Introduction
Oral surgery frequently causes craniofacial bone
defects in various situations, including trauma, infection, tumors,
congenital malformations or degenerative skeletal diseases. To
date, such bone defects have been repaired by the use of bone
autografts and allografts (1,2);
however, autografts and allografts require invasive surgery and the
subsequent use of immunosuppressive agents. Therefore, the use of
autologous osteogenic stem/progenitor cells may potentially
overcome the disadvantages of autografts and allografts.
Mesenchymal stem cells (MSCs) have been shown to be a potential
cell source for bone-tissue engineering (3). Briefly, MSCs undergo osteogenic
differentiation via a well-defined pathway following appropriate
stimulation (4,5). However, the harvesting of bone marrow
is an invasive procedure and therefore the burden on patients is
great (1,6). Recently, it was reported that the
tongue muscle may provide an additional source of autologous cells
for cardiac regeneration. Briefly, Shibuya et al (7) isolated Sca-1-positive cells, which
are considered stem cells, from tongue muscle and demonstrated that
these positive cells were able to differentiate into beating cells
similar to cardiomyocytes. Tongue muscle-derived Sca-1-positive
cells (TDSCs), as well as MSCs, may have multipotent
differentiation capacity and thus present a novel therapeutic tool
with the potential to replace autologous tissue grafting for bone
defects. However, to the best of our knowledge, few studies have
utilized tongue muscle cells as a cell source for bone-tissue
engineering. In the present study, TDSCs were isolated and their
potential as a cell source for bone-tissue engineering was
examined.
Materials and methods
Animals
Female Jcl-ICR mice (Japan SLC, Shizuoka, Japan) and
female athymic nude mice with CAnN.Cg-Foxnlnu/CrlCrlj genetic
backgrounds (CLEA Japan, Inc., Tokyo, Japan) were purchased at four
weeks of age and bred in the Animal Center of Yamaguchi University
(Ube, Japan). The animals were housed in plastic cages in a
pathogen-free environment and kept in an air-conditioned room at
23±2°C with a relative humidity of 55±10% under a 12-h light/dark
cycle. The mice were provided sterile water and food ad
libitum. All experiments were approved by the Institutional
Animal Care and Use Committee of Yamaguchi University. The study
conformed to the Guide for the Care and Use of Laboratory Animals
published by the US National Institutes of Health (NIH Publication
no. 85-23, revised 1996; Bethesda, MA, USA).
Immunohistochemistry
The 8-week-old mice were sacrificed through an
overdose of Somnopentyl (200mg/kg; Merchk and Co., Inc., Whitehouse
Station, NJ, USA), and the tongue of the mouse was resected using a
surgical knife and was then fixed in 10% formalin neutral-buffer
solution (Wako Pure Chemical Industries, Ltd., Osaka, Japan) and
embedded in paraffin (Wako Pure Chemical Industries, Ltd., Osaka,
Japan). Sections (4 μm) were prepared from the paraffin
blocks and mounted on slides. These sections were fixed and
processed for immunostaining with anti-Sca-1 mouse monoclonal
antibody (1:100; 130-092-529; BD Biosciences, Franklin Lakes, NJ,
USA), anti-osteocalcin rabbit polyclonal antibody (1:100, sc-30044;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA), anti-SPARC
(secreted protein acidic and rich in cysteine, also known as
osteonectin) rabbit polyclonal antibody (1:100; sc-25574; Santa
Cruz Biotechnology, Inc.), anti-osteopontin mouse monoclonal
antibody (1:100; sc-21742; Santa Cruz Biotechnology, Inc.) and the
appropriate peroxidase-conjugated goat anti-rabbit polyclonal or
-mouse monoclonal immunoglobulin G (IgG) secondary antibody (1:100;
sc-2005; Santa Cruz Biotechnology, Inc.). Negative controls were
performed using non-specific IgG (1:100; sc-2072 or sc-2025; Santa
Cruz Biotechnology, Inc.). The blocking and immunostaining were
performed using the Dako Envision kit (Dako, Glostrup, Denmark)
according to the manufacturer’s instructions. All specimens were
counterstained with hematoxylin. The slides were subsequently
examined under a bright-field microscope (BX51; Olympus Corp.,
Tokyo, Japan). A reddish-brown precipitate indicated a positive
reaction.
Isolation and expansion of TDSCs
TDSCs were obtained from the tongue muscle of
8-week-old female, Jcl-ICR mice. Briefly, mice were sacrificed and
the tongue was removed, as described above. Enzyme solution
containing 40 μg/ml Liberase Blendzyme 3 (Roche Diagnostics
GmbH, Mannheim, Germany) and 200 μg/ml DNase I solution
(Thermo Fisher Scientific, Waltham, MA, USA) in Dulbecco’s minimal
essential medium (D-MEM; Sigma-Aldrich, St. Louis, MO, USA) was
injected directly into the tongue with a 25-gauge needle (Terumo,
Tokyo, Japan). Mouse tongues were incubated by agitation at 37°C
for 1 h and dissociated with MACS Dissociator (Miltenyi Biotec,
Bergisch Gladbach, Germany) completely. The dissociated tissue was
centrifuged at 80.57 × g at 4°C for 2 min and the cell pellet was
resuspended in phosphate-buffred saline without calcium or
magnesium [PBS(-)]. The cell suspension was harvested and passed
through a 100 μm-mesh filter (Miltenyo Biotec). The cells
were washed by adding MACS buffer [PBS(-) containing 0.5% bovine
serum albumin and 2 mM EDTA; Miltenyi Biotec] and centrifuged at
80.57 × g at 4°C for 2 min. The supernatant was discarded and the
cell pellet was resuspended in 50 ml MACS buffer. The cell
suspension was passed through a 70 μm-mesh filter (Miltenyi
Biotec) and centrifuged at 1200 rpm at room temperature for 10 min.
The supernatant was discarded, the cell pellet was resuspended in
10 ml MACS buffer and a cell suspension of 3.0×106
nucleated cells in 80 μl MACS buffer was prepared. FcR
Blocking reagent (10 μl; Miltenyi Biotec) was added and
incubated at 4°C for 5 min. Subsequently, 10 μl
anti-Sca-1-fluorescein isothiocyanate (FITC) antibody was added and
incubated at 4°C for 10 min. The cells were washed by the addition
of 2 ml MACS buffer and centrifugation at 1200 rpm at 4°C for 10
min. The supernatant was discarded completely and the cell pellet
was resuspended in 500 μl MACS buffer. Sca-1 positive cells
were separated from the cell suspension using a magnetic cell
separation system with microbeads (autoMACS Pro Separator™;
Miltenyi Biotec). Propidium iodide solution (final concentration, 1
μg/ml) was added to the Sca-1 positive cells and the Sca-1
positivity of TDSCs was evaluated by flow cytometric analysis
(FACSCalibur™; BD Biosciences). TDSCs were cultured in culture
medium, which consisted of D-MEM supplemented with 5% fetal bovine
serum (FBS; Thermo Fisher Scientific), 5% growth-stimulating medium
(NH CFU-Medium®; Miltenyi Biotec), 100 U/ml penicillin,
100 μg/ml streptomycin (Thermo Fisher Scientific) and 12
μM l-glutamine (Invitrogen Life Technologies). Non-adherent
cells were removed following 24 h of culture by washing with PBS
and adding fresh culture medium. Every 3–4 days, cells were washed
and fresh culture medium was added for a period of four weeks.
Following four weeks of culture, cells were washed and trypsinized
by incubation in 2 ml of 0.25% trypsin/1 mM EDTA (Thermo Fisher
Scientific) for 2 min at 37°C. The trypsin/EDTA was neutralized by
the addition of 10 ml culture medium and the cells (passage one)
were replated in 10 ml culture medium in a 100-mm dish. Following
one week of culture, cells were trypsinized and subcultured
(passage two) at 1.0×105 cells/100-mm dish in culture
medium. The culture medium was replaced every three days. Following
one further week, cells were trypsinized and either frozen at −80°C
in TC-protecto cell freezing medium (DS Pharma Biomedical, Osaka,
Japan) or expanded further by plating at 1.0×105/100-mm
dish in culture medium. Identical conditions were used for the
subsequent passages.
Cell growth analysis
To evaluate cell growth, clonal populations (derived
from a single cell by limiting dilution) of TDSCs were plated at a
density of 5×103 cells/well and subcultured in a 96-well
culture plate (BD Biosciences). Following two, four, six, eight, 10
and 12 days of culture, MTT was added to each well (25
μl/well) and incubated for 4 h. The blue dye taken up by the
cells was dissolved in dimethyl sulfoxide (100 μl/well) and
the absorbance was measured with a spectrophotometer (BioRad
Laboratories, Hercules, CA, USA) at 490 nm. All assays were
performed in triplicate.
Osteogenic differentiation analysis
The osteogenic differentiation capacity of TDSCs was
assessed by the measurement of alkaline phosphatase (ALP) activity,
ALP staining, alizarin red S staining and Von Kossa staining under
a bright-field microscope (BX51; Olympus Corp.). The cells were
grown on six-well plates at a density of 5×105
cells/well in bone differentiation-inducing medium (NH OsteoDiff
Medium®; Miltenyi Biotec). The medium was replaced every two
days.
ALP activity
ALP activity was evaluated using fast
p-nitrophenyl phosphate tablets (Sigma-Aldrich) according to
the manufacturer’s instructions. Briefly, cells were seeded at a
density of 1×104 cells/well on six-well culture plates
(BD Biosciences) in DMEM supplemented with 10% FBS. After 24 h,
cells were treated with bone differentiation-inducing medium
(Miltenyi Biotec). The treated cells were collected and lysed with
1X radioimmunoprecipitation assay buffer (Thermo Fisher
Scientific). Subsequently, a Bradford protein assay sample (20
μl) was prepared. The sample was reacted with buffered
substrate (100 μl) and the reaction was stopped by the
addition of stop solution (0.2 M NaOH; 80 μl). The relative
quantity of reacted p-nitrophenyl was estimated from the
absorbance at 405 nm (BioRad Laboratories) at days one, three,
five, seven, 10, 14, 21 and 28 of culture. Each experiment was
performed in triplicate.
ALP staining
ALP staining was performed using a
tartrate-resistant acid phosphatase and ALP double-stain kit
(Takara Bio, Inc., Otsu, Japan) according to the manufacturer’s
instructions. Briefly, cells were fixed with fixation solution
(acetone/citrate buffer pH 5.4) for 5 min at room temperature and
stained with a substrate for ALP
(5-bromo-4-chloro-3-indolyl-phosphate/nitro blue tetrazolium;
Sigma-Aldrich) for 45 min at room temperature. Cells were
counterstained with methyl green for 5 min at room temperature.
Alizarin red S staining for mineralized
matrix
Cells were fixed with 70% ice-cold ethanol for 1 h
at −20°C and stained with 40 mM alizarin red S solution (pH 4.2;
Sigma-Aldrich) for 10 min at room temperature. Cells were dried
following rinsing under running tap water. Red staining indicated
mineral nodule formation.
Von Kossa staining
Cells were fixed with absolute ethanol and stained
using the Von Kossa method to detect mineral nodule formation in
vitro using a Von Kossa Stain kit (Diagnostic BioSystems,
Pleasanton, CA, USA) according to the manufacturer’s instructions.
Cells were fixed with 70% ice-cold ethanol for 1 h at −20°C and
stained with 5% silver nitrate for 60 min with exposure to
ultraviolet light. Following washing with distilled water, cells
were stained with 5% sodium thiosulfate for 3 min. Cells were
subsequently rinsed under running tap water and stained with
nuclear fast red stain for 5 min. Black staining was indicative of
mineral nodule formation.
Western blot analysis
Cells were lysed with radioimmunoprecipitation assay
buffer and whole cell lysates were subjected to 10% SDS-PAGE and
transferred onto polyvinylidene difluoride membranes (Thermo Fisher
Scientific). The membranes were incubated with the anti-Sca-1 mouse
monoclonal antibody (1:1,000), anti-c-kit rabbit polyclonal
antibody (1:1,000; sc-168; Santa Cruz Biotechnology, Inc.),
anti-osterix rabbit polyclonal antibody (1:1,000), anti-RUNX2
(runt-related transcription factor 2) rabbit polyclonal antibody
(1:,000; sc-10758; Santa Cruz Biotechnology, Inc.),
anti-fibronectin mouse monoclonal antibody (1:1,000; sc-59826;
Santa Cruz Biotechnology, Inc.), anti-osteocalcin rabbit polyclonal
antibody (1:1,000) and anti-osteopontin rabbit polyclonal antibody
(1:1,000; sc-20788; Santa Cruz Biotechnology, Inc.). The membranes
were then incubated with Novex®
alkaline-phosphatase-conjugated goat anti-rabbit polyclonal
secondary antibody (WP20006; Thermo Fisher Scientific), or goat
anti-mouse monoclonal secondary antibody (WP20007; Thermo Fisher
Scientific) according to manufacturer’s instructions. The
antibodies were detected using a chromogenic immunodetection system
(WesternBreeze; Thermo Fisher Scientific) according to the
manufacturer’s instructions. The anti-α-tubulin monoclonal antibody
(Santa Cruz Biotechnology, Inc.) was used for the normalization of
western blot analyses.
Three-dimensional (3D) cell culture and
histological evaluations
Cells were seeded into gelatin sponges of β-TCP
(MedGel® Scaffold; MedGel, Kyoto, Japan) by an agitated
seeding method as it had been previously demonstrated that this
method was effective for seeding cells homogeneously throughout
porous 3D scaffolds (8,9). The cell-seeded gelatin sponges of
β-TCP were cultured in tissue culture plates with bone
differentiation-inducing medium at 37°C in a 5% CO2-95%
air atmosphere for three weeks. Cell culture was performed for
three weeks until 90% confluence was reached and the medium was
replaced every three days. Subsequently, the cell-seeded gelatin
sponges of β-TCP were transplanted subcutaneously into nude mice.
In brief, a small incision was made using a knife into the dorsum
of nude mice under general anesthetic (Sonnopentyl injection, 20
mg/kg). The cell-seeded gelatin sponges of β-TCP were inserted
surgically beneath the skin and the incision sites were sutured
using 3-0 silk (Alfresa Pharma Corporation, Osaka, Japan). All mice
were monitored weekly. At 28 days post-transplantation, the
cell-seeded gelatin sponges of β-TCP were resected with a surgical
knife under general anesthesia (Somnopentyl, 20 mg/kg). The
cell-seeded gelatin sponges of β-TCP were fixed in 10%
neutral-buffered formalin solution, dehydrated, immersed in xylene
and embedded in paraffin. The samples were cut into 4-μm
sections and stained with hematoxylin and eosin to histologically
view on an optical microscope (BX51; Olympus Corp.).
Results
Sca-1-positive cells are located in the
tongue muscle and may be isolated at high purity
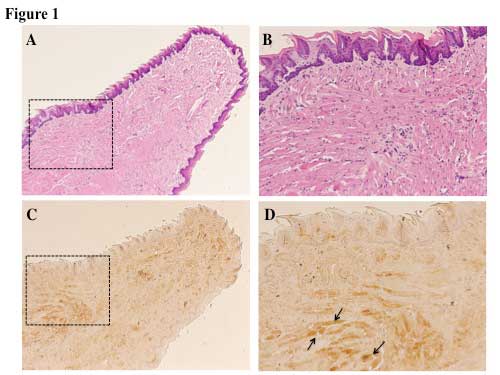
Sca-1 expression was detected in the muscle of
Jcl-ICR mouse tongues by immunohistochemical analysis. The majority
of the expression of Sca-1 was localized to the tongue muscle
rather than the tongue mucosa (Fig.
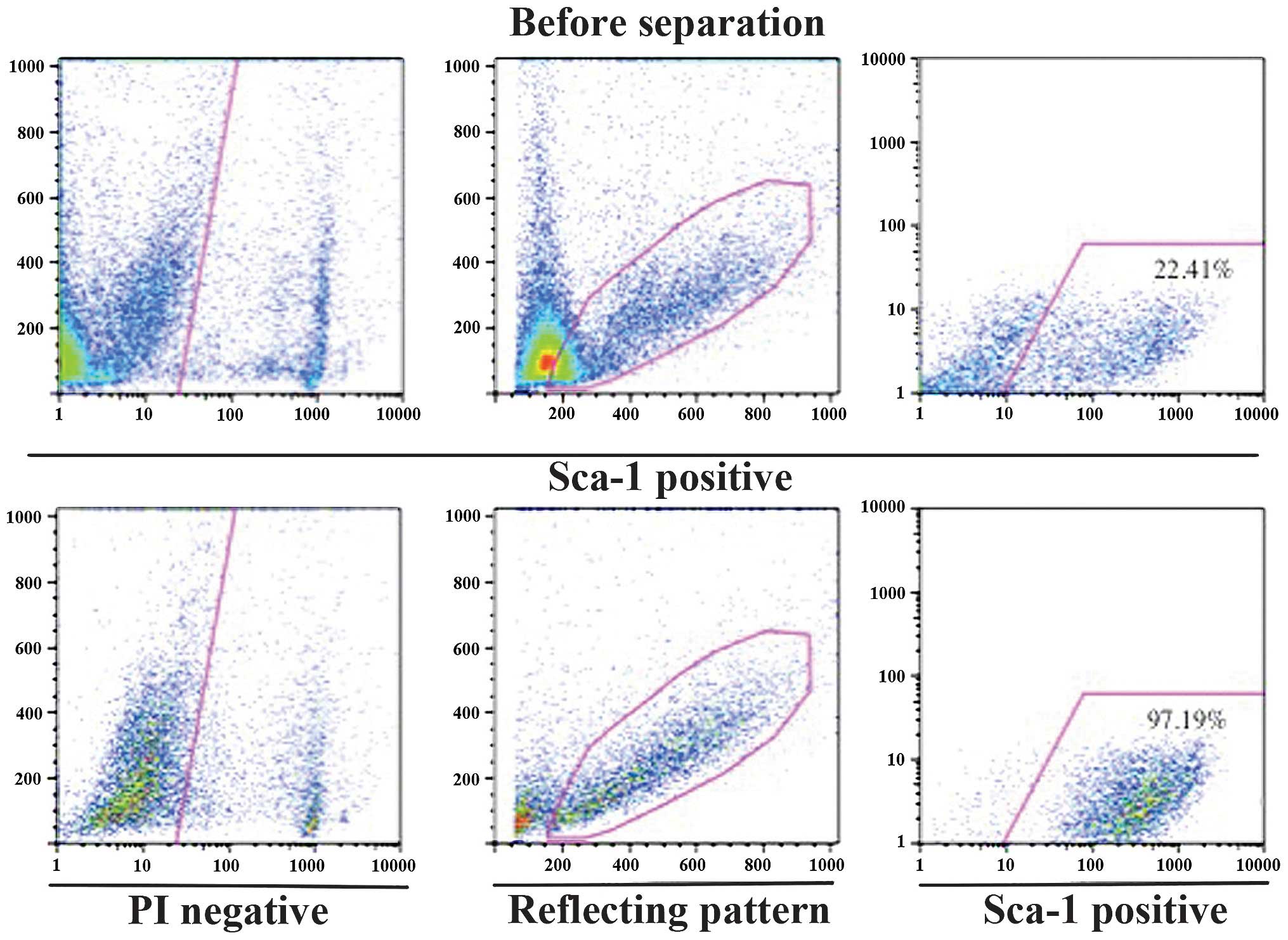
1). Cells were harvested from Jcl-ICR mouse tongue muscles and
highly pure populations of Sca-1 positive cells were obtained using
a magnetic cell separation system with microbeads. Briefly, 97.19%
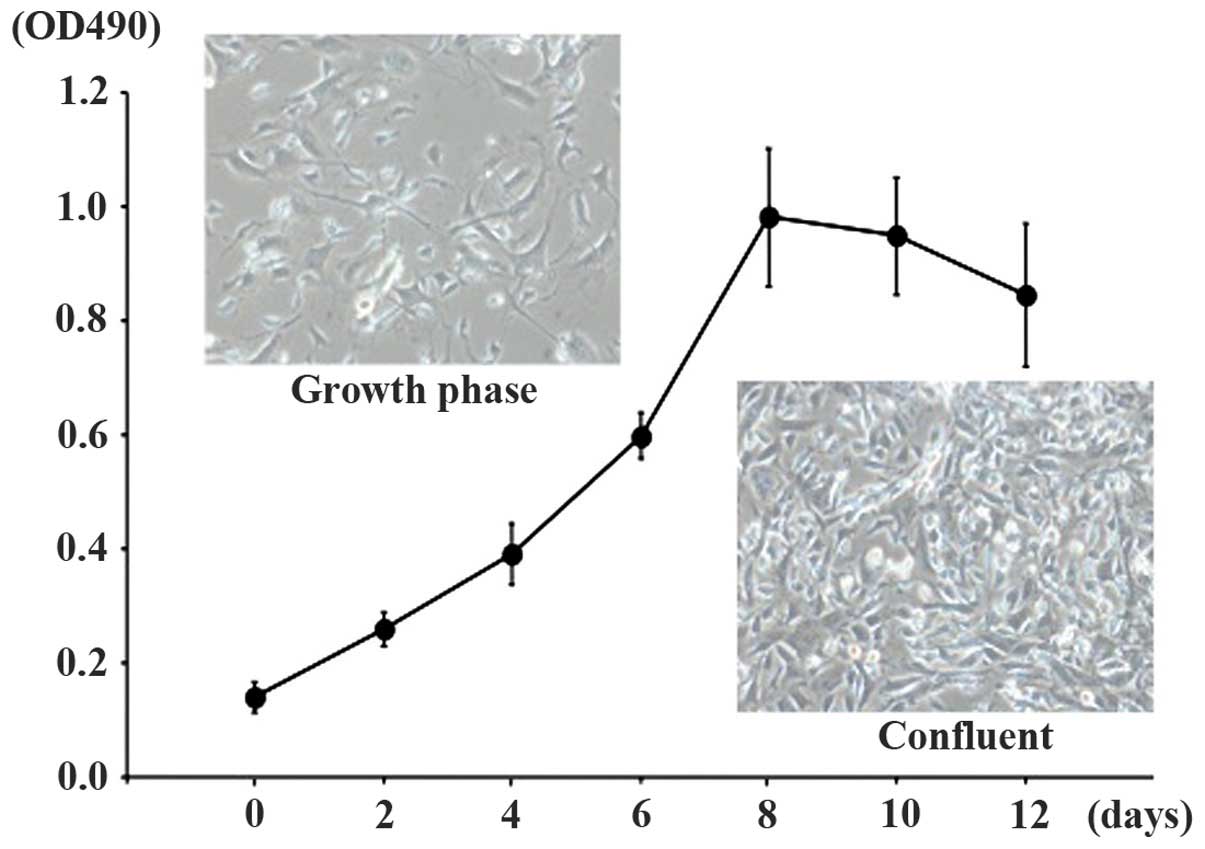
of cells in the Sca-1-positive fraction expressed Sca-1 (Fig. 2). TDSCs were able to be readily
expanded in expansion medium and the doubling time was ~48 h. TDSCs
exhibited a fusiform, fibroblast-like morphology (Fig. 3).
Cultured Sca-1-positive cells
differentiate into osteoblasts
The differentiation capacity of TDSCs was evaluated
for osteoblast differentiation. TDSCs were cultured in bone
differentiation-inducing medium supplemented with osteogenic
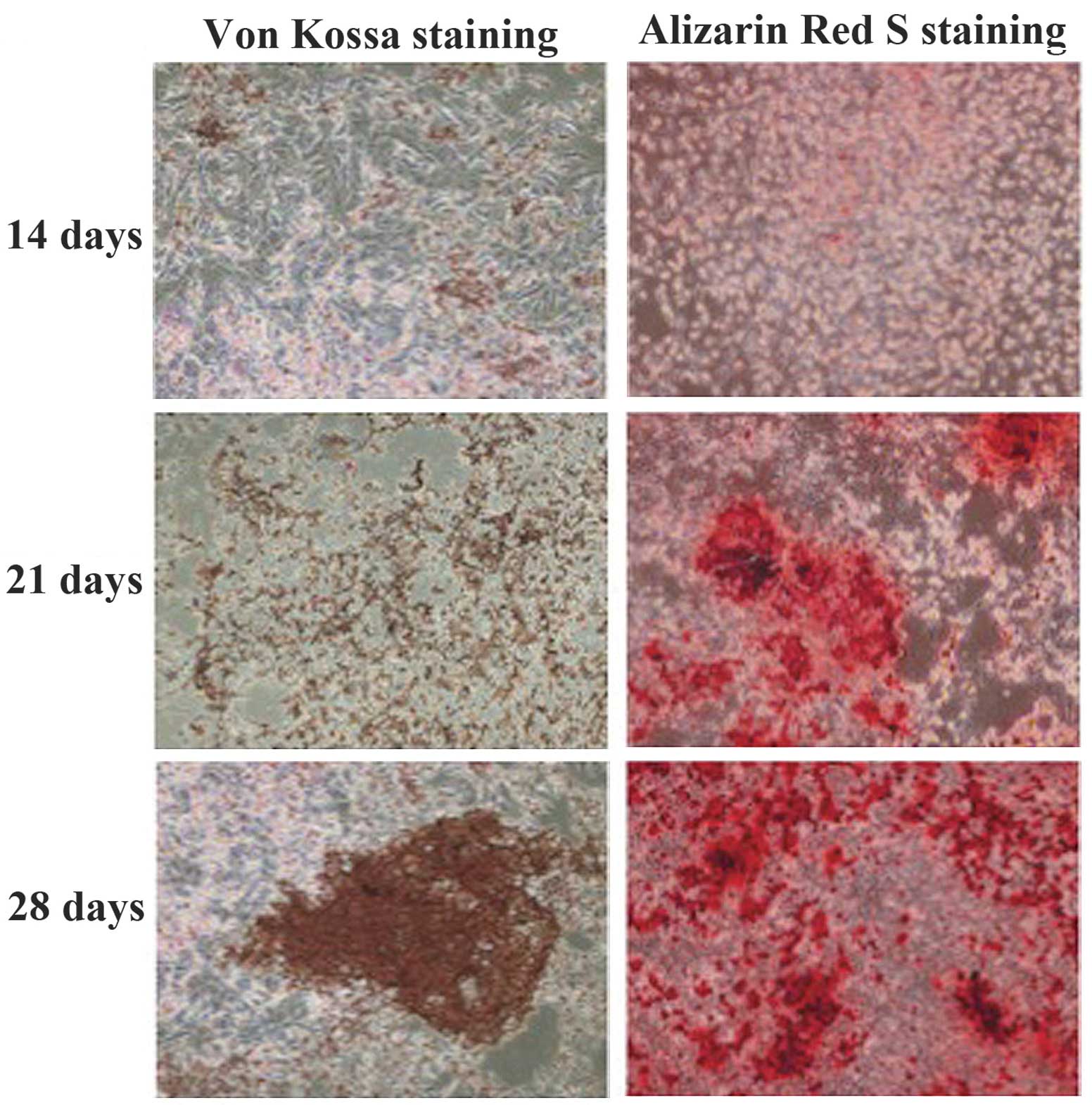
induction components. TDSCs began to develop distinct mineralized
nodules following 14 days in culture. The mineralized nodules were
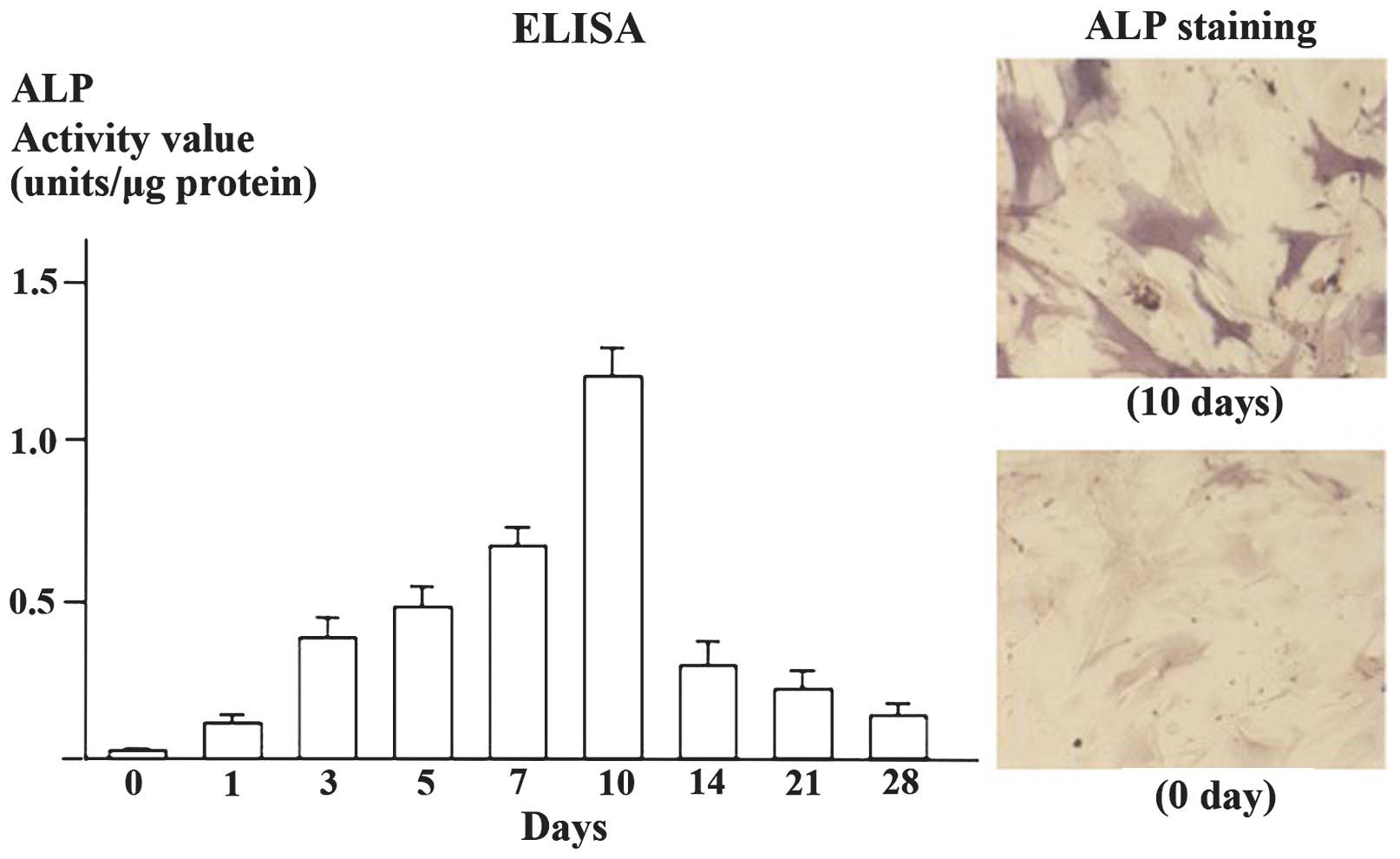
stained with alizarin red S or Von Kossa (Fig. 4). ALP activity was found to
increase following treatment with bone differentiation medium until
the activity peaked following 10 days of culture and subsequently
decreased as determined by an assay with fast p-nitrophenyl
phosphate tablets or cytochemical staining (Fig. 5).
Furthermore, western blot analysis of mesenchymal
stem cell marker expression (Sca-1 and c-kit) and osteoblastic
protein expression (osterix, Runx2, fibronectin, osteocalcin,
osteonectin and osteopontin) was performed. As indicated in
Fig. 6, TDSCs initially expressed
mesenchymal stem cell markers and the expression of Sca-1 decreased
following a peak at three days of culture. In addition, the
expression of c-kit also decreased after peaking at the seventh day
of culture. The expression of osteoblastic markers was increased by
treatment with bone differentiation medium, although the majority
began to decrease following a peak at 5–7 days of culture.
Expression of osteocalcin was observed in 14–21 day-cultured
cells.
Sca-1-positive cells differentiate into
bone in vivo
Subsequently, the in vivo differentiation
potential of TDSCs was investigated. TDSCs were seeded into the
gelatin sponges of β-TCP using an agitated seeding method and
transplanted subcutaneously into nude mice. At 28 days
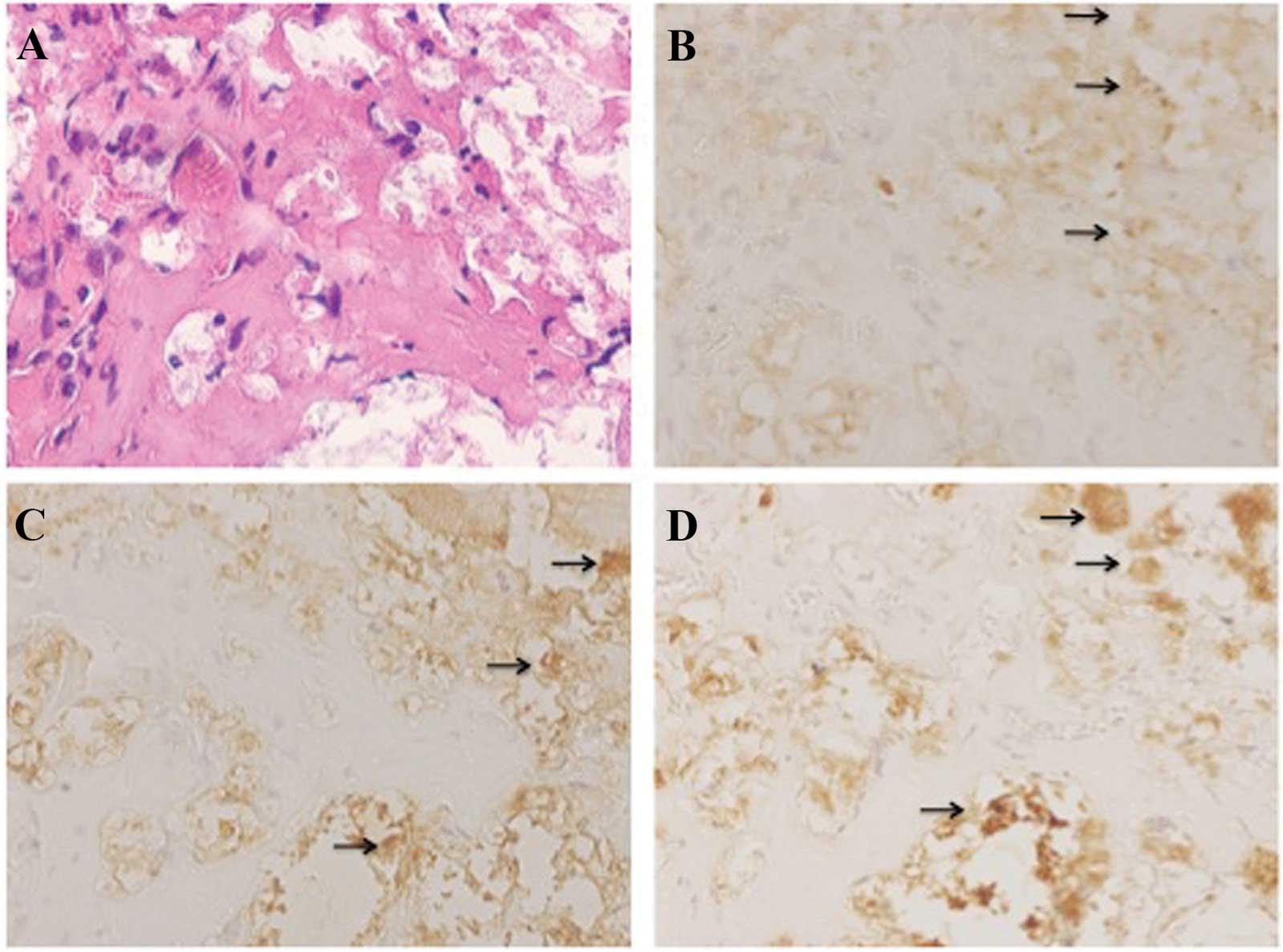
post-transplantation, areas with bone-like morphology were observed
in the gelatin sponges of β-TCP (Fig.
7A). Bone matrix expression was identified in the cells of
these areas by immunohistochemical analysis. Cells in these areas
produced osteocalcin, osteonectin and osteopontin (Fig. 7B–D).
Discussion
In the present study, the potential of the tongue
muscle as a cell source for bone-tissue engineering was evaluated,
as stem cells, including satellite cells and muscle side population
cells, exist in skeletal muscle tissue (10) and osterix overexpression enhances
osteoblast differentiation of muscle satellite cells in
vitro (11). Shibuya et
al (7) previously succeeded at
inducing cardiomyogenic differentiation of tongue muscle-derived
stem cells. They used Sca-1 as a stem cell marker for isolating
stem cells from mouse tongue muscle. Sca-1 was initially reported
to be a cell surface marker of hematopoietic stem cells (12). However, several studies have since
reported that multipotent stem cells derived from bone marrow
(13) or skeletal muscle (14,15)
also express Sca-1. Based on the results presented in the above
reports, Sca-1 was selected as a stem cell marker for isolating
stem cells from mouse tongue muscle in the present study. Notably,
high expression levels of Sca-1 proteins were detected in the
tongue muscles of Jcl-ICR mice. Following the method outlined by
Shibuya et al (7), highly
pure populations of Sca-1-positive cells were obtained from mouse
tongue muscle using a magnetic cell separation system with
microbeads.
It has been reported that osteoblasts synthesize the
macromolecules of the bone matrix, including osteocalcin,
osteonectin and osteopontin (16).
Osteoblasts also express high levels of the membrane-bound enzyme
ALP, which has a critical function in mediating matrix
mineralization. Early in the process of cellular commitment to the
osteoblastic phenotype ALP is expressed, whereas osteopontin and
osteocalcin are expressed later during osteoblast differentiation
(17–19). The treatment of TDSCs with bone
differentiation-inducing medium resulted in the loss of stem cell
markers c-kit and Sca-1. These TDSCs also began to express multiple
osteogenic markers, including osterix, Runx2, fibronectin,
osteocalcin, osteonectin and osteopontin, as well as the
osteoblastic functions of mineralization and enhanced ALP activity.
Furthermore, osteocalcin expression was found to be late in onset
during osteoblast differentiation in the present study. These
results demonstrated that the treatment of TDSCs with bone
differentiation-inducing medium induced osteoblast differentiation
in vitro.
TDSCs were able to form areas with bone-like
morphology using the gelatin sponges of β-TCP as a scaffold.
However, TDSCs were unable to form bone without the gelatin sponges
of β-TCP (data not shown). Whether the gelatin sponges of β-TCP
would be useful as a scaffold for bone formation remains to be
elucidated. Therefore, the identification of a suitable scaffold
for the formation of bone cells from TDSCs is required.
In conclusion, the present study provided evidence
that TDSCs contained lineage-committed populations of
pre-osteoblastic cells. TDSCs exhibited osteoblastic phenotypes in
in vitro differentiation assays. Furthermore, TDSCs
implanted subcutaneously into nude mice confirmed the results
obtained from the in vitro differentiation assays,
demonstrating that TDSCs were able to form areas with bone-like
morphology in nude mice. However, prior to the clinical application
of TDSCs, further experimental studies are required in order to
elucidate the full benefits and side effects of this potential
therapeutic approach.
Acknowledgments
The present study was supported in part by a
Grant-in-Aid from the Japanese Ministry of Education, Science and
Culture (Tokyo, Japan).
References
|
1
|
Betz RR: Limitations of autograft and
allograft: new synthetic solutions. Orthopedics. 25(5 Suppl):
S561–S570. 2002.PubMed/NCBI
|
|
2
|
Khan SN: The biology of bone grafting. J
Am Acad Orthop Surg. 13:77–86. 2005.PubMed/NCBI
|
|
3
|
Jones E and Yang X: Mesenchymal stem cells
and bone regeneration: current status, injury. 42:562–568.
2011.
|
|
4
|
Mao JJ, Giannobile WV, Helms JA, Hollister
SJ, Krebsbach PH, Longaker MT, et al: Craniofacial tissue
engineering by stem cells. J Dent Res. 85:966–979. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Caplan AI: Tissue engineering designs for
the future: new logics, old molecules. Tissue Eng. 6:1–8. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Joyce MJ: Safety and FDA regulations for
musculoskeletal allografts: perspective of an orthopaedic surgeon.
Clin Orthop Relat Res. 435:22–30. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shibuya M, Miura T, Fukagawa Y, Akashi S,
Oda T, Kawamura S, Ikeda Y and Matsuzaki M: Tongue muscle-derived
stem cells express connexin 43 and improve cardiac remodeling and
survival after myocardial infarction in mice. Circ J. 74:1219–1226.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim BS, Nikolovski J, Bonadio J, Smiley E
and Mooney DJ: Engineered smooth muscle tissues: regulating cell
phenotype with the scaffold. Exp Cell Res. 251:318–328. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Takahashi Y and Tabata Y: Homogeneous
seeding of mesenchymal stem cells into nonwoven fabric for tissue
engineering. Tissue Eng. 9:931–938. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Asakura A: Stem cells in adult skeletal
muscle. Trends Cardiovasc Med. 13:123–128. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sun S, Wang Z and Hao Y: Osterix
overexpression enhances osteoblast differentiation of muscle
satellite cells in vitro. Int J Oral Maxillofac Surg. 37:350–356.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Van de Rijn M, Heimfeld S, Spangrude GJ
and Weissman IL: Mouse hematopoietic stem-cell antigen Sca-1 is a
member of the Ly-6 antigen family. Proc Natl Acad Sci USA.
86:4634–4638. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gojo S, Gojo N, Takeda Y, Mori T, Abe H,
Kyo S, et al: In vivo cardiovasculogenesis by direct injection of
isolated adult mesenchymal stem cells. Exp Cell Res. 288:51–59.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qu-Petersen Z, Deasy B, Jankowski R,
Ikezawa M, Cummins J, Pruchnic R, et al: Identification of a novel
population of muscle stem cells in mice: potential for muscle
regeneration. J Cell Biol. 157:851–864. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Asakura A, Seale P, Girgis-Gabardo A and
Rudnicki MA: Myogenic specification of side population cells in
skeletal muscle. J Cell Biol. 159:123–134. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ozawa H and Amizuka N: Structure and
function of bone cells. Nihon Rinsho. 52:2246–2254. 1994.PubMed/NCBI
|
|
17
|
Beck GR Jr, Sullivan EC, Moran E and
Zerler B: Relationship between alkaline phosphatase levels,
osteopontin expression and mineralization in differentiating
MC3T3-E1 osteoblasts. J Cell Biochem. 68:269–280. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Standal T, Borset M and Sundan A: Role of
osteopontin in adhesion, migration, cell survival and bone
remodeling. Exp Oncol. 26:179–184. 2004.PubMed/NCBI
|
|
19
|
Watts NB: Clinical utility of biochemical
markers of bone remodeling. Clin Chem. 45:1359–1368.
1999.PubMed/NCBI
|