Introduction
X-linked lymphoproliferative disease type 1 (XLP1)
is a well-defined maternally-inherited immunodeficiency disease
caused by a mutation of the SH2D1A gene, which encodes for
signaling lymphocytic activation molecule (SLAM)-associated protein
(SAP), leading to functional defects in natural killer (NK) cells,
CD8+ T cells and natural killer T (NKT) cells (1). Clinical manifestations of this
genetic mutation vary, the most severe complications may include
hemophagocytic lymphohistiocytosis (HLH), lymphoproliferative
disorders, dysgammaglobulinemia and vasculitis, which are often
triggered by Epstein-Barr virus (EBV) infection (2). The present study reported the case of
a 4-year-old male patient with a maternal-onset de novo
SH2D1A mutation, who had suffered from lymphocytic choriomeningitis
(LCMV) infection and developed XLP1, together with other atypical
clinical manifestations.
Case report
The patient was a 4 year-old male who had
experienced intermittent high fevers and a mild cough for six
weeks. On physical examination the patient present with tachypnea,
but no other respiratory symptoms and the examination of the
central nervous system was normal. The study was approved by the
ethics committee of Shandong Provincial Hospital Affiliated to
Shandong University (Jinan, China). Written informed consent was
obtained from the patient’s family. Laboratory testing revealed
hypogammaglobulinemia, elevated C-reactive protein levels and mild
pancytopenia. Sputum and blood cultures were negative for bacteria,
fungi and Mycobacterium tuberculosis. Examinations,
including those for the serum fungal G test, galactomannan test,
toxoplasma antibodies, cryptococcal antigens, anti-nuclear
antibodies, double-stranded DNA, anti-neutrophil cytoplasmic
antibodies and extractable nuclear antigens, were all negative. In
addition, morphological examination of the bone marrow was normal.
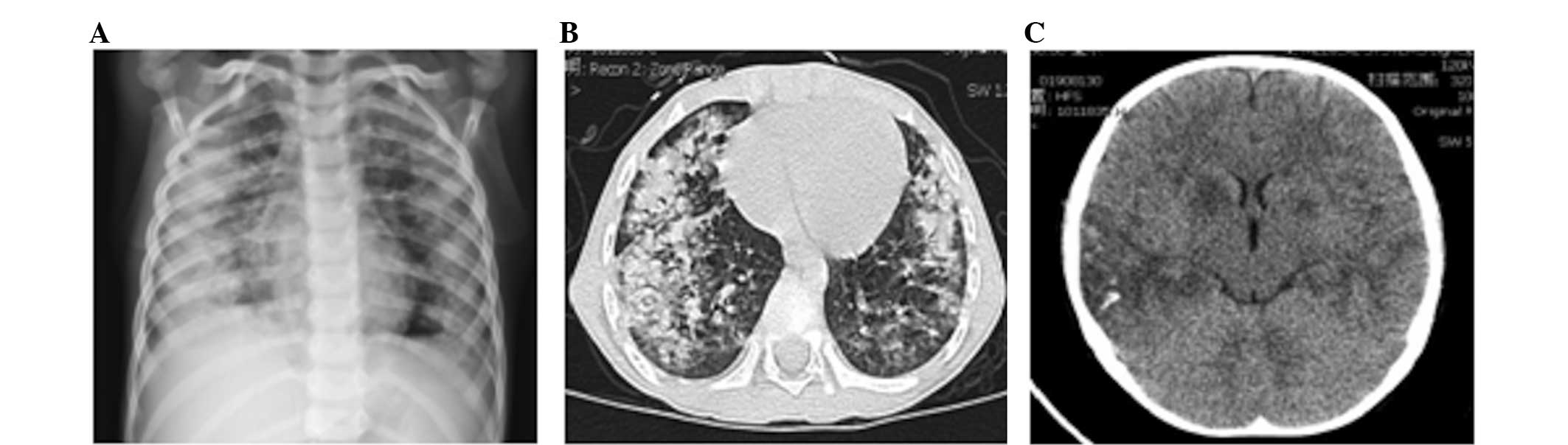
Chest X-ray and computerized tomography (CT) scans revealed diffuse
lung infiltrates (Fig. 1A and B).
Following admission, the patient was treated with ganciclovir and a
γ-globulin (400 mg/kg) infusion. High fevers and coughing
continued, and the patient developed lethargy, slurred speech and
convulsions. A head CT scan revealed multiple low density areas in
the left frontal lobe, right temporal lobe, parietal lobe, left
parietal-occipital lobe, bilateral basal ganglia and trigone of the
left lateral ventricle, with bleeding in the right temporal lobe
(Fig. 1C). The patient succumbed
to a pulmonary hemorrhage two weeks following admission. The
patient had been exposed to a cat in the home environment since
birth and had a history of recurrent respiratory tract
infections.
Detection of the human herpes virus (HHV)
and LCMV
Plasma DNA was screened for HHV 1-8 virus infections
using multiplex polymerase chain reaction (PCR), as previously
described (3,4), the results of which were negative. In
addition, reverse transcription-quantitative PCR was performed, as
previously described (5), in order
to quantitatively analyze LCMV in plasma. The results revealed that
the LCMV load was 7.8×105 copies/ml at the time of
admission and 2.3×106 copies/ml on the eleventh day of
admission when the patient began to develop lethargy, slurred
speech and convulsions.
SH2D1A gene mutation, pedigree analysis
and clone sequencing analysis
Genomic DNA extracted from peripheral blood
mononuclear cells (PBMCs) on the second day of admission was used
for analysis and primers were designed to amplify the coding
sequence of the SH2D1A gene. The PCR product was sequenced by
Sanger sequencing using an AB3130XL genetic analyzer (Applied
Biosystems®, Life Technologies, Grand Island, NY, USA)
and mutations were analyzed using VariantReporter V1.0 software
(Life Technologies).
PCR amplification and sequencing results
demonstrated that the proband carried both hemizygous SH2D1A
c.7G>T/p.A3S and c.228T>A/p.Y76X mutations and the p.Y76X
mutation was predicted to truncate SAP at tyrosine 76 (p.Y76X)
(Fig. 2A). The two mutations were
found to be carried by the proband’s mother, with the c.7G>T
mutation showing a typical heterozygous profile (Fig. 2B and C); however, the c.228T>A
mutation showed a chimeric heterozygous profile (Fig. 2C). The sister of the proband’s
mother carried only a heterozygous c.7G>T mutation (Fig. 2B and C) and their brother was not a
carrier for either mutation (Fig.
2B). The proband’s maternal grandfather was also found to carry
the hemizygous c.7G>T mutation, but not the c.228T>A mutation
(Fig. 2B and C). Neither mutation
was carried by the proband’s father or maternal grandmother.
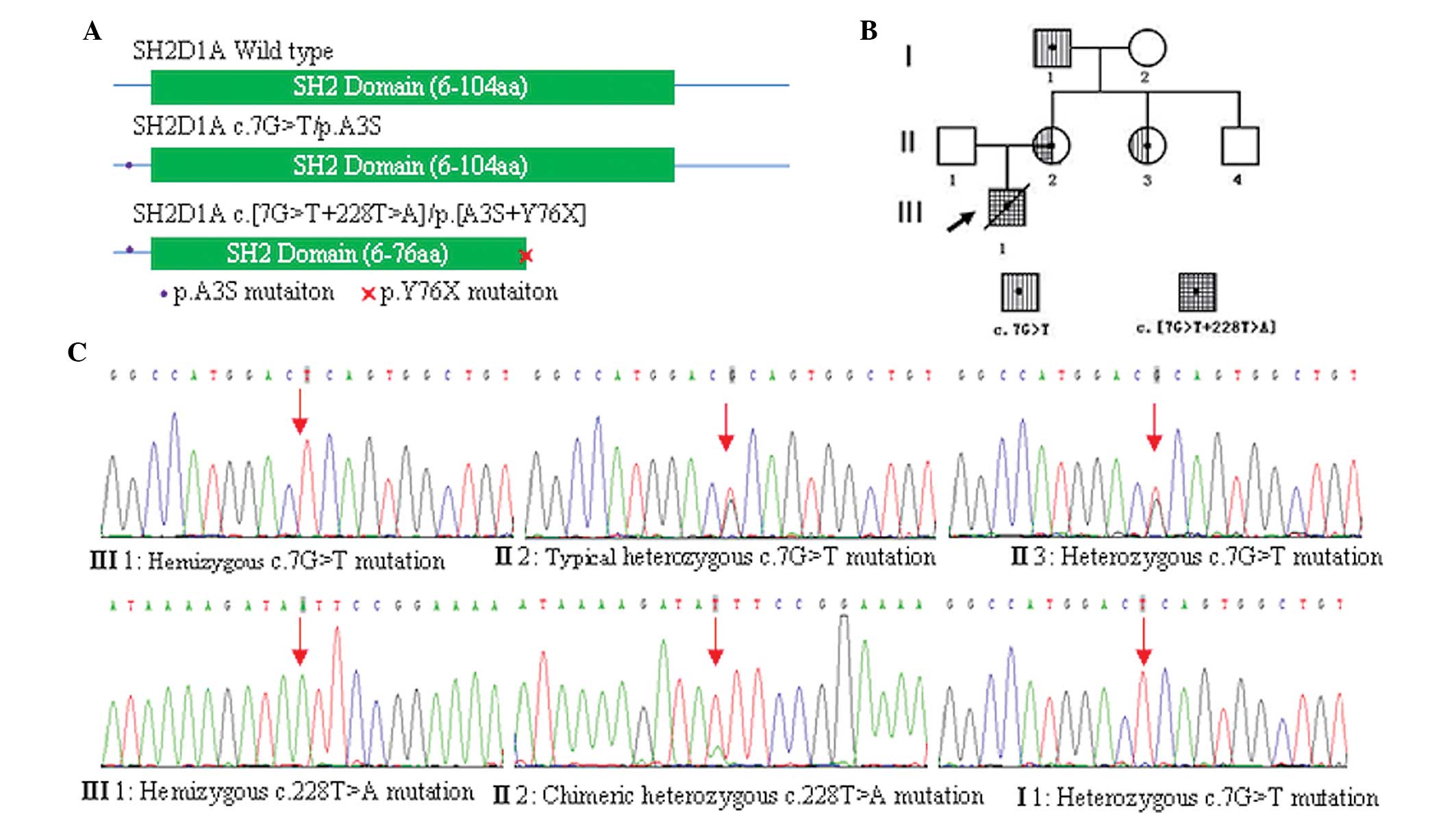 | Figure 2SH2D1A gene mutation, pedigree
analysis and clone sequencing analysis. (A) SH2D1A gene encodes the
128-amino acid protein SAP. Polymerase chain reaction amplification
and sequencing of the patient’s DNA revealed that the p.A3S
mutation was located at the N-terminal sequence of SAP and p.Y76X
mutation was predicted to truncate SAP at tyrosine 76; therefore,
greatly interfering with normal SAP function. (B) Pedigree of three
generations of the patient’s family for SH2D1A gene mutation(s).
Circles represent females, while squares indicate males. Arrow
signifies the proband and slash through symbol indicates mortality.
(C) Sequencing results of amplified genomic DNA from peripheral
blood mononuclear cells from the proband and their family showing:
hemizygous c.7G>T and c.228T>A mutations in the proband
(III1); typical heterozygous c.7G>T mutation and chimeric
heterozygous c.228T>A mutation in II2, heterozygous c.7G>T
mutation in II 3 and hemizygous c.7G>T mutation in I1. SAP,
signaling lymphocytic activation molecule-associated protein; III1,
proband; II1 and II2, proband’s father and mother, respectively;
II3 and II4, mother’s sister and brother, respectively; I1 and I2,
proband’s maternal grandfather and grandmother, respectively. |
Primers were designed for amplification of the
SH2D1A mRNA sequence, including the c.7G>T and c.228T>A
mutation loci. In addition, reverse transcription-PCR was performed
using total RNA extracted from PBMCs. The PCR product was purified,
cloned and then sequenced by Sanger sequencing. The results showed
that the two mutations were located on the same SH2D1A allele in
the proband’s mother, with 26/96 clones of the SH2D1A mRNA
molecules carrying the two mutations and 23/96 clones only carrying
the c.7G>T mutation. Clone sequencing of the genomic DNA PCR
product also revealed that 17/36 clones carried the c.7G>T
mutation and 13/49 clones carried the c.228T>A mutation.
Genotyping of all 15 short tandem repeat (STR) loci, using an AB
Identifier forensic kit, demonstrated that the proband’s mother
exhibited one or two genotypes at each STR loci, indicating that
she was not a chimera.
Discussion
XLP1 is a rare and often fatal X-linked
immunodeficiency disease caused by mutation of the SH2D1A gene,
this gene encodes for a 128-amino acid protein (SAP), which is
comprised of a 5-amino acid N-terminal sequence, a Src homology 2
(SH2) domain and a 25-amino acid C-terminal tail (6). To date, all reported families with
XLP1 have followed the regular inheritance pattern of X-linked
genetic disease. Furthermore, the reported probands carried only
one detrimental SH2D1A mutation or partial/whole SH2D1A gene
deletion, which was often triggered by an EBV infection (1,7).
In the present case report, the proband carried two
mutations in a single maternally inherited SH2D1A allele. The p.A3S
mutation was found to be inherited from the maternal grandfather,
not from the maternal grandmother, and was located at the
N-terminal of the SAP protein, outside of the SH2 domain (Fig. 2A). The proband’s maternal
grandfather carried a hemizygous p.A3S mutation, but maintained a
normal immune status, indicating that the p.A3S mutation was not
lethal. Therefore, it was hypothesized that the p.A3S mutation may
not have a significant adverse impact on the SH2 domain.
The p.Y76X mutation was predicted to truncate SAP at
tyrosine 76 (p.Y76X) (Fig. 2A),
thus resulting in severe deleterious effects on the function. A
previous patient was reported to carry an inherited germiline
p.Y76X mutation and suffered from hemophagocytic
lymphohistiocytosis, hypogammaglobulinemia and EBV infection
(8), which also corroborated the
pathogenicity of this mutation. In the present study, direct
sequencing and clone sequencing were used to determine the chimeric
mutation status of the proband’s mother. The results revealed that
the c.228T>A mutation was a de novo mutation, which
occurred in the same SH2D1A c.7G>T mutated allele at the time of
the first division of the fertilized egg. A previous study
indicated that humans have an intergenerational mutation rate of
~1.1×10−8 per position per haploid genome (9). In addition, comparative genomic
methods were used to estimate that any single conceptus has 1–3
novel deleterious mutations, which lead to an altered amino acid;
therefore, it was predicted that on average, one novel mutation
occurs every 10,000 genes/zygote (10). De novo mutations of rare
diseases have been reported occasionally (11); however, such mutations have not
been reported for XLP1. Spontaneous mutations may have severe
phenotypic consequences when they functionally affect relevant
bases. In addition, when these mutations are carried in germ cells,
they may be inherited.
The patient in the current report had a LCMV
infection, but no EBV or other HHV infection. LCMV, the prototype
of the Arenaviridae family, has been associated with the
natural reservoir Mus domesticus. In addition, LCMV has been
reported to cause meningitis and a flu-like illness. The prevalence
of LCMV in urban residents was found to be 1.0–3.6% (12). LCMV has also been shown to cause
pulmonary infiltrates and central nervous system disease in humans,
predominantly in people with immune deficiencies, including
leukemia (13) or in
post-transplantation patients (14). It was reported that LCMV infection
may result in the mortality of SAP-deficient mice, indicating that
SAP may have an important role in LCMV immunity (15).
In conclusion, it was therefore considered that in
the present case report, the LCMV infection may have resulted in
the onset of XLP1 and the aberrant clinical phenotype in the
patient.
Acknowledgements
The authors would like to thank Professor Zhao Ri Ge
Tu for the help in revising the manuscript and thank the parents
and relatives of the patient who participated in the study. This
study was supported by grants from the National Science Foundation
of China (no. 81000731), the Promotive Research Fund for Excellent
Young and Middle-Aged Scientisits of Shandong Province (no.
BS2010YY045) and the Shandong Provincial Natural Science Foundation
of China (no. ZR2011HM019).
References
|
1
|
Hislop AD, Palendira U, Leese AM, et al:
Impaired Epstein-Barr virus-specific CD8+ T-cell function in
X-linked lymphoproliferative disease is restricted to SLAM
family-positive B-cell targets. Blood. 116:3249–3257. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Booth C, Gilmour KC, Veys P, et al:
X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency:
a multicenter study on the manifestations, management and outcome
of the disease. Blood. 117:53–62. 2011. View Article : Google Scholar
|
|
3
|
Markoulatos P, Georgopoulou A,
Kotsovassilis C, Karabogia-Karaphillides P and Spyrou N: Detection
and typing of HSV-1, HSV-2 and VZV by a multiplex polymerase chain
reaction. J Clin Lab Anal. 14:214–219. 2000. View Article : Google Scholar
|
|
4
|
Pozo F and Tenorio A: Detection and typing
of lymphotropic herpesviruses by multiplex polymerase chain
reaction. J Virol Methods. 79:9–19. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McCausland MM and Crotty S: Quantitative
PCR technique for detecting lymphocytic choriomeningitis virus in
vivo. J Virol Methods. 147:167–176. 2008. View Article : Google Scholar :
|
|
6
|
Morra M, Simarro-Grande M, Martin M, et
al: Characterization of SH2D1A missense mutations identified in
X-linked lymphoproliferative disease patients. J Biol Chem.
276:36809–36816. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Coffey AJ, Brooksbank RA, Brandau O, et
al: Host response to EBV infection in X-linked lymphoproliferative
disease results from mutations in an SH2-domain encoding gene. Nat
Genet. 20:129–135. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Arico M, Imashuku S, Clementi R, et al:
Hemophagocytic lymphohistiocytosis due to germline mutations in
SH2D1A, the X-linked lymphoproliferative disease gene. Blood.
97:1131–1133. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Roach JC, Glusman G, Smit AF, et al:
Analysis of genetic inheritance in a family quartet by whole-genome
sequencing. Science. 328:636–639. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Eyre-Walker A and Keightley PD: High
genomic deleterious mutation rates in hominids. Nature.
397:344–347. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lindhurst MJ, Sapp JC, Teer JK, et al: A
mosaic activating mutation in AKT1 associated with the Proteus
syndrome. N Engl J Med. 365:611–619. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Riera L, Castillo E, Del Carmen Saavedra
M, et al: Serological study of the lymphochoriomeningitis virus
(LCMV) in an inner city of Argentina. J Med Virol. 76:285–289.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Al-Zein N, Boyce TG, Correa AG and
Rodriguez V: Meningitis caused by lymphocytic choriomeningitis
virus in a patient with leukemia. J Pediatr Hematol Oncol.
30:781–784. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fischer SA, Graham MB, Kuehnert MJ, et al;
LCMV in Transplant Recipients Investigation Team. Transmission of
lymphocytic choriomeningitis virus by organ transplantation. N Engl
J Med. 354:2235–2249. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu C, Nguyen KB, Pien GC, et al: SAP
controls T cell responses to virus and terminal differentiation of
TH2 cells. Nat Immunol. 2:410–414. 2001. View Article : Google Scholar : PubMed/NCBI
|