Introduction
Esophageal squamous cell carcinoma (ESCC) is a
highly malignant and lethal disease due to its late diagnosis, the
high incidence of post-surgical local-regional recurrence, and
frequent distant metastasis. Although therapeutic methods have been
improved, the 5-year survival rate is still only approximately 20%
(1–2). Currently, a combination of cisplatin
and 5-fluorouracil (5-FU) is frequently used to treat ESCC patients
in clinical practice. However, the outcome is unsatisfactory due to
its limited effects and side-effects, including nausea, vomiting
and myelosuppression. In addition, certain patients are resistant
to the radical chemotherapy (3).
Therefore, new drugs are required to improve the clinical outcome
and decrease the tolerance of ESCC to chemotherapy.
Epigenetic mechanisms including DNA methylation and
histone modification play significant roles in the initiation and
progression of cancer. The histone deacetylase (HDAC) is an enzyme
which removes acetyl groups from histone and non-histone proteins,
which will lead to chromatin remodeling. HDACs play crucial roles
in numerous biological processes, including cell cycle regulation,
cell proliferation and differentiation (4–5).
HDACs have been observed to be overexpressed in a number of tumor
types, suggesting that HDACs are potential targets for epigenetic
treatment (6–7).
HDACs are a multiclass family consisting of 18 human
HDACs, which are divided into four major classes: class I HDACs
including HDAC 1, 2, 3 and 8; class II HDACs including HDAC 4, 5,
6, 7, 9 and 10; class III HDACs including SIRT1, 2, 3, 4 and 5; and
class IV HDACs including HDAC 11 (8–9).
Trichostatin A (TSA), a known class I and II HDAC inhibitor, has
been demonstrated to exert multiple antitumor effects (10–12).
It is reported that TSA strongly inhibits cell proliferation and
induces cell cycle arrest, and subsequently induces cell apoptosis
(13–15). However, the effect of TSA on ESCC
cells has not been characterized.
In this study, we report on the inhibition of the
proliferation of ESCC cells by TSA through cell cycle arrest and
cell apoptosis. We further investigate the mechanism involved in
this process by analyzing cell cycle regulators p21 and p27 as well
as apoptotic protein markers Bcl-2 and Bax. In addition, we analyze
the expression of phosphatidylinositol-3-kinase (PI3K)/Akt and
extracellular signal-regulated kinase (ERK)1/2, and the level of
histone H4 acetylation before and after TSA treatment to reveal the
mechanism of epigenetic modification.
Materials and methods
Cell lines and cell culture
The EC9706 cell line was a gift from the State Key
Laboratory of Molecular Oncology, Chinese Academy of Medical
Science (Beijing, China). The EC1 cell line was kindly provided by
the University of Hong Kong (Hong Kong, China). The cell lines were
propagated in monolayer culture in RPMI-1640 medium supplemented
with 10% fetal bovine serum (56°C, 30 min), 1×105 U/l
penicillin and 100 mg/l streptomycin in a humidified atmosphere
with a 5% CO2 incubator at 37°C. The present study was
approved by the Medical Ethics Committee of Zhengzhou University
(Zhengzhou, China).
Reagents and treatment
TSA was purchased from Sigma (St. Louis, MO, USA).
It was dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich) as a 5
μM stock solution, and stored at −20°C. Control cells were
treated with DMSO in parallel in each experiment. Mouse monoclonal
antibodies to p21, p27, Bcl-2 and Bax, and rabbit monoclonal
antibodies to PI3K, p-Akt, Akt, ERK1/2 and acetyl-histone H4 (Lys8)
were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA); horse anti-mouse and goat anti-rabbit horseradish
peroxidase-conjugated secondary antibodies were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). The CCK-8 kit was
purchased from Dojindo Laboratories (Kumamoto, Japan). The Annexin
V-FITC kit was purchased from Beckman Coulter (Miami, FL, USA).
Cell viability assay
ESCC cell lines were seeded at a density of
5×103 in 96-well microtiter plates. After culturing for
24 h, cells were treated with TSA at various concentrations (0.1,
0.3, 05, 1.0, 3.0 and 5.0 μM) prepared from a stock solution
dissolved in DMSO for 24 and 48 h, respectively. Cells treated with
identical concentrations of DMSO (diluent for depsipeptide) were
used as control. Four hours before measuring the absorbance, 10
μl CCK-8 solution was added to each well and incubated. The
absorbance at 450 nm wavelength was determined for each well using
an enzyme-labeling instrument. All studies were performed in
triplicate independently.
Cell cycle analysis
Cells were treated with various concentrations of
TSA (0.3, 0.5 and 1.0 μM) for 48 h, then cells were
harvested with 2.5 g/l trypsin and fixed in absolute ethanol
overnight at 4°C. The cells were resuspended in phosphate-buffered
saline (PBS) containing 1% RNase and then 5 μg/ml propidium
iodide (PI) was added. Cells were incubated in the dark for 15 min
at room temperature. A total of 3×104 cells were counted
using a flow cytometer.
Apoptosis assay
Following incubation with or without TSA for 48 h,
ESCC cells were harvested with 2.5 g/l trypsin and washed twice
with PBS. A total of 1×105 cells were stained with
fluorescein isothiocyanate (FITC)-Annexin V-PI using the Annexin
V-FITC kit (Beckman Coulter) according to the manufacturer’s
instructions. At least 1.5×104 cells were counted by
flow cytometric analysis. Each experiment was performed in
triplicate. The percentage of apoptotic cells was calculated with
CellQuest 3.0 software (BD Biosciences, Franklin Lakes, NJ, USA).
Cells that were stained negatively with Annexin V and PI were
considered viable cells. Early apoptotic cells were positive for
Annexin V and negative for PI, and late apoptotic cells were
positive for Annexin V and PI.
Western blot analysis
Cells were washed with cold PBS twice and lysed in
lysis buffer. After 20 min on ice, the lysates were centrifuged at
14000 rpm at 4°C for 10 min. The supernatants were used as whole
cell extracts. The protein concentration of cells was analyzed
using the BCA protein assay kit (Pierce Biotechnology, Inc.,
Rockford, IL, USA). Cell lysate (50 μg) was mixed with 5X
sodium dodecyl sulfate (SDS) sample buffer, separated on a 10%
SDS-polyacrylamide gel and transferred to polyvinylidene fluoride
membranes. Membranes were blocked with 5% non-fat milk for 1 h and
incubated with the primary antibodies at 4°C overnight. The
following primary antibodies and dilutions were used: p21 (1:500),
p27 (1:500), Bcl-2 (1:1000), Bax (1:1000), PI3K (1:200), p-Akt
(1:500), Akt (1:500), p-ERK1/2 (1:1000), ERK1/2 (1:1000) and
acetylated histone 4 (AH4; 1:1000). The membranes were then washed
with PBS-Tween (0.1%) three times, each for 5 min. Next, membranes
were incubated with a 1:5000 dilution of horseradish peroxidase
conjugated with either goat antimouse or goat antirabbit for 2 h at
room temperature. Target proteins were detected with an enhanced
chemiluminescence detection kit (ECL, Pierce). Protein levels were
quantified with Quantity One® software (Bio-Rad,
Hercules, CA, USA).
Statistical analysis
Data were expressed as the means ± standard
deviation, and statistical analysis was performed by analysis of
variance using SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA).
One-way analysis of variance was used to compare differences among
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
TSA induces morphological change and
inhibits cell viability of ESCC cell lines
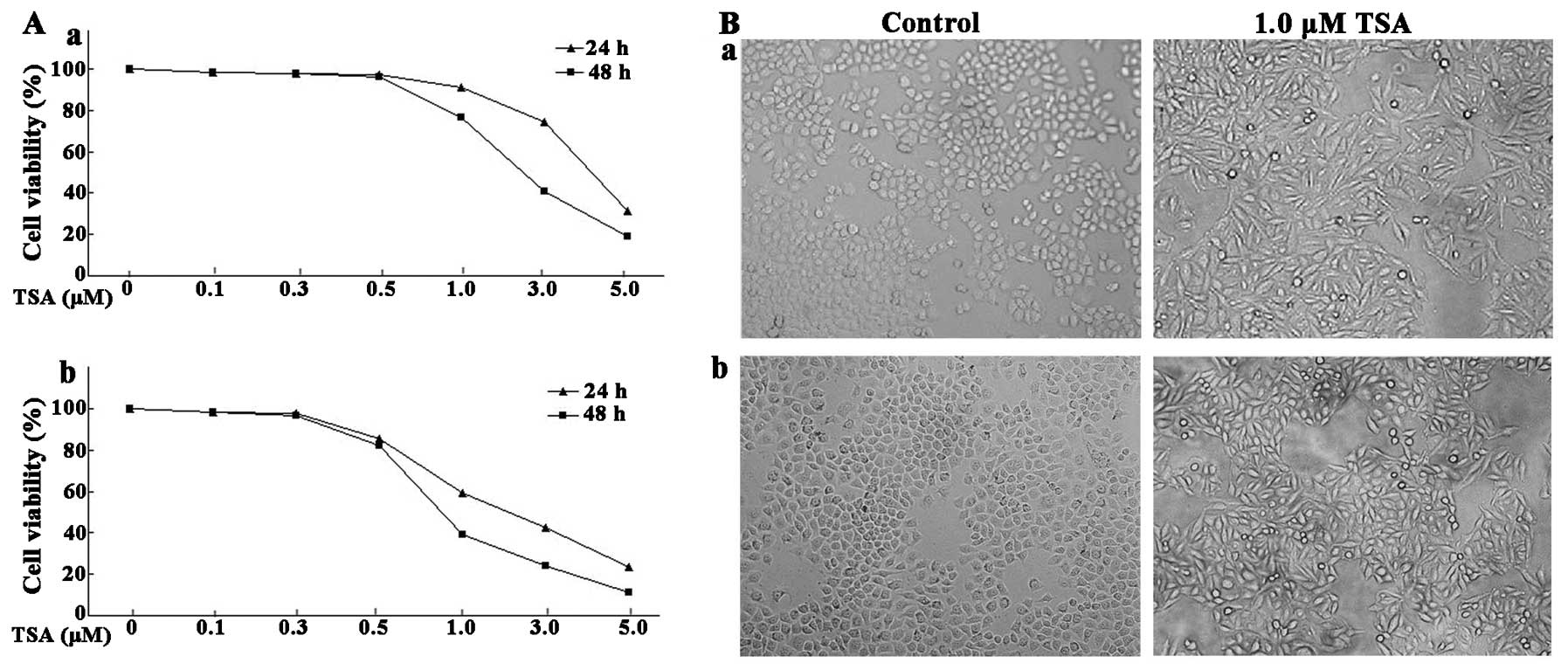
Morphological changes of ESCC cells were examined
via microscopy. As shown in Fig.
1B, ESCC cells treated with control (DMSO) grew in clusters and
were confluent. However, cells treated with 1.0 μM TSA lost
the normal cell morphology and had flattened and spindle shapes.
The proliferative ability of the ESCC cells treated with various
concentrations of TSA (0.1–5.0 μM) was assessed by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. The proliferation of EC9706 was not significantly inhibited
by TSA at doses of 0.1, 0.3 or 0.5 μM after 24 and 48 h.
However, the proliferation of EC9706 was notably inhibited at doses
over 1.0 μM (Fig. 1A). The
cell proliferation analysis revealed that EC1 cells were more
sensitive to TSA treatment than EC9706 cells. After 24 h of
treatment at the dose of 5.0 μM, the relative cell viability
of EC9706 and EC1 cells declined to 31.32 and 19.35%, respectively.
The cell viability declined to 18.92 and 12.33% after 48 h of
treatment with 5.0 μM TSA. These data indicated that TSA
exhibited its inhibitory effects in ESCC cells in a
concentration-dependent manner.
TSA suppresses proliferation of ESCCs by
cell cycle arrest
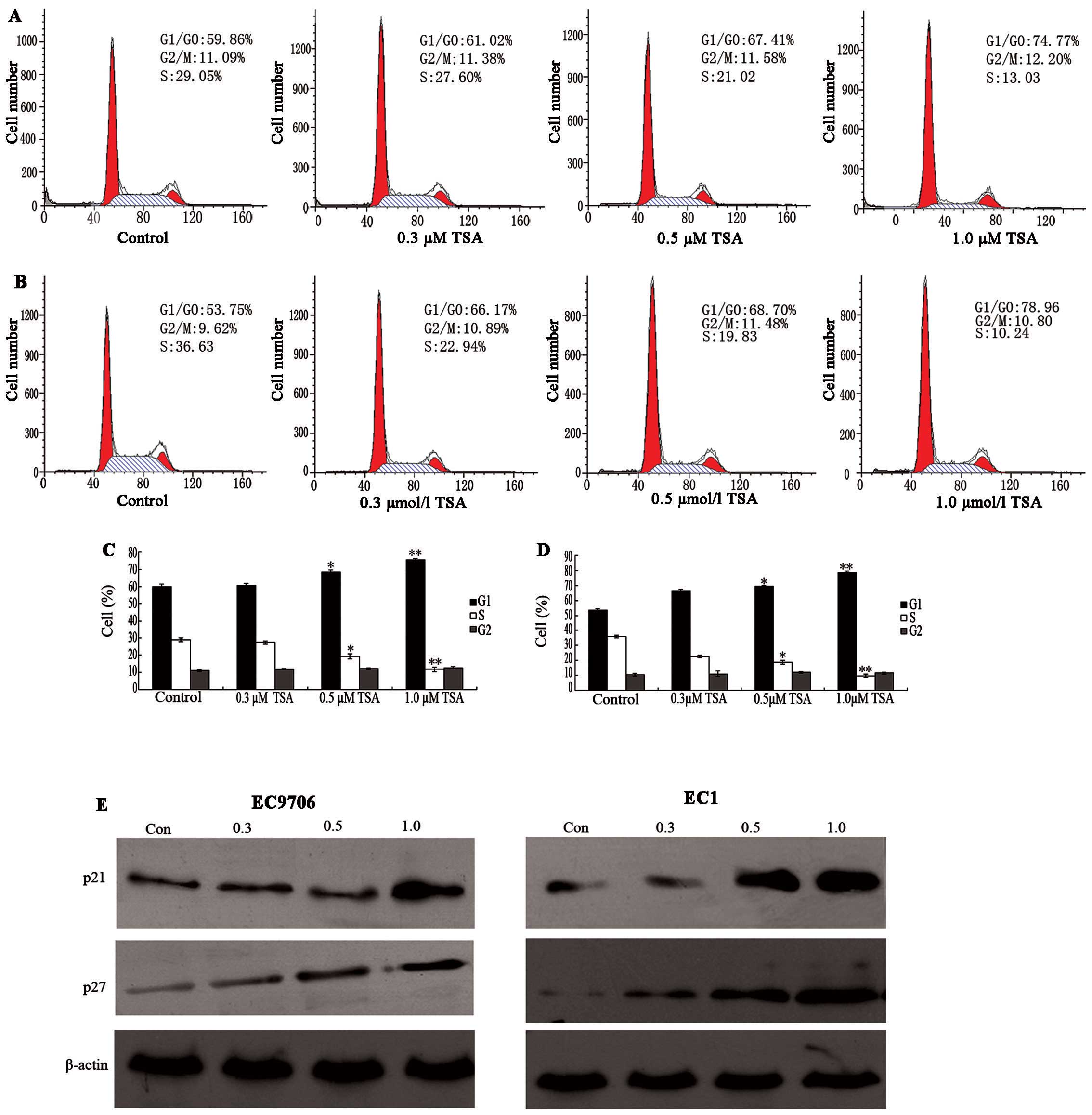
To determine whether the cell growth inhibition
effects of TSA were due to cell cycle arrest, the cell cycle phase
distribution of ESCC was examined following TSA treatment. There
was no notable change in the cell cycle phase distribution of
EC9706 at doses of 0.3 or 0.5. μM after 48 h of TSA
treatment. However, the percentage of untreated EC9706 cells in the
G1/G0 phase was 59.86%, which increased to 74.77% after 48 h 1.0
μM TSA treatment, while the percentage of S phase cells
decreased from 29.09 to 13.03% (Fig.
2A). In EC1 cells, the percentage of G1/G0 phase cells
increased from 53.75 to 78.96%, and S phase cells decreased from
36.63 to 10.24%. These results indicated that a significant G1/G0
arrest was induced in ESCC cells compared with control cells, with
a corresponding decrease of cells in the S phase after 48 h of
treatment (P<0.05; Fig.
2B).
With these results, it was assumed that TSA might
selectively affect the expression of G1 cell cycle components. To
investigate the mechanism of cell cycle arrest in ESCC cells, we
analyzed the protein levels of p21 and p27 via western blot
analysis following treatment with control and TSA (0.3 to 1.0
μM). As shown in Fig. 2E,
it was observed that TSA induced a significant increase in p21 and
p27 protein levels at the dose of 1.0 μM. It has been
previously demonstrated that p21 and p27 participate in negative
control of the cell checkpoint by blocking cyclin-dependent kinase
(CDK) activity (16). Thus,
increases in cellular p21 and p27 correlate with increased
inactivation of CDKs. These results suggest that TSA inhibits the
proliferation of ESCC lines EC9706 and EC1 by G1 phase cell cycle
arrest through an induction of p21 and p27.
TSA suppresses proliferation of ESCCs by
cell apoptosis
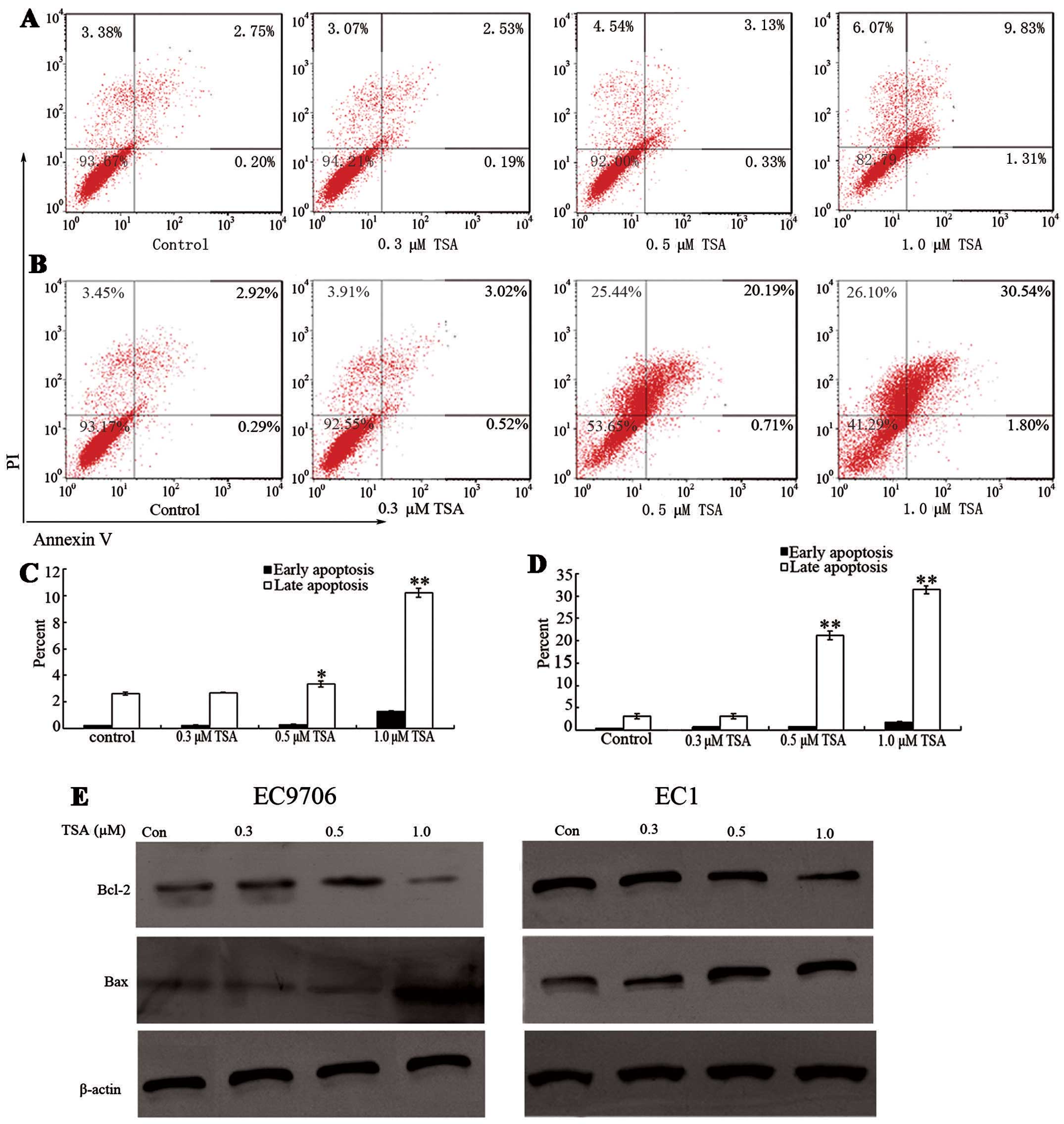
In addition to modulating cell cycle regulatory
proteins to suppress cell growth, it has been previously
demonstrated that TSA is capable of inducing cell apoptosis in
other tumor cell lines (17). To
determine whether TSA induces cell apoptosis in ESCC cell lines,
EC9706 and EC1 cells were treated with various doses of TSA for 48
h. As shown in Fig. 3A and B, the
percentage of early apoptosis was not notably increased at doses of
0.3 and 0.5 μM TSA. However, 1.0 μM TSA treatment
significantly induced early apoptosis compared with that of the
control groups (P<0.05). Furthermore, the percentages of late
apoptosis in both EC9706 and EC1 cells increased in a
concentration-dependent manner.
To investigate the underlying mechanism of cell
apoptosis, the protein levels of apoptosis-related proteins Bcl-2
and Bax were measured before and after TSA treatment. Bcl-2 is an
anti-apoptotic protein which inhibits the activity of cysteine
proteases known as the ICE family proteases. Significant Bcl-2
downregulation was induced in EC9706 cells and EC1 cells after 48 h
TSA exposure relative to the control groups (P<0.05).
Furthermore, Bcl-2 downregulation occurred earlier in EC1 cells
than in EC9706 cells (Fig. 3E).
These responses are in accordance with cell apoptosis induced by
TSA. Decreased Bcl-2 protein levels may cause an increase in ICE
protease activity; thus, ESCC cells may undergo apoptosis. Bax is a
pro-apoptotic protein which regulates programmed cell death. The
level of Bax significantly increased following 1.0 μM TSA
treatment. These results indicate that decreased anti-apoptotic
protein Bcl-2 and increased pro-apoptotic protein Bax are
underlying apoptotic mechanisms of cell apoptosis.
TSA suppresses the PI3K/Akt and
mitogen-activated protein kinase (MAPK)/ERK1/2 signaling pathways
to inhibit ESCC proliferation
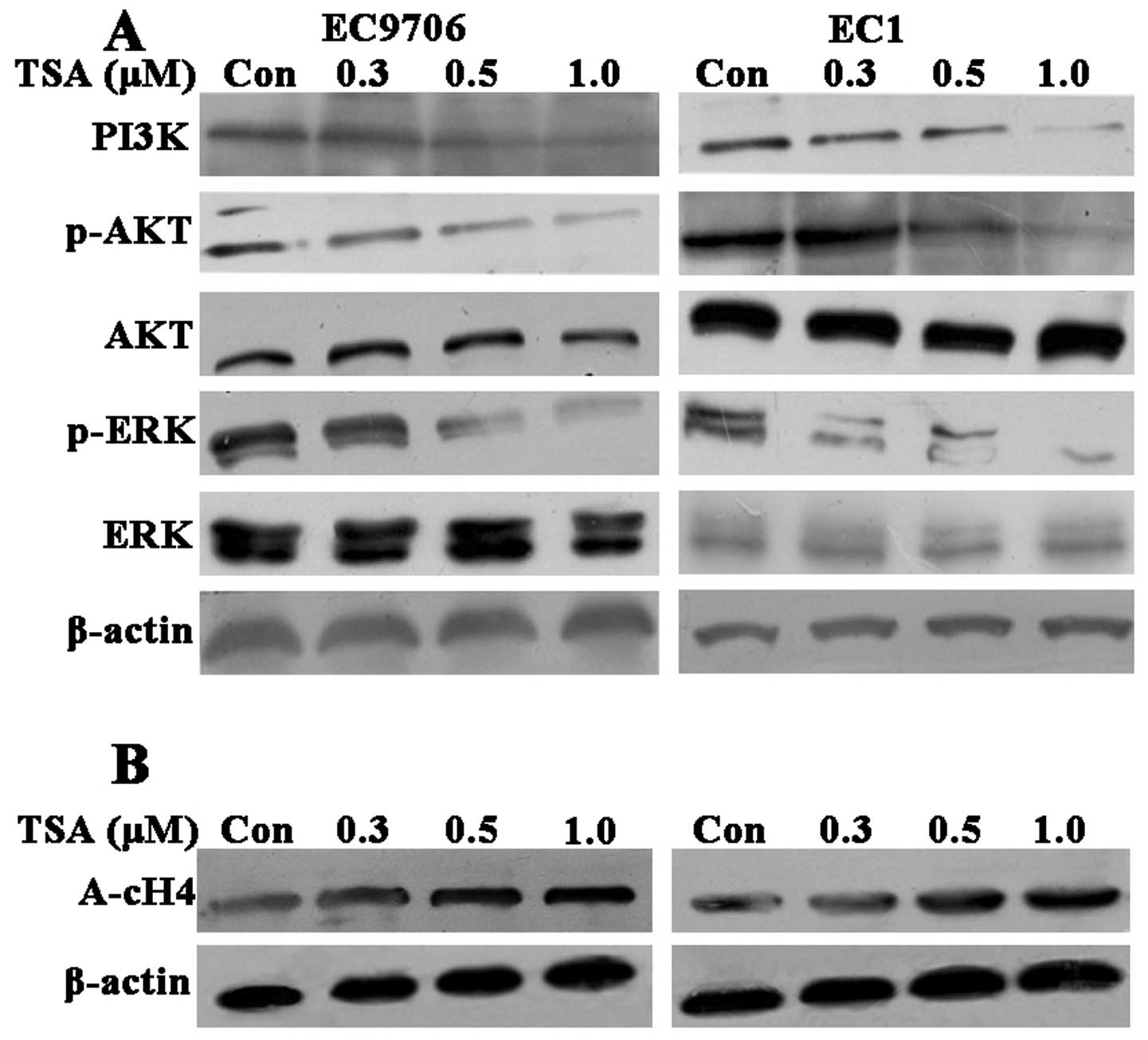
The PI3K/Akt and MAPK signaling pathways including
the ERK pathways play a key role in regulating cell growth,
proliferation and survival. To investigate the underlying mechanism
of TSA on the biological behavior of ESCC cells, we examined the
expression of PI3K and the phosphorylation of Akt and ERK1/2
(Fig. 4A). TSA decreased the
protein level of PI3K as well as the phosphorylation of Akt and
ERK1/2 in a dose-dependent manner without affecting their total
protein levels following TSA treatment for 60 min in the two ESCC
cell lines. These results indicated that TSA potently inhibited the
proliferation of ESCC by suppressing the PI3K/Akt and ERK1/2
pathways.
To further examine whether the observed effects of
ESCC induced by TSA are due to direct modification of histone
acetylation we also evaluated the acetylation level of histone H4
by western blot analysis. The acetylation of histone H4 increased
in a concentration-dependent manner following TSA treatment for 48
h in both EC9706 cells and EC1 cells (Fig. 4B). These results suggested that TSA
inhibited their deacetylation. Therefore, the effects of TSA on
ESCC proliferation are partly owing to modification of histone
acetylation.
Discussion
ESCC is one of the most aggressive types of
malignant cancer with poor prognosis. Epigenetic regulation of gene
expression via acetylation of histone and other essential cellular
proteins is a potentially useful therapeutic strategy. The
HDAC-mediated epigenetic mechanism has a central role in regulating
gene expression through chromatin remodeling. HDACs are often
overexpressed in numerous types of cancer (18–20).
For these reasons, HDAC inhibitors are promising antitumor drugs
(21–23). In this study, we evaluated the
effects of pan-HDAC inhibitor TSA on cell proliferation, cell cycle
regulation and cell apoptosis in ESCC cell lines. Moreover, we also
demonstrated that inhibition of HDAC activity with TSA dramatically
suppressed the PI3K/Akt and MAPK signaling pathways.
TSA was observed to exert a potent antitumor
activity on human colon carcinoma cells and breast adenocarcinoma
cells (24,25). TSA was also reported to inhibit the
growth of prostate cancer cells through the induction of cell cycle
arrest and cell apoptosis (26).
In this study, we demonstrated that TSA exhibits its inhibitory
effects in ESCC cells in a time- and dose-dependent manner. To
further investigate the anti-proliferation mechanisms of TSA toward
ESCC, we analyzed the effects of TSA on the cell cycle. G0/G1
arrest was observed in treated EC9706 and EC1 cells. The cell cycle
in mammals is controlled by cyclins and CDKs (27–29).
CDK inhibitors including p21 and p27 are essential regulators of
the cell cycle, inhibiting the activity of cyclin D1/CDK4/6 and
cyclin E/CDK2 complexes and blocking cell-cycle transition from G1
phase to S phase (30–32). In our study, p21 and p27 were
continually upregulated after 48 h incubation with TSA. These
results further demonstrate that TSA exerts its inhibitory effects
on cell cycle progression, possibly by inducing p21 and p27, and
this may be the underlying mechanism of the subsequent growth
inhibition effect of TSA in ESCC.
In addition to blocking cell-cycle transition, HDAC
inhibitors induce apoptosis via the death-receptor apoptosis
pathway or the mitochondrial-mediated apoptosis pathway. The
mitochondrial pathway disrupts the mitochondrial membrane, causing
the release of proteins, including cytochrome c and other
pro-apoptotic molecules, into the cytoplasm (33,34).
In our study, TSA enhanced the expression of pro-apoptotic protein
Bax and decreased the expression of anti-apoptotic protein Bcl-2,
thereby activating the mitochondrial-mediated apoptosis
pathway.
As is well known, the PI3K/Akt and MAPK signaling
pathways are closely related to cell proliferation, differentiation
and survival (35–37). It was demonstrated that HDAC
inhibitor caused Akt dephosphorylation in three diffuse large
B-cell lymphoma cell lines (17).
We demonstrated that TSA decreased the expression of PI3K and the
phosphorylation of Akt in two ESCC cell lines. We further
investigated whether the MAPK signaling pathway is part of the
mechanism triggered by TSA to inhibit cell proliferation in ESCC.
In our study, TSA reduced the phosphorylation of ERK1/2 in a
dose-dependent manner in both EC9706 and EC1 cell lines. These
results indicated that suppressing the PI3K/Akt and MAPK signaling
pathways in ESCC is partly responsible for the cell growth
inhibition induced by TSA.
In conclusion, we demonstrated that the HDAC
inhibitor TSA has an anti-proliferative effect in ESCC cell lines
via the induction of cell cycle arrest and apoptosis. TSA induces
cell cycle arrest in the G1/G0 phases through the induction of p21
and p27. TSA induced apoptosis by decreasing the anti-apoptotic
protein Bcl-2 and increasing the pro-apoptotic protein Bax. TSA
induces a significant decrease in ESCC cell growth by potently
inhibiting the PI3K/Akt and ERK1/2 pathways. Overall, our results
suggest that HDAC inhibitors are promising drugs to treat ESCC.
Acknowledgments
This study was supported by the National Basic
Research Program of China (grant no. 2012CB933300), the Natural
Science Foundation of the Henan Province of China (grant nos.
12B310023 and 81372269) and the Natural Science Foundation of the
Henan province of China (grant nos. 13HASTIT022 and 12B310022).
References
|
1
|
Zhu H, Huo X, Chen L, Wang H and Yu H:
Clinical experience with radio-, chemo- and hyperthermotherapy
combined trimodality on locally advanced esophageal cancer. Mol
Clin Oncol. 1:1009–1012. 2013.
|
|
2
|
Jemal A, Center MM, DeSantis C and Ward
EM: Global patterns of cancer incidence and mortality rates and
trends. Cancer Epidemiol Biomarkers Prev. 19:1893–1907. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Minsky BD, Pajak TF, Ginsberg RJ, et al:
INT 0123 (Radiation Therapy Oncology Group 94–05) phase III trial
of combined-modality therapy for esophageal cancer: high-dose
versus standard-dose radiation therapy. J Clin Oncol. 20:1167–1174.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Minucci S and Pelicci PG: Histone
deacetylase inhibitors and the promise of epigenetic (and more)
treatments for cancer. Nat Rev Cancer. 6:38–51. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Reichert N, Choukrallah MA and Matthias P:
Multiple roles of class I HDACs in proliferation, differentiation
and development. Cell Mol Life Sci. 69:2173–2187. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ellis L, Atadja PW and Johnstone RW:
Epigenetics in cancer: targeting chromatin modifications. Mol
Cancer Ther. 8:1409–1420. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ellis L and Pili R: Histone deacetylase
inhibitors: advancing therapeutic strategies in hematological and
solid malignancies. Pharmaceuticals (Basel). 3:2411–2469. 2010.
View Article : Google Scholar
|
|
8
|
Abbas A and Gupta S: The role of histone
deacetylases in prostate cancer. Epigenetics. 3:300–309. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bonfils C, Walkinshaw DR, Besterman JM,
Yang XJ and Li Z: Pharmacological inhibition of histone
deacetylases for the treatment of cancer, neurodegenerative
disorders and inflammatory diseases. Expert Opin Drug Discov.
3:1041–1065. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ropero S and Esteller M: The role of
histone deacetylases (HDACs) in human cancer. Mol Oncol. 1:19–25.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cress WD and Seto E: Histone deacetylases,
transcriptional control and cancer. J Cell Physiol. 184:1–16. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Meng F, Sun G, Zhong M, Yu Y and Brewer
MA: Inhibition of DNA methyltransferases, histone deacetylases and
lysine-specific demethylase-1 suppresses the tumorigenicity of the
ovarian cancer ascites cell line SKOV3. Int J Oncol. 43:495–502.
2013.PubMed/NCBI
|
|
13
|
Ueki N, Lee S, Sampson NS and Hayman MJ:
Selective cancer targeting with prodrugs activated by histone
deacetylases and a tumour-associated protease. Nat Commun.
4:27352013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wilson PM, Labonte MJ, Martin SC, et al:
Sustained inhibition of deacetylases is required for the antitumor
activity of the histone deactylase inhibitors panobinostat and
vorinostat in models of colorectal cancer. Invest New Drugs.
31:845–857. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen X, Xiao W, Chen W, Luo L, Ye S and
Liu Y: The epigenetic modifier trichostatin A, a histone
deacetylase inhibitor, suppresses proliferation and
epithelial-mesenchymal transition of lens epithelial cells. Cell
Death Dis. 4:e8842013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Stein C, Riedl S, Rüthnick D, Nötzold RR
and Bauer UM: The arginine methyltransferase PRMT6 regulates cell
proliferation and senescence through transcriptional repression of
tumor suppressor genes. Nucleic Acids Res. 40:9522–9533. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cai Y, Cui W, Chen W, et al: The effects
of a histone deacetylase inhibitor on biological behavior of
diffuse large B-cell lymphoma cell lines and insights into the
underlying mechanisms. Cancer Cell Int. 13:572013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Spiegel S, Milstien S and Grant S:
Endogenous modulators and pharmacological inhibitors of histone
deacetylases in cancer therapy. Oncogene. 31:537–551. 2012.
|
|
19
|
Mutze K, Langer R, Becker K, et al:
Histone deacetylase (HDAC) 1 and 2 expression and chemotherapy in
gastric cancer. Ann Surg Oncol. 17:3336–3343. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jurkin J, Zupkovitz G, Lagger S, et al:
Distinct and redundant functions of histone deacetylases HDAC1 and
HDAC2 in proliferation and tumorigenesis. Cell Cycle. 10:406–412.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hoshino I and Matsubara H: Recent advances
in histone deacetylase targeted cancer therapy. Surg Today.
40:809–815. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Noureen N, Rashid H and Kalsoom S:
Identification of type-specific anticancer histone deacetylase
inhibitors: road to success. Cancer Chemother Pharmacol.
66:625–633. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Marson CM: Histone deacetylase inhibitors:
design, structure-activity relationships and therapeutic
implications for cancer. Anticancer Agents Med Chem. 9:661–692.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rezaei PF, Fouladdel S, Hassani S, et al:
Induction of apoptosis and cell cycle arrest by pericarp
polyphenol-rich extract of Baneh in human colon carcinoma HT29
cells. Food Chem Toxicol. 50:1054–1059. 2012. View Article : Google Scholar
|
|
25
|
Ward CS, Eriksson P, Izquierdo-Garcia JL,
Brandes AH and Ronen SM: HDAC inhibition induces increased choline
uptake and elevated phosphocholine levels in MCF7 breast cancer
cells. PLoS One. 8:e626102013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Watson JA, McKenna DJ, Maxwell P, et al:
Hyperacetylation in prostate cancer induces cell cycle aberrations,
chromatin reorganization and altered gene expression profiles. J
Cell Mol Med. 14:1668–1682. 2010. View Article : Google Scholar
|
|
27
|
Gerard C and Goldbeter A: From quiescence
to proliferation: Cdk oscillations drive the mammalian cell cycle.
Front Physiol. 3:4132012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yagi Y, Fushida S, Harada S, et al:
Effects of valproic acid on the cell cycle and apoptosis through
acetylation of histone and tubulin in a scirrhous gastric cancer
cell line. J Exp Clin Cancer Res. 29:1492010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang C, Fu M, Mani S, Wadler S,
Senderowicz AM and Pestell RG: Histone acetylation and the
cell-cycle in cancer. Front Biosci. 6:D610–D629. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Aguero MF, Facchinetti MM, Sheleg Z and
Senderowicz AM: Phenoxodiol, a novel isoflavone, induces G1 arrest
by specific loss in cyclin-dependent kinase 2 activity by
p53-independent induction of p21WAF1/CIP1. Cancer Res.
65:3364–3373. 2005.PubMed/NCBI
|
|
31
|
Queiroz AB, Focchi G, Dobo C, Gomes TS,
Ribeiro DA and Oshima CT: Expression of p27, p21 (WAF/Cip1) and p16
(INK4a) in normal oral epithelium, oral squamous papilloma and oral
squamous cell carcinoma. Anticancer Res. 30:2799–2803.
2010.PubMed/NCBI
|
|
32
|
Tula-Sanchez AA, Havas AP, Alonge PJ, et
al: A model of sensitivity and resistance to histone deacetylase
inhibitors in diffuse large B cell lymphoma: Role of
cyclin-dependent kinase inhibitors. Cancer Biol Ther. 14:949–961.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
You BR and Park WH: Trichostatin A induces
apoptotic cell death of HeLa cells in a Bcl-2 and oxidative
stress-dependent manner. Int J Oncol. 42:359–366. 2013.
|
|
34
|
Hacker S, Karl S, Mader I, et al: Histone
deacetylase inhibitors prime medulloblastoma cells for
chemotherapy-induced apoptosis by enhancing p53-dependent Bax
activation. Oncogene. 30:2275–2281. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang J, Roberts TM and Shivdasani RA:
Targeting PI3K signaling as a therapeutic approach for colorectal
cancer. Gastroenterology. 141:50–61. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen CS, Weng SC, Tseng PH, Lin HP and
Chen CS: Histone acetylation-independent effect of histone
deacetylase inhibitors on Akt through the reshuffling of protein
phosphatase 1 complexes. J Biol Chem. 280:38879–38887. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu B and Kuang A: Genetic alterations in
MAPK and PI3K/Akt signaling pathways and the generation,
progression, diagnosis and therapy of thyroid cancer. Sheng Wu Yi
Xue Gong Cheng Xue Za Zhi. 29:1221–1225. 2012.In Chinese.
|