicity
models. Phytother Res. 27:731–736. 2013. View Article : Google Scholar
|
|
32
|
D’Argenio G, Mazzone G, Ribecco MT, et al:
Garlic extract attenuating rat liver fibrosis by inhibiting TGF-β1.
Clin Nutr. 32:252–258. 2013. View Article : Google Scholar
|
|
33
|
Li J, Li J, Li S, et al: Ameliorative
effect of grAPE seed proanthocyanidin extract on
thioacetamide-induced mouse hepatic fibrosis. Toxicol Lett.
213:353–360. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lang Q, Liu Q, Xu N, et al: The
antifibrotic effects of TGF-β1 siRNA on hepatic fibrosis in rats.
Biochem Biophys Res Commun. 409:448–453. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu X, Yang Y, Zhang X, et al: Compound
Astragalus and Salvia miltiorrhiza extract inhibits cell invasion
by modulating transforming growth factor-beta/Smad in HepG2 cell. J
Gastroenterol Hepatol. 25:420–426. 2010. View Article : Google Scholar
|
|
36
|
Tian XP, Yin YY and Li X: Effects and
mechanisms of Acremoniumterricola milleretal mycelium on liver
fibrosis induced by carbon tetrachloride in rats. Am J Chin Med.
39:537–550. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cai Y, Zhou CH, Fu D and Shen XZ:
Overexpression of Smad ubiquitin regulatory factor 2 suppresses
transforming growth factor-beta mediated liver fibrosis. J Dig Dis.
13:327–334. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sferra R, Vetuschi A, Catitti V, et al:
Boswellia serrata and Salvia miltiorrhiza extracts reduce
DMN-induced hepatic fibrosis in mice by TGF-beta1 downregulation.
Eur Rev Med Pharmacol Sci. 16:1484–1498. 2012.PubMed/NCBI
|
|
39
|
Freudlsperger C, Bian Y, Contag WS, et al:
TGF-β and NF-κB signal pathway cross-talk is mediated through TAK1
and SMAD7 in a subset of head and neck cancers. Oncogene.
32:1549–1559. 2013. View Article : Google Scholar
|
|
40
|
Bian EB, Huang C, Wang H, et al:
Repression of Smad7 mediated by DNMT1 determines hepatic stellate
cell activation and liver fibrosis in rats. Toxicol Lett.
224:175–185. 2014. View Article : Google Scholar
|
|
41
|
Hamzavi J, Ehnert S, Godoy P, et al:
Disruption of the Smad7 gene enhances CCI4-dependent liver damage
and fibrogenesis in mice. J Cell Mol Med. 12:2130–2144. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dooley S, Hamzavi J, Breitkopf K, et al:
Smad7 prevents activation of hepatic stellate cells and liver
fibrosis in rats. Gastroenterology. 125:178–191. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ji H, Tang H, Lin H, Mao J, Gao L, Liu J
and Wu T: Rho/Rock cross-talks with transforming growth
factor-β/Smad pathway participates in lung fibroblast-myofibroblast
differentiation. Biomed Rep. 2:787–792. 2014.PubMed/NCBI
|
|
44
|
Lin Z, Jiang L, Yuan C, et al: Structural
basis for recognition of urokinase-type plasminogen activator by
plasminogen activator inhibitor-1. J Biol Chem. 286:7027–7032.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang B, Li W, Chen Y, et al: Coexpression
of Smad7 and UPA attenuates carbon tetrachloride-induced rat liver
fibrosis. Med Sci Monit. 18:R394–R401. 2012. View Article : Google Scholar
|
|
46
|
Tang LX, He RH, Yang G, Tan JJ, Zhou L,
Meng XM, Huang XR and Lan HY: Asiatic acid inhibits liver fibrosis
by blocking TGF-beta/Smad signaling in vivo and in vitro. PLoS One.
7:e313502012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Salas AL, Montezuma TD, Farina GG,
Reyes-Esparza J and Rodriguez-Fragoso L: Genistein modifies liver
fibrosis and improves liver function by inducing uPA expression and
proteolytic activity in CCl4-treated rats. Pharmacology. 81:41–49.
2008. View Article : Google Scholar
|
|
48
|
Ghosh AK and Vaughan DE: PAI-1 in tissue
fibrosis. J Cell Physiol. 227:493–507. 2012. View Article : Google Scholar
|
|
49
|
Tuan TL, Zhu JY, Sun B, Nichter LS, Nimni
ME and Laug WE: Elevated levels of plasminogen activator
inhibitor-1 may account for the altered fibrinolysis by keloid
fibroblasts. J Invest Dermatol. 106:1007–1011. 1996. View Article : Google Scholar : PubMed/NCBI
|