Introduction
Environmental or occupational exposure to asbestos
fibers increase the risk for chronic respiratory diseases,
including interstitial lung fibrosis (for example, asbestosis),
lung cancer, and pleural malignant mesothelioma (1–4).
Asbestos fibers are naturally found in rocks and soils and consist
of six distinct types: Crocidolite, amosite, anthophyllite,
tremolite, actinolite and chrysotile (4,5).
Chrysotile asbestos is the sole serpentine type of asbestos
(6,7), which has been widely used as an
industrial material in China. The mechanisms of injury to the cells
of the lung and pleura, resulting in lung diseases, following
asbestos exposure have not yet been fully established, despite
extensive investigations over several decades (1,2,4,7). All
forms of asbestos, including chrysotile, are carcinogenic, and have
been previously shown to promote iron-derived free radical
formation in vitro, injure lung target cells, and induce
asbestosis, lung cancer, and mesothelioma in humans (1,2,4,7). As
previously reviewed, accumulating evidence firmly implicates
alveolar epithelial cell (AEC) apoptosis as an important early
event in the pathophysiology of idiopathic pulmonary fibrosis (IPF)
and asbestosis (2,4). Previous studies have shown that
asbestos-induced pulmonary toxicity is mediated in part by lung
epithelial cell mitochondrial dysfunction, mitochondrial reactive
oxygen species production, DNA damage, p53 activation and
mitochondria-regulated apoptosis (2,4,7,8).
Previous studies on lung epithelial cell mitochondria-regulated
(intrinsic) apoptosis have focused solely on the effects of
amphibole asbestos, including amosite and crocidolite, while the
effects of chrysotile asbestos on lung epithelial cells is
currently unknown (2–4,9–11).
Asbestos pulmonary toxicity, including apoptosis,
occurs partly by the activation of epidermal growth factor receptor
and other receptors, resulting in the activation of the
mitogen-activated protein kinase (MAPK) pathway, which includes
p38, c-Jun N-terminal kinase (JNK) and extracellular signal
regulated kinase (ERK 1/2) (12,13).
The MAPK p38 has been previously implicated in the chronic immune
response elicited by asbestos in rat mesothelial cells (14). Furthermore, fibroblast growth
factor-10 has been shown to decrease asbestos-induced DNA damage
and apoptosis in AECs by modulating MAPK-ERK-dependent signaling
that affects the mitochondria-regulated apoptosis pathway (15). Asbestos induces AEC plasticity
through the MAPK-ERK signaling pathway (16). Protein kinase delta
(PKCδ)-dependent mechanisms have been implicated in mediating AEC
intrinsic apoptosis, in part through PKCδ phosphorylation of JNK
which triggers pro-apoptotic Bcl-2-like protein 11 (Bim) expression
(8). Whereas the ERK1/2-related
anti-apoptotic pathways have been shown to be activated at lower
asbestos concentrations as a survival response in tumor cells
(8).
In the present study, it was hypothesized that
chrysotile asbestos may induce AEC intrinsic apoptosis via the
MAPK-JNK signaling pathway. To address this hypothesis, the effects
of chrysotile asbestos on cell viability and DNA fragmentation were
analyzed in human A549 bronchoalveolar carcinoma cells, with
alveolar epithelial type II-like features. Chrysotile-induced A549
cell expression of activated JNK, ERK1/2, p38, B cell lymphoma-2
(Bcl-2) associated X protein (Bax), Bcl-2 homologous antagonist
killer (Bak), cytochrome c, caspase-9 and poly (ADP-ribose)
polymerase (PARP) proteins was also assessed by western blotting.
Furthermore, the effects of the JNK inhibitor SP600125, on
chrysotile asbestos-induced A549 cell apoptosis and JNK-PARP
signaling were determined. The present study showed that chrysotile
asbestos can induce intrinsic apoptosis in A549 cells through the
JNK-dependent signaling pathway.
Materials and methods
Reagents
Dulbecco’s modified Eagle’s medium (DMEM) and fetal
bovine serum (FBS) were obtained from Gibco-BRL (Carlsbad, CA,
USA). Primary rabbit polyclonal antibodies against human
phospho-JNK1/2, phospho-ERK1/2, phospho-p38, phospho-p53, JNK1/2,
ERK1/2, p38, cytochrome c, PARP, Bax, Bak, and caspase-9
were purchased from Cell Signaling Technology, Inc. (Danvers, MA,
USA), and an antibody against β-actin was purchased from Santa Cruz
Biotechnology Inc. (Santa Cruz, CA, USA). The antibodies were used
at a 1:2,000 dilution. The JNK inhibitor SP600125, was purchased
from Calbiochem® (Merck Millipore, La Jolla, CA, USA).
Polyvinylidene difluoride membrane (PVDF) was purchased from
Millipore (Billerica, MA, USA). Cellular DNA Fragmentation ELISA
kits were purchased from Roche (Basel, Switzerland), the DeadEnd™
Fluorometric terminal deoxynucleotidyl transferase-mediated dUTP
nick-end labeling (TUNEL) system was purchased from Promega
Corporation (Madison, WI, USA), and the PrimeScript Reverse
Transcription (RT) Enzyme Mix kits and SYBR® Green
polymerase chain reaction (PCR) reagent were purchased from Takara
Biotechnology Co., Ltd. (Osaka, Japan). All the reagents used
throughout the study were of analytical or cell culture grade
purity. Chrysotile asbestos was obtained from Mangya Moutain
(Qinghai Province, China).
Pretreatment of chrysotile asbestos
Chrysotile asbestos fibers used in the following
experiments were mined from Qinghai, China. The chrysotile asbestos
used in the treatments had an average length of 7.8 μm, and an
average diameter of 0.2 μm, which was confirmed using transmission
electron microscopy. The fibers were prepared as described by
previous methods (17). Briefly,
fiber samples were weighed and crushed into an ultrafine powder,
using a mechanical crusher (Xulang Machinery Equipment Co., Ltd,
Guangzhou, China). Following ultra-sonication at 20 kHz for 10 min
using a bath-type sonicator (Q700; QSonica LLC, Newtown, CT, USA),
the mixtures were centrifuged at 2,000 × g for 10 min, the
supernatants were removed and the pellets were washed with 2 ml
distilled water. Each chrysotile asbestos sample was then
re-suspended in phosphate-buffered saline (PBS) for the cell
treatment. A stock solution of the fibers (5 mg/ml) was sterilized
by autoclaving and mixed to ensure a uniform suspension prior to
dilution with tissue culture medium, ready for the cell
treatment.
Cell culture
A549 human bronchoalveolar carcinoma-derived cells,
with some features characteristic of alveolar epithelial type II
cells, were obtained from the American Type Culture Collection
(Manassas, VA, USA). The cells were cultured in a humidified
chamber containing 5% CO2 at 37°C and maintained in DMEM
supplemented with 10% FBS and antibiotics (100 U/ml penicillin and
100 g/ml streptomycin). For the experiments, A549 cells were plated
in 35 mm diameter dishes (2×105 cells/dish). Following
24 h in culture, the medium was refreshed with 4 ml medium
containing chrysotile fibers at the indicated final
concentrations.
Measurement of cell viability by trypan
blue exclusion method
The trypan blue exclusion method is a classic cell
procedure used for assessing cell viability. The cells were divided
and cultured in six-well plates (1×105 cells/well) for
24 h, followed by treatment with chrysotile asbestos at various
concentrations for 24 h. The cells were harvested and centrifuged
(5810R; Eppendorf, Hamburg, Germany) to remove the medium. The
cells were then washed in PBS three times and resuspended,
resulting in a suspension of 1×106 cells/ml. The
suspension was mixed with 0.4% trypan blue dye (Sigma-Aldrich, St.
Louis, MO, USA) for 5 min at 25°C. The unstained (viable) and
stained (non-viable) cells were counted using a hemacytometer
(Neubauer Improved; Marienfeld, Lauda-Königshofen, Germany) within
5 min in four microscope fields, at magnification ×40, per well
(>100 cells/field).
Detection of apoptotic cells
Experiments were performed using a cellular DNA
fragmentation ELISA kit according to the manufacturer’s
instructions (Roche). A549 cells were labeled with 10 μM
bromodeoxyuridine (BrdU; Roche) at 1×105 cells/ml.
BrdU-labeled cells (1×104) in 100 μl were treated with
varying concentrations (100, 150, 200 μg/cm2) of cell
extract for a period of 4 h. Following treatment, the cells were
lysed with lysis buffer (20 mM Tris, pH 7.6; 1% Triton X-100; 137
mM NaCl; 2 mM EDTA; 1 mM Na3O4V; 10 mM NaF; 1
mM DTT; 1 mM phenylmethylsulfonyl fluoride; 10 μg/ml leupeptin and
10 μg/ml aprotinin) for 30 min at 25°C, and the supernatants
containing apoptotic fragments were obtained following
centrifugation at 1,500 × g for 10 min. The recovered samples (100
μl) were transferred onto anti-DNA-coated 96-well flat-bottom
microplates (Corning Inc., Corning, NY, USA). The plates were
incubated for 90 min at 15–25°C, and the wells were washed three
times with washing buffer, which was provided in the DNA
fragmentation ELISA kit, for 2–3 min per wash. The DNA bound to the
coated microplates was denatured by nuclease treatment (exonuclease
III solution, 37°C for 30 min; Takara Biotechnology Co., Ltd),
followed by the addition of 100 μl anti-BrdU-peroxidase (POD;
Roche) conjugate solution. The plates were incubated for an
additional 90 min and were washed again with washing buffer.
Following washing, 100 μl of the substrate solution was added, and
the plates were shaken until color development was deemed
sufficient. The absorbance was measured at 450 nm following the
addition of 25 μl of stop solution (Roche).
TUNEL assay
DNA damage was assessed by TUNEL assay using an
in situ Cell Death Detection kit with fluorescein-dUTP as a
label, according to the manufacturer’s instructions (Promega
Corporation). A549 cells were plated onto confocal petri dishes and
grown to confluence over 24 h in DMEM supplemented with 10% FBS,
followed by treatment with chrysotile asbestos for 24 h. Following
incubation, the culture medium was removed and the cells were
washed three times in PBS. The cells were then fixed with 4%
paraformaldehyde (Sigma-Aldrich), permeabilized with 0.1% Triton
X-100 (Sigma-Aldrich), and incubated in the dark at 37°C for 1 h in
a TUNEL reaction mixture containing 50 μl of a mixture of terminal
deoxynucleotidyl transferase and dUTP. DAPI was added at 25°C for
10 min as a non-specific stain of the cellular nuclei. Five fields
per dish of cells were randomly analyzed using a Leica TCS SP5 II
confocal microscope (Leica, Wetzlar, Germany)(>100cells/field).
Each DAPI-stained cell was categorized as apoptotic if green
nuclear fluorescence was observed, or normal if no green
fluorescence was observed.
Western blot analysis
The treated cells were rinsed with ice-cold PBS and
incubated with radioimmunoprecipitation assay lysis buffer
containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton-X 100,
1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 1 mM
ethylenediaminetetraacetic acid, 1 mM sodium fluoride, 1 mM
phenylmethanesulfonyl fluoride, 10 μg/ml aprotinin, 1 μg/ml
leupeptin, and 1 μg/ml pepstatin for 20 min. The cell lysates were
then centrifuged at 12,000 × g for 15 min, and protein
concentrations were determined using the Bicinchoninic Acid Protein
Assay kit (Beyotime, Jiangsu, China). Total cell protein (20
μg/lane) was separated by 10 or 12% SDS-PAGE followed by transfer
to PVDF membranes. The membranes were blocked for 1 h in
Tris-buffered saline containing 0.05% Tween-20 (TBST), with 5%
nonfat dry milk. The membranes were then incubated with rabbit
polyclonal antibodies against phospho-JNK1/2, phospho-ERK1/2,
phospho-p38, phospho-p53, JNK1/2, ERK1/2, p38, cytochrome c,
Bax, Bak, caspase-9, PARP or β-actin overnight at 4°C. Following
primary antibody incubation the membranes were washed with TBST and
incubated for 1 h with goat anti-rabbit immunoglobulin G-conjugated
horseradish peroxidase-conjugated secondary antibody. The
antibody-reactive bands were revealed using an enhanced
chemiluminescence reagent (GE Healthcare, Little Chalfont, UK) and
exposed to radiographic film.
Real-time quantitative polymerase chain
reaction (PCR) (qPCR) analysis
The cells were treated with chrysotile asbestos for
the indicated times, followed by extraction of total RNA using
TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA)
according to the manufacturer’s instructions. Total RNA (1 μg) was
reverse-transcribed into cDNA using PrimeScript RT Enzyme mix at
37°C for 15 min, followed by an 85°C incubation for 5 s. Specific
primers for real-time qPCR are detailed in Table 1. GAPDH was used as an internal
control.
 | Table IPrimer seqences used for quantitative
polymerase chain reaction. |
Table I
Primer seqences used for quantitative
polymerase chain reaction.
| Gene | Primer sequence
(5′-3′) |
|---|
| Bax | F:
AAGCTGAGCGAGTGTCTCAAG
R: CAAAGTAGAAAAGGGCGACAAC |
| Bak | F:
AGGACACAGAGGAGGTTTTCC
R: ATAGCGTCGGTTGATGTCGT |
| JNK | F:
CTTTGCCAAGTGATTCAGATGGA
R: TTACTGGGCTTTAAGTCCCGATG |
| Caspase-9 | F:
CTAACAGGCAAGCAGCAAAGT
R: GACATCACCAAATCCTCCAGA |
| PARP | F:
AGGACGACAAGGAAAACAGGTA
R: CATAGTCAATCTCCAGGGGGTA |
| GAPDH | F:
AGAAGGCTGGGGCTCATTTG
R: AGGGGCCATCCACAGTCTTC |
Amplification was performed in a 20 μl total
reaction volume using real-time SYBR® Green PCR reagent
in a LightCycler® 480II Real-Time thermal cycler
(Roche). The cycling conditions were as follows: 95°C for 30 s,
followed by 40 cycles of 95°C for 5 s and 60°C for 20 s.
Melting-curve analysis was performed for each primer set, to ensure
that no primer dimers or nonspecific amplification was present
under the optimized cycling conditions. The fold difference in mRNA
expression was determined using the relative quantification method,
with normalization to GAPDH mRNA, by comparing the relative cycle
threshold (Ct) changes between the control and the experimental
samples. The fold change was calculated from the mean of the
control group for each individual sample, including individual
control samples, to assess variability within the groups.
Statistical analysis
The data are presented as the means ± standard
deviation. Statistical significance was evaluated using a Student’s
t-test. When more than one group was compared with a control, the
significance was evaluated according to a one-way analysis of
variance, and a Duncan’s post hoc test was applied to
identify group differences. A P<0.05 was considered to indicate
a statistically significant difference. The statistical package
SPSS, version 11.0 for Windows (SPSS Inc., Chicago, IL, USA) was
used for statistical analyses.
Results
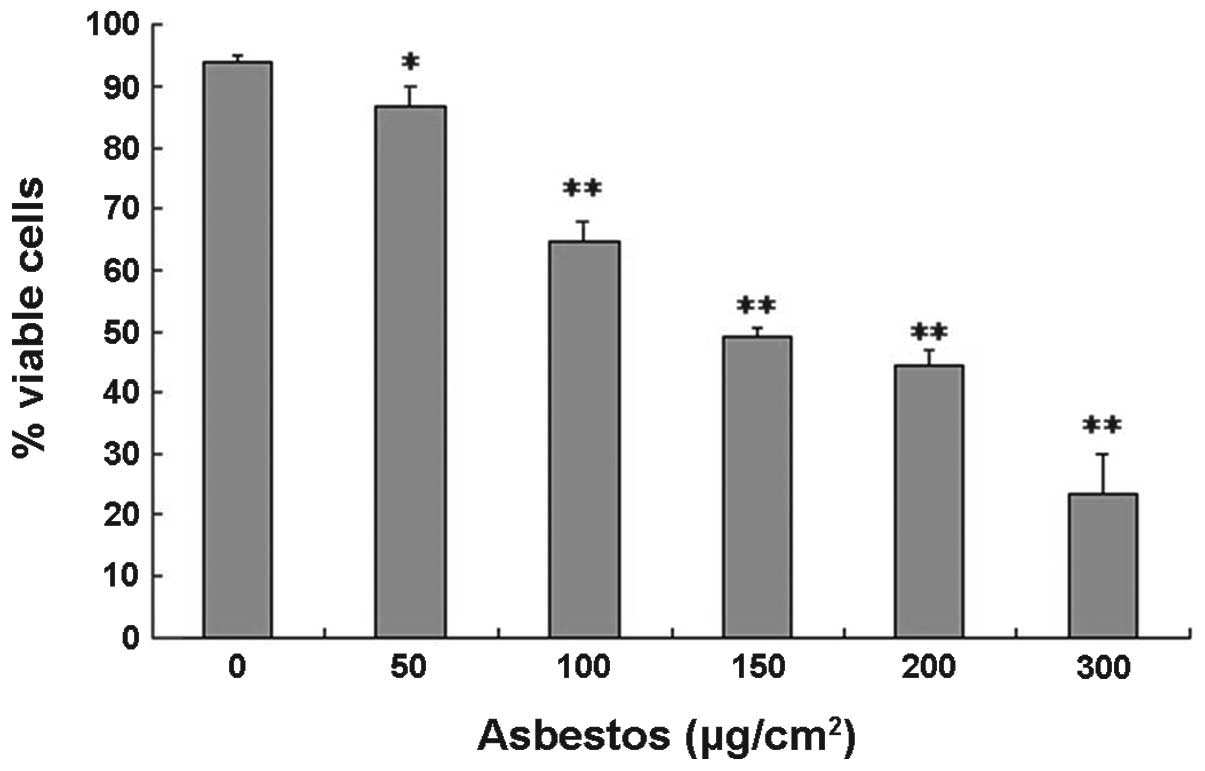
Chrysotile asbestos decreases cell
viability and induces apoptosis in A549 cells
To examine chrysotile asbestos-induced cytotoxicity
on A549 cells, the effects of chrysotile asbestos on cell survival
were determined. The cells were treated for 24 h with increasing
concentrations of chrysotile asbestos, ranging from 50–300
μg/cm2. As the concentration of chrysotile asbestos
increased there was a significant decrease in the number of viable
cells, as assessed using the trypan blue exclusion assay. For the
control untreated A549 cells, the viability was 93.87%, and for the
cells treated with chrysotile asbestos (50, 100, 150, 200, 300
μg/cm2), viability was 86.5, 64.93, 49.1, 44.43, and
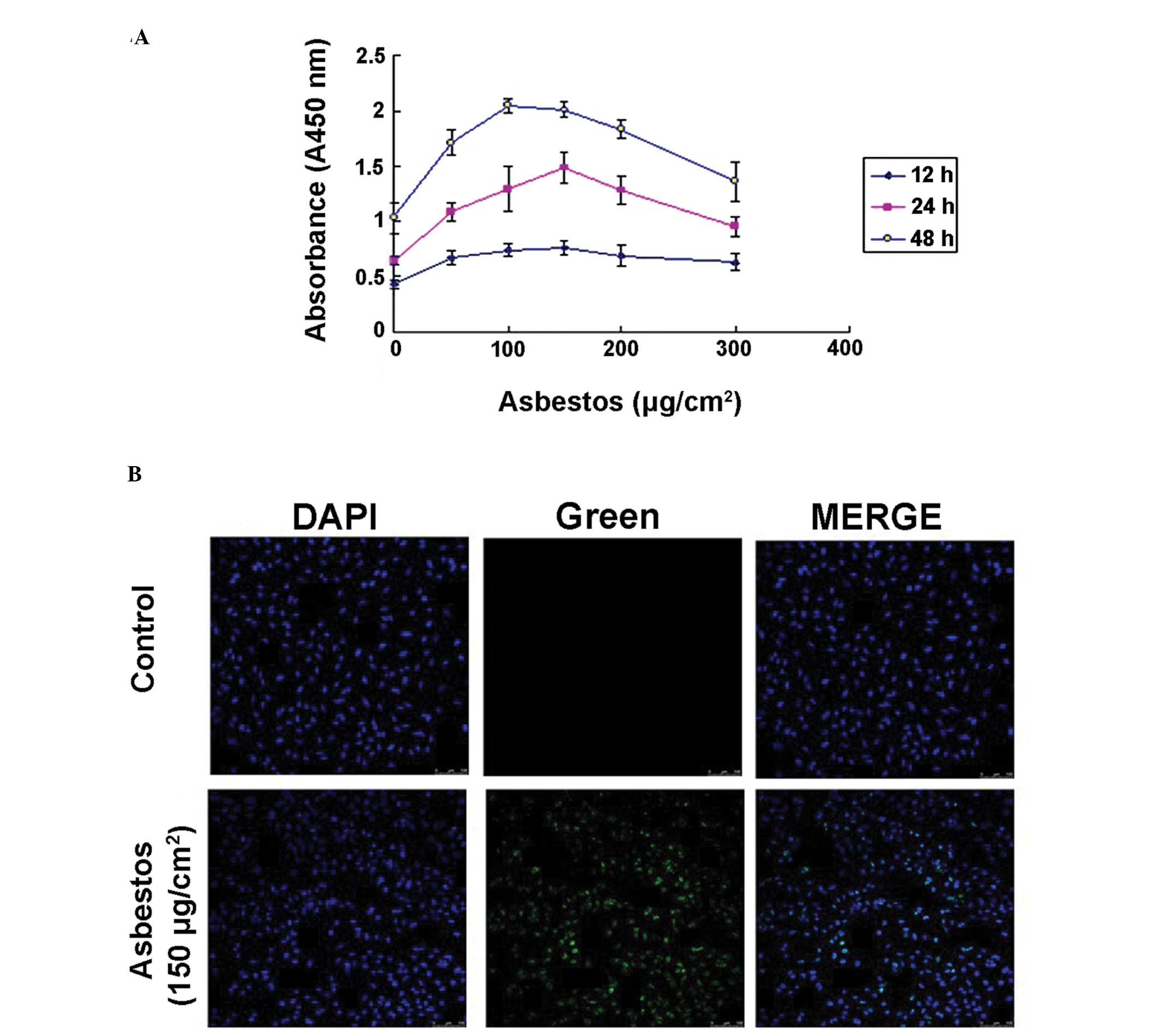
23.63%, respectively (Fig. 1). To
further investigate whether chrysotile asbestos induces apoptosis
in A549 cells, the levels of cellular DNA fragmentation, a hallmark
of apoptotic cell death, were assessed. The results indicated that
chrysotile asbestos induced DNA fragmentation in a dose- and
time-dependent manner, with the levels peaking at ~100–150
μg/cm2 chrysotile asbestos (Fig. 2A). To verify these findings, DNA
cleavage was assayed by TUNEL staining, following 24 h exposure of
A549 cells to chrysotile asbestos. Chrysotile asbestos led to an
increased intensity of green fluorescence in the nuclear region of
the cells (Fig. 2B), further
confirming that chrysotile asbestos may induce apoptotic cell
death.
Chrysotile asbestos induces apoptosis in
A549 cells, via the JNK-MAPK signaling pathway
MAPKs have an important role in certain apoptotic
signaling pathways. Therefore, the possible role of MAPKs in
chrysotile asbestos-induced A549 cell apoptosis was determined. As
compared with the control untreated cells, the chrysotile
asbestos-treated A549 cells had significantly increased levels of
phosphorylated (activated) JNK1/2 protein, but not of
phosphorylated ERK1/2 or p38 (Fig.
3A). The effects of chrysotile asbestos on the JNK1/2 pathway
could be reversed by pre-treatment of the cells with the JNK
inhibitor SP600125 (Fig. 3B). A
marked increase in the relative expression of JNK mRNA was observed
following chrysotile asbestos exposure (150 μg/cm2)
(Fig. 3C). To investigate the role
of JNK in asbestos-induced A549 cell death, apoptosis levels were
assessed following treatment with SP600125 (Fig. 3D). Chrysotile asbestos-induced
apoptosis was partially decreased by treatment with SP600125. These
results further suggest that JNK has a role in chrysotile
asbestos-induced apoptosis of A549 cells.
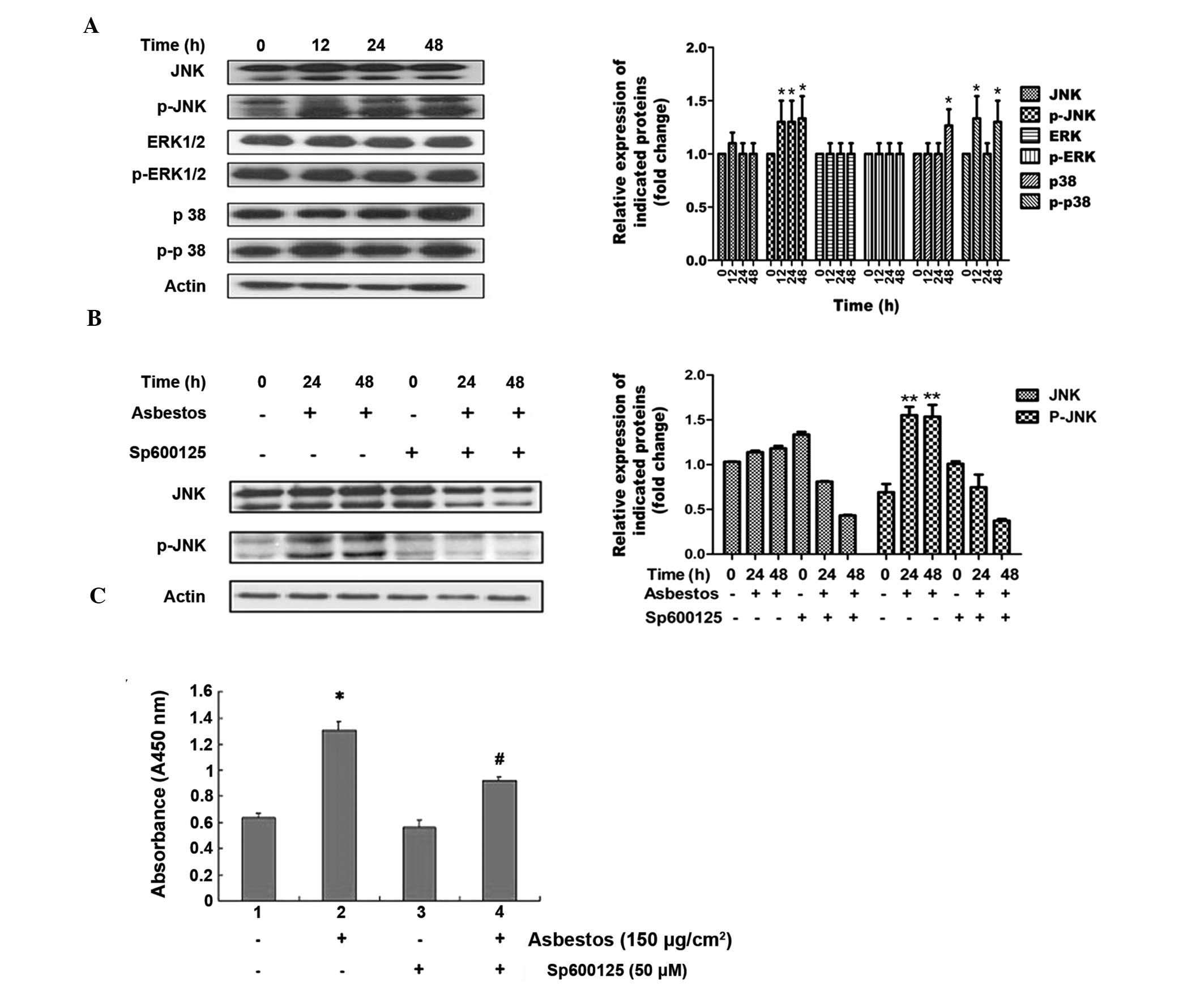 | Figure 3Effects of chrysotile asbestos on
mitogen-activated protein kinase (MAPK) activation in A549 human
bronchoalveolar carcinoma cells. (A) A549 cells were left untreated
or were treated with chrysotile asbestos (150 μg/cm2)
for 12, 24 or 48 h, and the phosphorylation of c-Jun N-terminal
kinase (JNK)-1/2, extracellular signal-regulated kinase (ERK)-1/2
and p38-MAPK were assessed by western blotting. The results are
representative of three independent experiments. (B) A549 cells
were treated with chrysotile asbestos (150 μg/cm2) for
24 or 48 h in the absence or presence of the JNK specific inhibitor
SP600125 (50 μM for 5 h prior to chrysotile asbestos treatment),
and the phosphorylation of JNK protein was examined by western
blotting. The results are representative of three independent
experiments. (C) A549 cells were pretreated with 50 μM SP600125 for
5 h prior to 150 μg/cm2 asbestos treatment. Following 24
h incubation, the apoptotic fragmentation was assessed with a
cellular DNA fragmentation ELISA. The values shown represent the
means ± standard deviation from three independent experiments. The
statistical significance of the results was analyzed by a Student’s
t-test, *P<0.05 vs. untreated control;
#P<0.05 vs. asbestos-treated cells. H, hours; JNK,
c-Jun N-terminal kinase; ERK, extracellular signal-regulated
kinase; nm, nanometers. |
Chrysotile asbestos induces intrinsic
apoptosis in A549 cells, and inhibition of JNK is protective
The Bcl-2 family of pro- and anti-apoptotic proteins
modulate the stability of the mitochondrial membrane, which
determines whether or not the intrinsic apoptosis pathway is
activated (18). To investigate
whether the apoptotic response to chrysotile asbestos is mediated
through this pathway, levels of pro-apoptotic Bax and Bak were
assessed, using western blotting and qPCR. As shown in Fig. 4A, chrysotile asbestos treatment
induced a time-dependent increase in the levels of Bax and Bak
protein. As confirmation, the release of cytochrome c from
the mitochondria into the cytosol was also shown to be markedly
increased in response to chrysotile asbestos treatment. The
increases in Bax/Bak and cytochrome c expression levels were
reversed by pretreatment with SP600125, suggesting that JNK
signaling regulates the effects on mitochondrial stability by
chrysotile asbestos (Fig. 4B).
Furthermore, 150 μg/cm2 chrysotile asbestos induced an
increase in the mRNA expression of Bax/Bak (Fig. 4C and D), thus suggesting that the
regulation may occur at the transcriptional level.
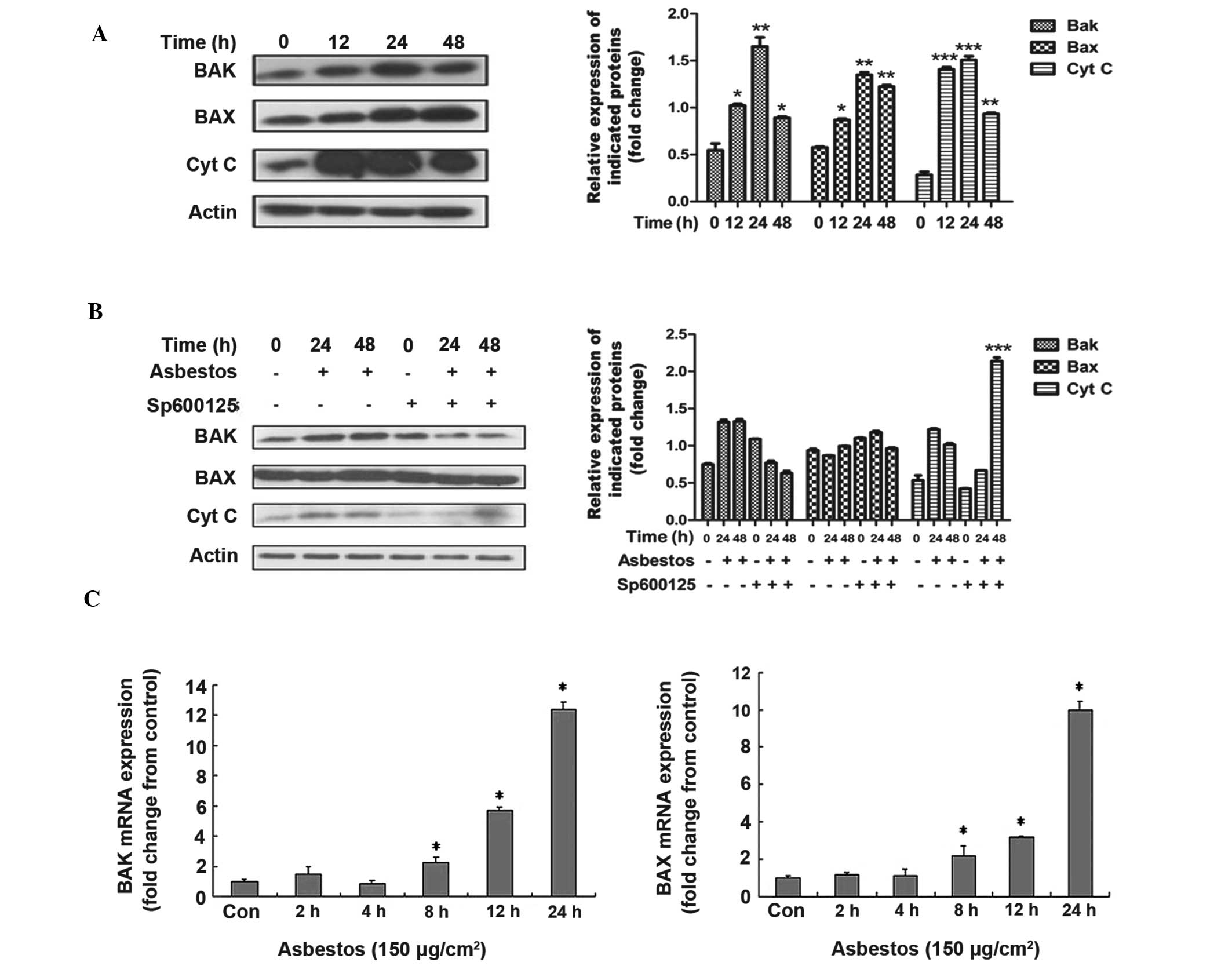 | Figure 4Analysis of mitochondrial dysfunction
in chrysotile asbestos-treated A549 human bronchoalveolar carcinoma
cells. (A) The cells were either left untreated or were treated
with chrysotile asbestos (150 μg/cm2) for 12, 24 or 48
h, and the expression of pro-apoptotic genes Bak and Bax, and
cytochrome c was assessed by western blotting. The results are
representative of three independent experiments. (B) c-Jun
N-terminal kinase (JNK) inhibitor SP600125 (50 μM for 5 h) was
added to A549 cells prior to treatment with chrysotile asbestos
(150 μg/cm2 for 0, 24 or 48 h), Bax/Bak, and cytochrome
c levels were examined by western blotting. The results are
representative of three independent experiments. (C) The mRNA
levels of Bak and Bax were assessed by quantitative polymerase
chain reaction following the treatment of A549 cells with
chrysotile asbestos (150 μg/cm2 for 0, 2, 4, 8, 12 or
24h). The data represent the means ± standard deviation from three
independent experiments. The statistical significance of the
results was analyzed by a Student’s t-test, *P<0.05
vs. untreated control.Bax, B cell lymphoma-2 (Bcl-2) associated X
protein; Bak, Bcl-2 homolohous antagonist killer; Cyt C, cytochrome
c; h, hours; Con, control. |
Chrysotile asbestos induces A549 cell
cleaved caspase-9 and PARP, and inhibition of JNK is
protective
Caspase-9 and PARP, a substrate of caspase-3,
represent two additional cellular proteins that are known to be
activated during apoptotic signaling (19). To determine whether these proteins
are activated by chrysotile asbestos, the relative protein and mRNA
expression levels were determined following treatment. As shown in
Fig. 5A and B, a marked increase
in protein and mRNA expression is observed for both caspase-9 and
PARP, following chrysotile asbestos treatment. Furthermore,
caspase-9 and PARP activation were blocked by the JNK inhibitor
SP600125, verifying the role for JNK in mediating chrysotile
asbestos-induced cell death (Fig.
5C).
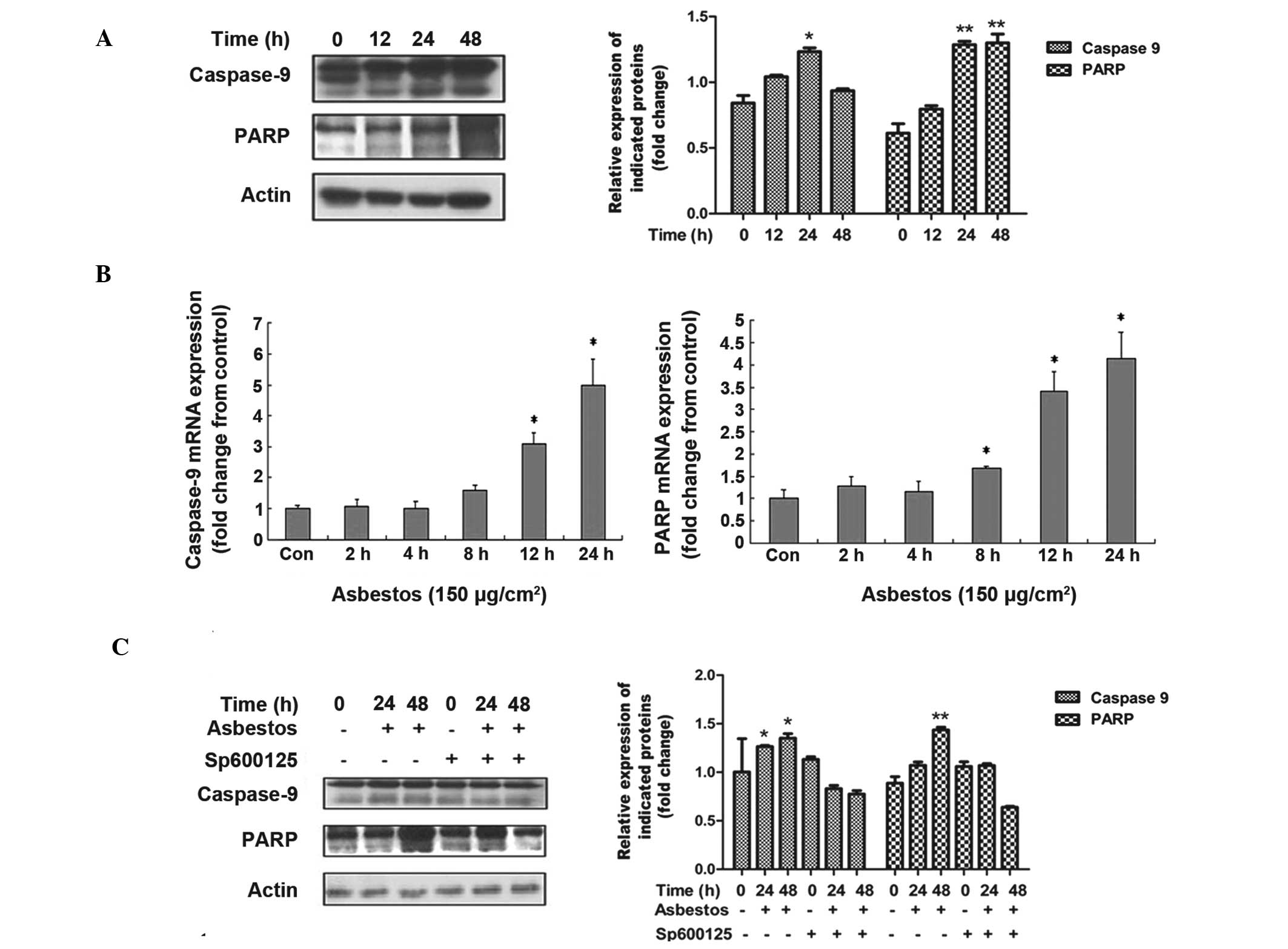 | Figure 5Chrysotile asbestos induces caspase
activation and cleavage of poly (ADP-ribose) polymerase (PARP) in
A549 human bronchoalveolar carcinoma cells. (A) The cells were
treated with chrysotile asbestos (150 μg/cm2) for 12, 24
or 48 h, and the cleavage of caspase-9 and PARP was assessed by
western blotting. The results are representative of three
independent experiments. (B) The cells were treated with chrysotile
asbestos (150 μg/cm2) for 0, 2, 4, 8, 12 or 24 h, and
caspase-9 and PARP mRNA expression was detected by quantitative
polymerase chain reaction. The values shown represent the means ±
standard deviation from three independent experiments. (C) A549
cells were treated with chrysotile asbestos (150 μg/cm2)
for 0, 24 or 48 h in the absence or presence of c-Jun N-terminal
kinase (JNK) inhibitor SP600125 (50 μM for 5 h), and caspase-9 and
PARP were then examined by western blotting. The results are
representative of three independent experiments. The statistical
significance of the results was analyzed by a Student’s t-test
(*p<0.05 vs. untreated control). Con, control;h,
hours. |
Discussion
Asbestos-related pulmonary diseases remain an
important long-term health concern world-wide. This is in part due
to the vast amount of fibers mined and used for numerous industrial
purposes and the long lag phase between exposure to the fibers and
initiation of disease, which can be between 20–40 years (20). The U.S. Environmental Protection
Agency has banned the production and use of all types of asbestos
(20). However, in some developing
countries, including China, asbestos fibers, especially chrysotile,
are still widely used as a construction material. Although there
are numerous pathophysiological mechanisms accounting for
asbestos-induced pulmonary toxicity, AEC apoptosis is widely
implicated but by mechanisms which are not yet fully understood
(1,2,4,7).
Furthermore, the role of chrysotile asbestos in mediating human AEC
apoptosis is not well characterized. The present study showed that
chrysotile asbestos induced intrinsic apoptosis in human A549 lung
epithelial cells, in a dose- and time-dependent manner, as assessed
by cell viability, DNA fragmentation, and expression of Bax-Bak,
cleaved caspase-9 and PARP activation. Furthermore, it was
determined that chrysotile asbestos-induced A549 cell intrinsic
apoptosis was mediated by JNK activation. Overall, these results
suggest that chrysotile-induced AEC JNK activation is a novel
signaling pathway which may be relevant in the pathophysiology of
asbestos-related pulmonary diseases.
Apoptosis, also known as programmed cell death, is
an important mechanism by which cells with DNA damage are
eliminated without the elicitation of an inflammatory response
(21). Apoptotic cells are
characterized by nuclear chromatid condensation, endonuclease
activation resulting in DNA fragmentation, translocation of
phosphatidylserine to the outer plasma membrane, and the generation
of double-stranded DNA breaks (22–24).
Asbestos has been shown to trigger apoptosis in all relevant lung
target cells (25). Previous
studies have shown that low levels of asbestos (<0.5
μg/cm2) promote the entry of cells into the S-phase of
the cell cycle without inducing apoptosis, whereas higher levels of
asbestos (1.0–5.0 μg/cm2) impede entry into the S-phase
and induce apoptosis (25–28). The results of the present study
indicated that chrysotile asbestos induced the greatest extent of
apoptosis in A549 cells at a concentration of 150 μg/cm2
for 48 h, likely due to the lower cytotoxicity of chrysotile
asbestos as compared with other types of asbestos (20). A previous study showed that
asbestos induces protein kinase C δ (PKCδ)-dependent protein kinase
D (PKD) phosphorylation in lung epithelial cells (8). PKCδ-dependent PKD phosphorylation by
asbestos is causally linked to a cellular pathway that involves the
phosphorylation of both ERK1/2 and JNK1/2, which have opposing
roles in the apoptotic response induced by asbestos (8). Furthermore, numerous studies have
previously shown that AEC apoptosis, via caspase-3 and -9
activation, mediates asbestos-induced lung injury (11, 29,
30). The results of the present
study showed that, in high doses, chrysotile asbestos triggers
apoptosis of A549 cells by inducing the release of cytochrome
c. This release was shown to be accompanied by a marked
increase in the activation of Bax/Bak and caspase-9. Furthermore,
the exposure of A549 cells to chrysotile asbestos resulted in a
significant increase in the relative expression levels of Bax/Bak
and Caspase-9 mRNA. These results indicate that chrysotile asbestos
induces A549 cell apoptosis through the mitochondria-dependent
pathway.
The MAPKs (p38, ERK, and JNK) are activated in
response to various cellular stressors or stimuli, including
oxidative stress, lipopolysaccharide and tumor necrosis factor-α
(31–33). Generally, the JNK and p38 MAPK
pathways are considered as “stress-activated protein kinases”,
which have an essential role in inflammation and apoptosis
(34). The ERK pathway has been
shown to be important in cellular differentiation and
proliferation, as well as in cell survival (34). To evaluate the involvement of the
MAPK signaling pathways in chrysotile asbestos-induced A549 cell
apoptosis, the expression of MAPKs, following treatment with
chrysotile asbestos, was determined in the present study (12, 24
and 48h). Chrysotile asbestos led to a significant time-dependent
activation of the JNK protein, whereas there were no significant
changes in the activation of p38 and ERK (Fig. 3). JNK has previously been shown to
phosphorylate mitochondrial membrane proteins, such as the Bcl-2
family members, activating their apoptotic function (35,36).
Furthermore, caspases transduce the signals of most
apoptosis-inducing factors (37),
and PARP is one of the main cleavage targets of caspase-3 and a
main effector in cell apoptosis (19). Therefore, to determine the
molecular mechanisms of chrysotile asbestos-induced apoptosis in
A549 cells, the expression levels of Bax/Bak, caspase-9 and PARP
were determined, following the treatment of A549 cells with
chrysotile asbestos. Treatment with chrysotile asbestos increased
the expression of each of these proteins at different time points.
Moreover, the JNK-specific inhibitor SP600125 substantially
inhibited chrysotile asbestos-induced activation. Apoptosis was
also significantly decreased by SP600125 pre-treatment (Fig. 4 and 5), suggesting that the JNK signaling
pathway is functionally involved in chrysotile asbestos-induced
A549 cell apoptosis through these downstream effectors.
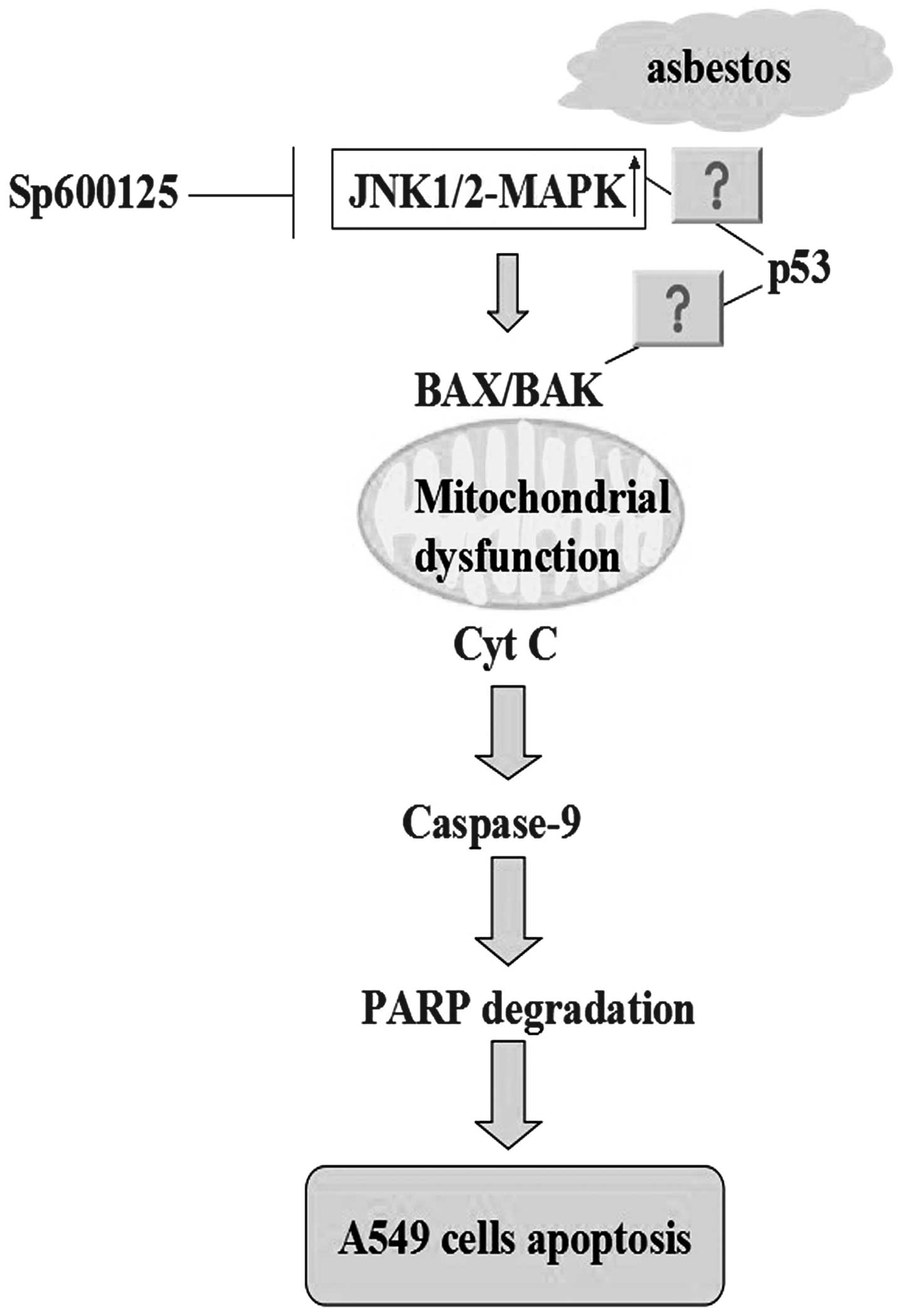
In conclusion, the results of the present study
revealed that chrysotile asbestos significantly decreased the
viability of human A549 cells. Furthermore, chrysotile asbestos
triggered mitochondrial dysfunction and intrinsic apoptotic
cascades through the activation of JNK1/2 phosphorylation, which
was shown to be reversed by the specific JNK inhibitor SP600125. A
hypothetical model illustrating the role of the JNK signaling
pathway in chrysotile asbestos-induced apoptosis is shown in
Fig 6. The precise molecular
mechanisms by which chrysotile-induced JNK activation triggers AEC
intrinsic apoptosis, as well as the in vivo relevance of the
present in vitro findings, requires further study.
Chrysotile asbestos-induced intrinsic AEC apoptosis through a
JNK-dependent mechanism may be a novel target for the modulation of
chrysotile asbestos-related lung diseases.
Acknowledgements
This work was supported by grants from the Natural
Science Foundation of China (no. 81172615) and by the Natural
Science Foundation of Guangdong Province (no. 2012B031800223), the
Science and Technology Project of Guangdong Province (no.
S2012010008299), the Science and Technology Plan Projects of
Zhanjiang City (no. 20201Z01101), and the VA Merit (DWK) (no.
RO1ES020357).
References
|
1
|
Mossman BT, Kamp DW and Weitzman SA:
Mechanisms of carcinogenesis and clinical features of
asbestos-associated cancers. Cancer Invest. 14:466–480. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kamp DW: Asbestos-induced lung diseases:
an update. Transl Res. 153:143–152. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu G, Beri R, Mueller A and Kamp DW:
Molecular mechanisms of asbestos-induced lung epithelial cell
apoptosis. Chem Biol Interact. 188:309–318. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu G, Cheresh P and Kamp DW: Molecular
basis of asbestos-induced lung disease. Annu Rev Pathol. 8:161–187.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin Z, Liu T, Kamp DW, Wang Y, He H, Zhou
X, Li D, Yang L, Zhao B and Liu G: AKT/mTOR and c-Jun N-terminal
kinase signaling pathways are required for chrysotile
asbestos-induced autophagy. Free Radic Biol Med. 72:296–307. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Heintz NH, Janssen-Heininger YM and
Mossman BT: Asbestos, lung cancers, and mesotheliomas: from
molecular approaches to targeting tumor survival pathways. Am J
Respir Cell Mol Biol. 42:133–139. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang SX, Jaurand MC, Kamp DW, Whysner J
and Hei TK: Role of mutagenicity in asbestos fiber-induced
carcinogenicity and other diseases. J Toxicol Environ Health B Crit
Rev. 14:179–245. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Buder-Hoffmann SA, Shukla A, Barrett TF,
MacPherson MB, Lounsbury KM and Mossman BT: A protein kinase
Cdelta-dependent protein kinase D pathway modulates ERK1/2 and
JNK1/2 phosphorylation and Bim-associated apoptosis by asbestos. Am
J Pathol. 174:449–459. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Aljandali A, Pollack H, Yeldandi A, Li Y,
Weitzman SA and Kamp DW: Asbestos causes apoptosis in alveolar
epithelial cells: role of iron-induced free radicals. J Lab Clin
Med. 137:330–339. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kamp DW, Panduri V, Weitzman SA and
Chandel N: Asbestos-induced alveolar epithelial cell apoptosis:
role of mitochondrial dysfunction caused by iron-derived free
radicals. Mol Cell Biochem. 234–235:153–160. 2002. View Article : Google Scholar
|
|
11
|
Panduri V, Weitzman SA, Chandel N and Kamp
DW: The mitochondria-regulated death pathway mediates
asbestos-induced alveolar epithelial cell apoptosis. Am J Respir
Cell Mol Biol. 28:241–248. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zanella CL, Posada J, Tritton TR and
Mossman BT: Asbestos causes stimulation of the extracellular
signal-regulated kinase 1 mitogen-activated protein kinase cascade
after phosphorylation of the epidermal growth factor receptor.
Cancer Res. 56:5334–5338. 1996.PubMed/NCBI
|
|
13
|
Baldys A and Aust AE: Role of iron in
inactivation of epidermal growth factor receptor after asbestos
treatment of human lung and pleural target cells. Am J Respir Cell
Mol Biol. 32:436–442. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Swain WA, O’Byrne KJ and Faux SP:
Activation of p38 MAP kinase by asbestos in rat mesothelial cells
is mediated by oxidative stress. Am J Physiol Lung Cell Mol
Physiol. 286:L859–L865. 2004. View Article : Google Scholar
|
|
15
|
Upadhyay D, Panduri V and Kamp DW:
Fibroblast growth factor-10 prevents asbestos-induced alveolar
epithelial cell apoptosis by a mitogen-activated protein
kinase-dependent mechanism. Am J Respir Cell Mol Biol. 32:232–238.
2005. View Article : Google Scholar
|
|
16
|
Tamminen JA, Myllärniemi M, Hyytiäinen M,
Keski-Oja J and Koli K: Asbestos exposure induces alveolar
epithelial cell plasticity through MAPK-Erk signaling. J Cell
Biochem. 113:2234–2247. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hei TK, Piao CQ, He ZY, Vannais D and
Waldren CA: Chrysotile fiber is a strong mutagen in mammalian
cells. Cancer Res. 52:6305–6309. 1992.PubMed/NCBI
|
|
18
|
Adams JM and Cory S: The Bcl-2 protein
family: arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Soldani C and Scovassi AI:
Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: An update.
Apoptosis. 7:321–328. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ramazzini C: Asbestos is still with us:
repeat call for a universal ban. Am J Ind Med. 54:168–173. 2011.
View Article : Google Scholar
|
|
21
|
Li P, Nijhawan D, Budihardjo I,
Srinivasula SM, Ahmad M, Alnemri ES and Wang X: Cytochrome c and
dATPdependent formation of Apaf-1/caspase-9 complex initiates an
apoptotic protease cascade. Cell. 91:479–489. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Franco R and Panayiotidis MI:
Environmental toxicity, oxidative stress, human disease and the
“black box” of their synergism: how much have we revealed? Mutat
Res. 674:1–2. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kroemer G, Galluzzi L and Brenner C:
Mitochondrial membrane permeabilization in cell death. Physiol Rev.
87:99–163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Upadhyay D and Kamp DW: Asbestos-induced
pulmonary toxicity: role of DNA damage and apoptosis. Exp Biol Med
(Maywood). 228:650–659. 2003.
|
|
25
|
Shukla A, Jung M, Stern M, Fukagawa NK,
Taatjes DJ, Sawyer D, Van Houten B and Mossman BT: Asbestos induces
mitochondrial DNA damage and dysfunction linked to the development
of apoptosis. Am J Physiol Lung Cell Mol Physiol. 285:L1018–L1025.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shukla A, Gulumian M, Hei TK, Kamp D,
Rahman Q and Mossman BT: Multiple roles of oxidants in the
pathogenesis of asbestos-induced diseases. Free Radic Biol Med.
34:1117–1129. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shukla A, Stern M, Lounsbury KM, Flanders
T and Mossman BT: Asbestos-induced apoptosis is protein kinase C
delta-dependent. Am J Respir Cell Mol Biol. 29:198–205. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yuan Z, Taatjes DJ, Mossman BT and Heintz
NH: The duration of nuclear extracellular signal-regulated kinase 1
and 2 signaling during cell cycle reentry distinguishes
proliferation from apoptosis in response to asbestos. Cancer Res.
64:6530–6536. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kido T, Morimoto Y, Asonuma E, Yatera K,
Ogami A, Oyabu T, Tanaka I and Kido M: Chrysotile asbestos causes
AEC apoptosis via the caspase activation in vitro and in vivo.
Inhal Toxicol. 20:339–347. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Soberanes S, Panduri V, Mutlu GM, Ghio A,
Bundinger GR and Kamp DW: p53 mediates particulate matter-induced
alveolar epithelial cell mitochondria-regulated apoptosis. Am J
Respir Crit Care Med. 174:1229–1238. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Frazier WJ, Xue J, Luce WA and Liu Y: MAPK
signaling drives inflammation in LPS-stimulated cardiomyocytes: the
route of crosstalk to G-protein-coupled receptors. PLoS One.
7:e500712012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sim YS, Kim SY, Kim EJ, Shin SJ and Koh
WJ: Impaired expression of MAPK is associated with the
downregulation of TNF-α, IL-6, and IL-10 in Mycobacterium abscessus
lung disease. Tuberc Respir Dis (Seoul). 72:275–283. 2012.
View Article : Google Scholar
|
|
33
|
Soga M, Matsuzawa A and Ichijo H:
Oxidative stress-induced diseases via the ASK1 signaling pathway.
Int J Cell Biol. 2012:4395872012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Aoki H, Kang PM, Hampe J, Yoshimura K,
Noma T, Matsuzaki M and Izumo S: Direct activation of mitochondrial
apoptosis machinery by c-Jun N-terminal kinase in adult cardiac
myocytes. J Biol Chem. 277:10244–10250. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang H, Yang YB, Shen HM, Gu J, Li T and
Li XM: ABT-737 induces Bim expression via JNK signaling pathway and
its effect on the radiation sensitivity of HeLa cells. PLoS One.
7:e524832012. View Article : Google Scholar
|
|
37
|
Xiao F, Liu B and Zhu QX: c-Jun N-terminal
kinase is required for thermotherapy-induced apoptosis in human
gastric cancer cells. World J Gastroenterol. 18:7348–7356. 2012.
View Article : Google Scholar
|