Introduction
Neuropathic pain is a chronic and complex type of
pain (1), which occurs due to
dysfunction or damage of the nerve fibers in the peripheral nervous
system (PNS) or central nervous system (CNS) (1). Neuropathic pain is characterized by
increased sensitivity to painful stimuli (hyperalgesia), the
perception of innocuous stimuli as painful (allodynia) and
spontaneous pain. These symptoms are associated with:
Hyperexcitability in the affected dorsal root ganglion (DRG)
neurons (1); atrophic changes and
a switch in neurotransmitter phenotype in the central afferent
terminals (2); aberrant
myelination (splitting, detachment and loss of myelin) (3,4);
alterations in synaptic plasticity (5), and excitatory and inhibitory
mechanisms in the spinal cord (6);
loss of inhibitory interneurons and modifications of the brain stem
input to the spinal cord (7).
These changes mainly result from de novo gene transcription,
post-translational modifications (8), alterations in ion channel
conductivity and receptor function (9,10),
neuroimmune interactions (11) and
neuronal apoptosis (12).
Treatments for neuropathic pain have improved, however, patients
are often unresponsive to the entire range of therapeutic
modalities, as the key mechanisms controlling the induction and
maintenance of neuropathic pain remain to be elucidated (13).
Notch signaling is a highly conserved pathway in
evolution, which regulates the determination of cell-fate during
nervous system development (14,15),
and is important in synaptic plasticity (16) in the adult CNS. Numerous studies
have demonstrated that the notch signaling pathway is crucial for
several biological processes, including development (16), immunology (17), inflammation (18), vasculogenesis (19), tumor formation (20) and memory (21). Previous findings have suggested
that the activation of notch signaling contributes to neuronal
death (22,23), microglial cell (24) and astrocyte generation and
activation (25), neurite growth
inhibition (26), increased
dendritic branching (26),
oligodendrocyte progenitor cell differentiation (27) and demyelination (28) in the PNS and CNS (14). In addition, the notch signaling
pathway controls the determination between excitatory and
inhibitory cell-fates in the developing spinal cord, and activation
of the notch signaling pathway promotes the generation of
excitatory neurons from the sensory interneuron progenitors
(29). Furthermore, the expression
and activity levels of the notch signaling pathway are
significantly increased following nerve injury (30).
Therefore, it was hypothesized that activation of
notch signaling may contribute to the induction and maintenance of
neuropathic pain. The present study aimed to investigate the
effects of the γ-secretase notch signaling inhibitor, DAPT,
administered at different time-points and concentrations, on
mechanical allodynia in a rat model of spared nerve injury
(SNI)-induced neuropathic pain. In addition, the present study
examined the effects of the notch signaling activator peptide,
Jagged-1 (JAG-1), on mechanical allodynia in normal rats. The
results of these investigations may provide a novel therapeutic
target for the treatment of neuropathic pain.
Materials and methods
Animals
All experiments were performed on 180 (6 per group)
adult male Sprague-Dawley rats, weighing 200–250 g (Laboratory
Animal Center of the Fourth Military Medical University, Xi’an,
China). The animals were housed in plastic boxes at 22–26°C with
access to food and water ad libitum. A 12-h light/dark
cycle, with lights on at 08:00, was maintained, and assessments
were performed between 09:00 and 18:00. Prior to experimental
manipulation, the animals were allowed to acclimate to the housing
facilities and were handled daily for at least 3 days. All
experimental and animal handling procedures were approved by the
Institutional Animal Care and Use Committee of General Hospital of
Tianjin Medical University (Tianjin, China), and were performed in
accordance with the guidelines for the ethical treatment of animals
established by the International Association for the Study of Pain.
All efforts were made to minimize animal suffering and to reduce
the number of animals used. The study was approved by the ethics
committee of General Hospital of Tianjin Medical University
(Tianjin, China).
Intrathecal (i.t.) catheterization and
drug delivery
A permanent i.t. catheter (PE-10 polyethylene tube;
BD Biosciences, Franklin Lakes, NJ, USA) was inserted into each
animal through the space between the T3 and
T4 vertebrae and extended slowly into the subarachnoid
space of the lumbar enlargement (L4 and L5)
following intraperitoneal administration of pentobarbital sodium
anesthesia (40 mg/kg; Sigma-Aldrich, St. Louis, MO, USA) (31). The catheter was filled with sterile
saline (~4 μl; Otsuka Pharmaceutical Co., Ltd., Tianjin,
China) and the outer end was plugged. The cephalic portion of the
catheter was externalized through the dorsal skin, where it was
relatively inaccessible to the paws of the animal. All the animals
appeared to be free of infection on gross inspection. The
cannulated animals were housed separately, then allowed to recover
for 4 days. Motoric integrity was assessed in all animals using a
righting reflex and inclined plane test (32). Animals exhibiting any neurological
deficits (~4.4% rats) were excluded from the subsequent
experiments.
Subsequently, drugs were injected over a period of 1
min via the catheter at a volume of 10 μl, followed by 5
μl sterile saline for flushing. DAPT (Sigma-Aldrich, St.
Louis, MO, USA) and JAG-1 peptide (CDDYYYGFGCNKFCRPR; AnaSpec,
Inc., San Jose, CA, USA), as a potent inhibitor and activator of
notch signaling pathway, respectively, were freshly dissolved in
dimethyl sulfoxide (DMSO; Sigma-Aldrich) (23) at a concentration of DAPT (5, 15, 50
or 150 μM) and at a concentration of JAG-1 peptide (1, 10 or
100 μM). Drugs were injected over a period of 1 min via the
catheter at a volume of 10 μl.. The DMSO or scrambled peptide
(SC)-JAG-1 (RCGPDCFDNYGRYKYCF; AnaSpec, Inc.) were used as a
control. The location of the distal end of this catheter was
verified at the end of each experiment by injection of pontamine
sky blue (Sigma-Aldrich) into the catheter. The location of the
distal end of this catheter was verified at the end of each
experiment by injection of 2% pontamine sky blue solution (1
μl) via the i.t. catheter.
Spared nerve injury (SNI) model
The neuropathic pain was induced using a left SNI
model, as previously described (33). In brief, under 1% pentobarbital
sodium anesthesia (40 mg/kg, i.p.), an incision was made on the
lateral thigh and the underlying muscle was separated to expose the
sciatic nerve. The three terminal branches of the sciatic nerve
(tibial, common peroneal and sural nerves) were then carefully
separated. Following separation, the tibial and common peroneal
nerves were tightly ligated with 5.0 silk (Ethicon, Somerville, NJ,
USA), and 2–3 mm of the nerves distal to the ligation was removed.
The muscle and skin incisions were then closed separately. A sham
group was included, in which identical surgery was performed,
without the passage of ligatures or transection of the nerves.
Assessment of mechanical allodynia
Mechanical allodynia was assessed using a von Frey
test, as previous described (34).
In brief, the mechanical allodynia of rats was determined by
measuring the paw withdrawal threshold (PWT) of the rats in
response to mechanical stimuli, produced by a calibrated series of
von Frey filaments (Stoelting Co., Chicago, IL, USA). The rats were
adapted to the assessment situation for at least 30 min prior to
the initiation of stimulation. During the assessment, the rats were
placed on a metal mesh floor, covered with the same plastic box
used for housing, and von Frey filaments were applied from
underneath the metal mesh floor to the lateral plantar surface of
the paw, which is the area innervated by the sural nerve. Each
filament was presented perpendicularly against the paw, with
sufficient force to cause slight bending, and held for 2–3 sec. The
filament was applied only when the rats were stationary and
standing on all four paws. A withdrawal response was considered
valid only if the hindpaw was completely removed from the
customized platform; lifting of the paw due to normal locomotor
behavior was ignored. The monofilaments were applied with
increasing force until the rat withdrew the paw. Each filament was
applied 10 times at 5 sec intervals. The bending force value of the
von Frey filament, which led to a paw withdrawal reflex at an
occurrence of 50% on stimulation 10 times was expressed as the PWT.
Following determination of the threshold for the left hindpaw, the
same procedure was repeated on the right hindpaw at 5 min
intervals. The assessment trial was run a second and third time for
the two hindpaws and, if the PWT in the second or third trials did
not match that of the previous trial in a given hindpaw, the next
large filament in the series was used. This was repeated until the
PWT in the three successive trials matched. In order to avoid
inter-experimental differences and subjective bias, all behavioral
observations were observed by one assessor, in a
blinded-manner.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. Statistical comparisons between the groups were performed
using univariate or repeated measures analysis of variance. All
statistical analyses were performed using SPSS 16.0 software (SPSS,
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Administration of DAPT prior to
appearance of pain sensitivity prevents the development of
mechanical allodynia in SNI-induced neuropathic pain
The effects of pretreatment with DAPT, a notch
signaling pathway inhibitor, on mechanical allodynia were
investigated in a rat model of SNI-induced neuropathic pain.
Mechanical allodynia in all animals was measured 24 h prior to SNI
surgery (baseline) and 7, 14, 21 and 28 days following SNI or sham
surgery. The results demonstrated that the SNI-induced animals
developed a marked hypersensitivity to innocuous mechanical
stimulation of the lateral surface of the hindpaw (sural nerve skin
area) compared with the sham group (Fig. 1). The hindpaw contralateral to the
surgery was assessed over the entire period, and no statistically
significant changes in mechanical PWT were observed from the
baseline (data not shown). In addition, the mechanical PWT between
7 and 28 days after SNI decreased significantly compared with the
corresponding time-point in the sham group (P<0.05; Fig. 1). A single administration of DAPT
(50 μM; 10 μl) 0.5 h prior to SNI surgery
significantly improved the mechanical PWT for >28 days following
surgery, compared with the SNI group (P<0.05;Fig. 1A). In addition, a single
administration of DAPT (50 μM; 10 μl) 0.5 h following
SNI surgery significantly improved the mechanical PWT for ~21 days
following surgery, compared with the SNI group (P<0.05;Fig. 1B). Furthermore, a once-daily
administration of DAPT (50 μM; 10 μl) for three
consecutive days, beginning 0.5 h following SNI surgery,
significantly improved the mechanical PWT for >28 days following
surgery compared with the SNI group (P<0.05; Fig. 1C). These results suggested that
early inhibition of the notch signaling pathway may prevent the
induction of mechanical allodynia in neuropathic pain.
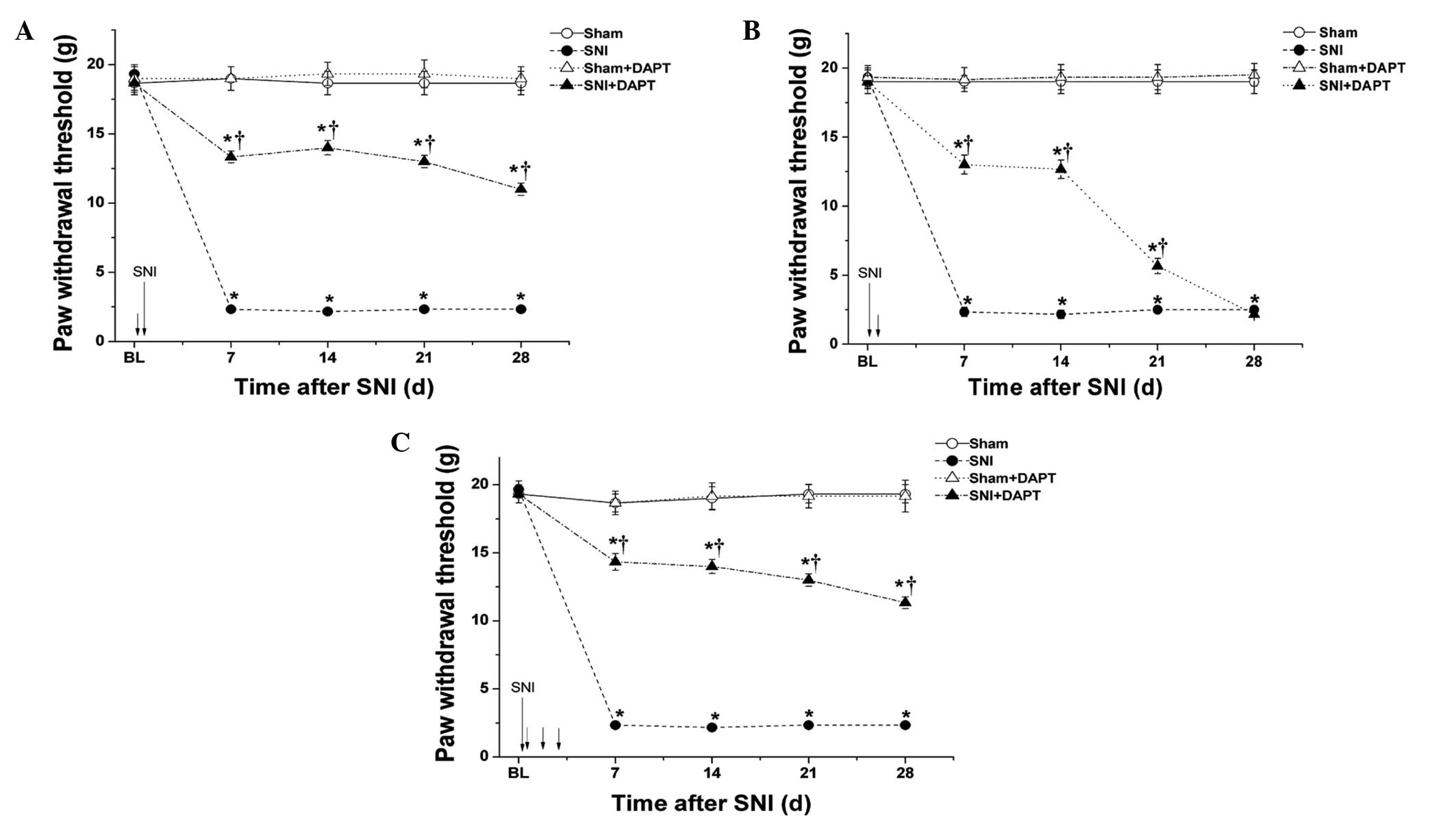 | Figure 1DAPT, a notch signaling pathway
inhibitor, administered prior to appearance of pain sensitivity
prevents the development of mechanical allodynia in SNI-induced
neuropathic pain. (A) Single administration of DAPT (50
μM;10 μl) 0.5 h prior to SNI surgery. (B) Single
administration of DAPT (50 μM; 10 μl) 0.5 h following
SNI surgery. (C) Once-daily administration of DAPT (50 μM;
10 μl) for three consecutive days, beginning 0.5 h after SNI
surgery. The mechanical paw withdrawal threshold was measured at 24
h prior to (BL) and 7, 14, 21 and 28 days following SNI or sham
surgery. The short arrows indicate points of DAPT administration.
Data are presented as the mean ± standard error of the mean (n=6
per group). *P<0.05, vs. Sham group;
†P<0.05, vs. SNI group. SNI, spared nerve injury; BL,
baseline; d, days. |
Administration of DAPT following the
appearance of pain sensitivity significantly reverses the
mechanical allodynia in SNI-induced neuropathic pain
The effects of post-treatment administration of the
notch signaling pathway inhibitor, DAPT on mechanical allodynia in
SNI-induced neuropathic pain were examined. In a preliminary
experiment, the animals developed significant mechanical allodynia
3 days after SNI surgery (data not shown). A single administration
of DAPT (50 μM; 10 μl) following the appearance of
pain sensitivity significantly increased the mechanical PWT between
1.5 and 72 h after DAPT administration compared with the SNI group
(P<0.05; Fig. 2A). In addition,
a once-daily administration of DAPT (50 μM; 10 μl)
following the appearance of pain sensitivity for 7 days
consecutively significantly increased the mechanical PWT compared
with the SNI group (P<0.05); however, the duration of its
antinociceptive action was only ~3 days following the final DAPT
administration (Fig. 2B). These
results suggested that late inhibition of the notch signaling
pathway may reverse the mechanical allodynia of neuropathic
pain.
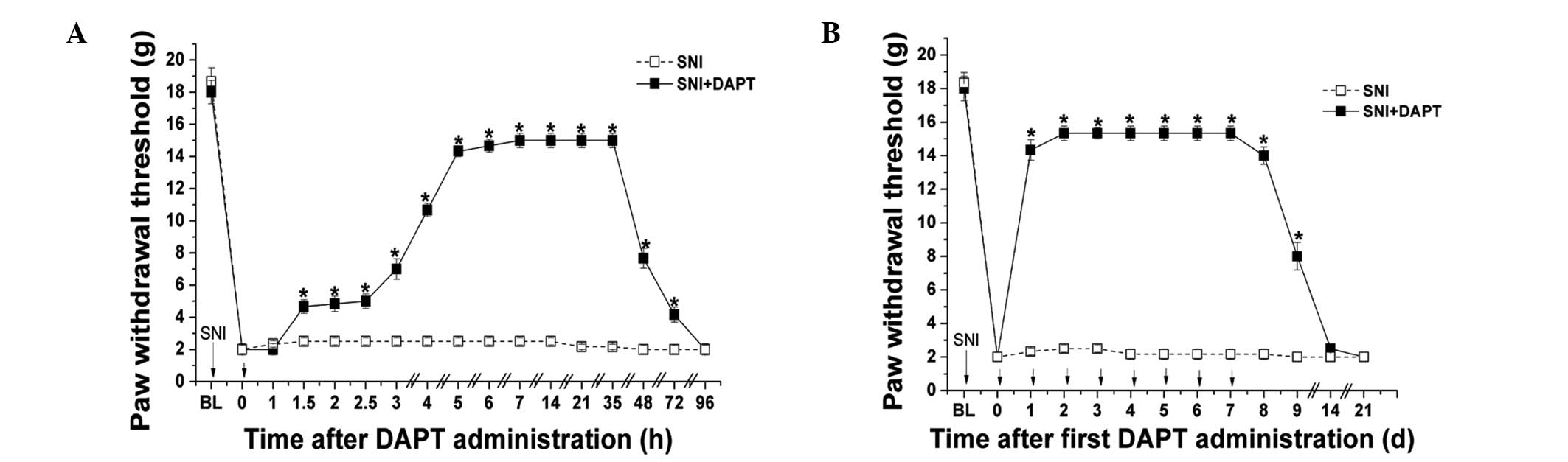 | Figure 2Administration of DAPT following
observation of pain sensitivity significantly reverses mechanical
allodynia in SNI-induced neuropathic pain. (A) DAPT (50 μM;
10 μl) or DMSO were administrated once following the
appearance of SNI-induced mechanical allodynia. Mechanical PWT was
measured prior to SNI surgery (BL), prior to administration (0 h)
and 1, 1.5, 2, 2.5, 3, 4, 5, 6, 7, 14, 21, 35, 48, 72 and 96 h
following DAPT or DMSO administration. (B) DAPT (50 μM; 10
μl) or DMSO were administrated once a day for seven
consecutive days following the appearance of SNI-induced mechanical
allodynia. Mechanical PWT was measured prior to SNI surgery, prior
to administration (0 h) and 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 14 and
21 days following DAPT or DMSO administration. Short arrows
indicate points of administration. Data are presented as the mean ±
standard error of the mean (n=6 per group). *P<0.05,
vs. SNI group. SNI, spared nerve injury; PWT, paw withdrawal
threshold; DMSO, dimethyl sulfoxide; BL, baseline; d, days. |
Administration of DAPT dose-dependently
attenuates mechanical allodynia in SNI-induced neuropathic
pain
The antinociceptive effects of different
concetrations of DAPT on mechanical allodynia were determined in
SNI-induced neuropathic pain. Different doses of DAPT (5, 15, 50
and 150 μM) were administered 0.5 h prior to SNI surgery,
and the mechanical PWT was evaluated 7 days after surgery. As shown
in Fig. 3A, administration of DAPT
prior to SNI surgery dose-dependently increased the mechanical PWT
of the rats (P<0.05). Furthermore, different concentrations of
DAPT (5, 15, 50 and 150 μM) were administered following the
appearance of pain sensitivity, and the mechanical PWT was
evaluated 24 h after DAPT administration. As shown in Fig. 3B, administration of DAPT following
the appearance of pain sensitivity dose-dependently increased the
mechanical PWT of the rats (P<0.05). These results indicated
that inhibition of the notch signaling pathway prevented and
reversed the mechanical allodynia of neuropathic pain in a
dose-dependent manner.
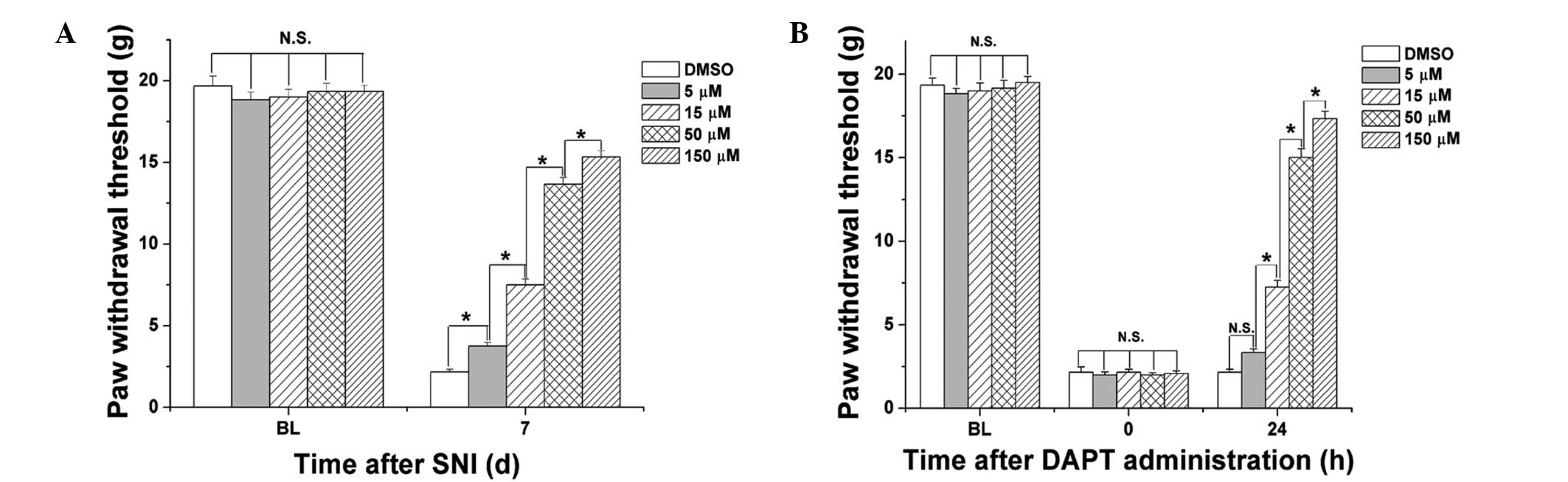 | Figure 3Administration of DAPT
dose-dependently attenuates mechanical allodynia in SNI-induced
neuropathic pain. (A) Various doses of DAPT (5, 15, 50 and 150
μM) or DMSO were administrated 0.5 h prior to SNI surgery.
Mechanical PWT was measured prior to (BL) and 7 days after SNI
surgery. (B) Different doses of DAPT (5, 15, 50 and 150 μM)
or DMSO were administred following the appearance of mechanical
allodynia. Mechanical PWT was measured prior to SNI surgery (BL),
prior to administration (0 h) and 24 h following DAPT or DMSO
administration. Data are presented as the mean ± standard error of
the mean (n=6 per group). *P<0.05; N.S, no
significant differences. SNI, spared nerve injury; PWT, paw
withdrawal threshold; DMSO, dimethyl sulfoxide; BL, baseline; d,
days; N.S. not significant. |
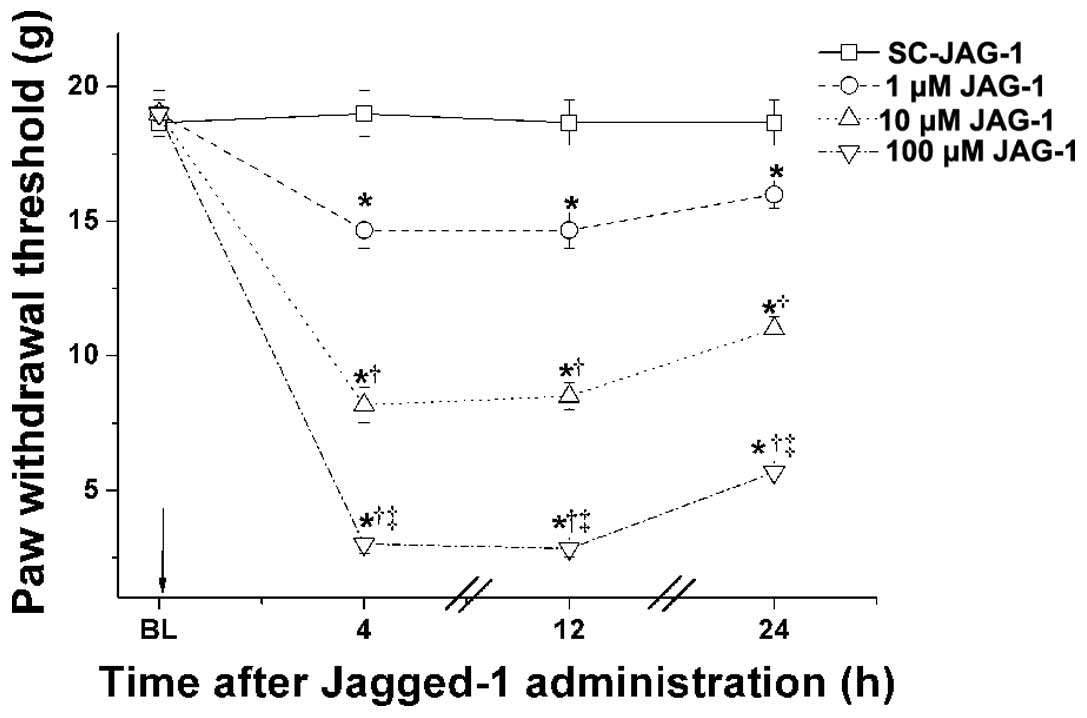
Administration of JAG-1 peptide, a ligand
of the notch signaling pathway, dose-dependently induces
neuropathic pain-like behavior in normal rats
In order to further investigate the roles of the
notch signaling pathway in neuropathic pain, normal rats were
administered with different concentrations of the JAG-1 peptide (1,
10 and 100 μM), and the mechanical PWT was measured 4, 12
and 24 h after treatment. SC-JAG-1 peptide was administered as a
negative control. As shown in Fig.
4, a single administration of the JAG-1 peptide
dose-dependently decreased the mechanical PWT of the normal animals
compared with the SC-JAG-1 peptide group (P<0.05). In addition,
the administration of DAPT had no effect on the mechanical PWT of
normal rats (data not shown). These results further suggested that
activation of the notch signaling pathway contributed to the
induction and maintenance of mechanical allodynia in neuropathic
pain.
Discussion
In the present study, it was found that early
inhibition of the notch signaling pathway prior to the appearance
of pain sensitivity prevented the induction of mechanical allodynia
in a rat model of SNI-induced neuropathic pain. In addition, late
inhibition of the notch signaling pathway following appearance of
pain sensitivity reversed the mechanical allodynia of neuropathic
pain. Early and late inhibition of the notch signaling pathway
produced antinociceptive effects in a dose-dependent manner.
Furthermore, activation of the notch signaling pathway
dose-dependently induced mechanical allodynia in normal animals.
Therefore, the activation of notch signaling contributed to the
induction and maintenance of neuropathic pain.
Chronic neuropathic pain can result from tissue
damage, inflammation or injury of nervous system, and symptoms
include hyperalgesia, allodynia and spontaneous pain (2,13).
It is well established that mechanical allodynia is characteristic
of neuropathic pain (13). The SNI
model has proven to be robust, with substantial and prolonged
changes in mechanical sensitivity and thermal responsiveness that
resemble those of clinical neuropathic pain (33,34).
Neuropathic pain affects areas innervated by the sural nerve and,
to a lesser extent, the saphenous nerve; however, the contralateral
hindpaw is unaffected (33,34).
The SNI model also exhibits marked hypersensitivity to normally
innocuous mechanical stimuli (33), and may assist in elucidating the
mechanisms underlying the development of neuropathic pain and be
used to screen for the efficacy of novel therapeutic agents. In the
present study, all the animals developed significant mechanical
allodynia following the induction of SNI neuropathic pain, which
was consistent with previous studies (33,34).
Numerous mechanisms have been investigated, which
may be involved in the pathogenesis of neuropathic pain. Several
mechanisms at various sites may operate independently, in
combination or at different time-points, which result in the onset
of the characteristic symptoms of neuropathic pain (13). These mechanisms may include changes
in terminal and peripheral sensitization (1); phenotypic switches and excitability
of injured axons; collateral sprouting in the periphery or central
terminal sprouting (2);
hyperexcitability in the affected DRG neurons (1); splitting, detachment and loss of
myelin (3,4); synaptic plasticity in the spinal cord
(5,6); loss of inhibitory interneurons
(7); and modifications of brain
stem input to the spinal cord (7).
In addition, excitatory and inhibitory interneurons integrate and
transduce sensory information from the periphery in the spinal
dorsal horn, therefore, alterations in the number or transmission
properties of these interneurons are considered to be major
contributors to the development of chronic sensory neuropathies,
including hyperalgesia and allodynia (35,36).
These changes result predominantly from de novo gene
transcription (8),
post-translational modifications (8), alterations in ion channel
conductivity and receptor function (9,10),
neuroimmune interactions (11) and
neuronal apoptosis (12). However,
the primary mechanisms involved in the induction and maintenance of
neuropathic pain remain to be elucidated.
Notch signaling is an evolutionarily conserved
pathway, which is essential for numerous biological processes,
including development (16),
immunology (17), inflammation
(18), vasculogenesis (19), tumor formation (20), and learning and memory (21). Notch is a cell-surface receptor,
which is involved in the regulation of cell-fate decisions during
nervous system development (14,15),
and is essential for synaptic plasticity (16) in the adult CNS. Proteolytic
cleavage of the notch extracellular and transmembrane domains is
mediated by the binding of ligands, including Delta and Jagged
(17,18). The latter cleavage is induced by
the γ-secretase enzyme complex, resulting in the release of a notch
intracellular domain, which translocates into the nucleus and
regulates transcription (17,18).
All the components of the notch signaling pathway, including
ligands, receptors and enzymes involved in notch receptor cleavage,
are expressed in the adult CNS, and are significantly increased
following nerve injury, suggesting that they are involved in its
repair (16,30). Previous findings have suggested
that the activation of notch signaling may contribute to neuronal
death (22,23), microglial cell (24) and astrocyte generation and
activation (25), neurite growth
inhibition (26), increased
dendritic branching (26),
oligodendrocyte progenitor cell differentiation (27) and demyelination (28) in the PNS and CNS (14). In addition, the notch signaling
pathway regulates the excitatory or inhibitory cell-fate decision
in the developing spinal cord; therefore, activation of the notch
signaling pathway was reported to promote the generation of
excitatory neurons from the sensory interneuron progenitors
(29). Therefore, notch signaling
activation may contribute to the generation and maintenance of
neuropathic pain.
In order to investigate the roles of the notch
signaling pathway in neuropathic pain, a γ-secretase enzyme (a key
enzyme of notch signaling pathway) inhibitor, DAPT, was
administered prior to or following the appearance of pain
sensitivity in SNI-induced neuropathic pain model rats. The results
revealed that early or late inhibition of notch signaling prevented
or reversed mechanical allodynia in rats with neuropathic pain, in
a dose-dependent manner. In addition, administration of JAG-1
peptide, a ligand of notch signaling pathway, induced mechanical
allodynia in normal rats, in a dose-dependent manner. These results
indicated that activation of notch signaling may contribute to the
induction and maintenance of neuropathic pain.
However, the roles of the notch signaling pathway in
thermal or cold allodynia of neuropathic pain, and the involvement
of notch signaling in inflammatory pain remain to be elucidated.
Furthermore, the mechanisms underlying notch signaling in
neuropathic pain require further investigation.
In conclusion, the results of the present study
demonstrated that early and late inhibition of the notch signaling
pathway led to the prevention and reversal of mechanical allodynia
in neuropathic pain, respectively; activation of notch signaling
induced mechanical allodynia in normal animals; and inhibition of
notch signaling produced an antinociceptive effect, in a
dose-dependent manner. These results suggest a novel therapeutic
target for the treatment of patients with neuropathic pain.
Acknowledgments
This study was supported by research grants from the
National Natural Science Foundation of China (no. 30872434 to Dr
Yanyan Sun; and no. 30972847 to Dr Guolin Wang) and the Natural
Science Foundation of Tianjin (no. 08JCYBJC08000 to Dr Guolin
Wang).
Abbreviations:
|
CNS
|
central nervous system
|
|
DMSO
|
dimethyl sulfoxide
|
|
DRG
|
dorsal root ganglion
|
|
i.t
|
intrathecal
|
|
JAG-1
|
Jagged-1
|
|
PWT
|
paw withdrawal threshold
|
|
SC-JAG-1
|
scrambled Jagged-1
|
|
SNI
|
spared nerve injury
|
References
|
1
|
Scholz J and Woolf CJ: The neuropathic
pain triad: neurons, immune cells and glia. Nat Neurosci.
10:1361–1368. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zimmermann M: Pathobiology of neuropathic
pain. Eur J Pharmacol. 429:23–37. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Willis WD: Role of neurotransmitters in
sensitization of pain responses. Ann N Y Acad Sci. 933:142–156.
2001. View Article : Google Scholar
|
|
4
|
Woolf CJ: Evidence for a central component
of post-injury pain hypersensitivity. Nature. 306:686–688. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Moore KA, Kohno T, Karchewski LA, Scholz
J, Baba H and Woolf CJ: Partial peripheral nerve injury promotes a
selective loss of GABAergic inhibition in the superficial dorsal
horn of the spinal cord. J Neurosci. 22:6724–6731. 2002.PubMed/NCBI
|
|
6
|
Porreca F, Ossipov MH and Gebhart GF:
Chronic pain and medullary descending facilitation. Trends
Neurosci. 25:319–325. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gillespie CS, Sherman DL, Fleetwood-Walker
SM, Cottrell DF, Tait S, Garry EM, Wallace VC, Ure J, Griffiths IR,
Smith A, et al: Peripheral demyelination and neuropathic pain
behavior in periaxin-deficient mice. Neuron. 26:523–531. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Woolf CJ and Costigan M: Transcriptional
and posttranslational plasticity and the generation of inflammatory
pain. Proc Natl Acad Sci USA. 96:7723–7730. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wood JN, Boorman JP, Okuse K and Baker MD:
Voltage-gated sodium channels and pain pathways. J Neurobiol.
61:55–71. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu XM and Salter MW: Src, a molecular
switch governing gain control of synaptic transmission mediated by
N-methyl-D-aspartate receptors. Proc Natl Acad Sci USA.
96:7697–7704. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marchand F, Perretti M and McMahon SB:
Role of the immune system in chronic pain. Nat Rev Neurosci.
6:521–532. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Scholz J, Broom DC, Youn DH, Mills CD,
Kohno T, Suter MR, Moore KA, Decosterd I, Coggeshall RE and Woolf
CJ: Blocking caspase activity prevents transsynaptic neuronal
apoptosis and the loss of inhibition in lamina II of the dorsal
horn after peripheral nerve injury. J Neurosci. 25:7317–7323. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Baron R, Binder A and Wasner G:
Neuropathic pain: diagnosis, pathophysiological mechanisms and
treatment. Lancet Neurol. 9:807–819. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Louvi A and Artavanis-Tsakonas S: Notch
signalling in vertebrate neural development. Nat Rev Neurosci.
7:93–102. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Latasa MJ, Cisneros E and Frade JM: Cell
cycle control of Notch signaling and the functional regionalization
of the neuroepithelium during vertebrate neurogenesis. Int J Dev
Biol. 53:895–908. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Costa RM, Drew C and Silva AJ: Notch to
remember. Trends Neurosci. 28:429–435. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kopan R and Ilagan MX: The canonical Notch
signaling pathway: unfolding the activation mechanism. Cell.
137:216–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fortini ME: Notch signaling: the core
pathway and its posttranslational regulation. Dev Cell. 16:633–647.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Woodhoo A, Alonso MB, Droggiti A, Turmaine
M, D’Antonio M, Parkinson DB, Wilton DK, Al-Shawi R, Simons P, Shen
J, et al: Notch controls embryonic Schwann cell differentiation,
postnatal myelination and adult plasticity. Nat Neurosci.
12:839–847. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gridley T: Notch signaling in vascular
development and physiology. Development. 134:2709–2718. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shih IeM and Wang TL: Notch signaling,
gamma-secretase inhibitors and cancer therapy. Cancer Res.
67:1879–1882. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Arumugam TV, Cheng YL, Choi Y, Choi YH,
Yang S, Yun YK, Park JS, Yang DK, Thundyil J, Gelderblom M, et al:
Evidence that {gamma}-secretase-mediated Notch signaling induces
neuronal cell death via the NF{kappa}B-Bim pathway in ischemic
stroke. Mol Pharmacol. 80:23–31. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Arumugam TV, Chan SL, Jo DG, Yilmaz G,
Tang SC, Cheng A, Gleichmann M, Okun E, Dixit VD, Chigurupati S, et
al: Gamma secretase-mediated Notch signaling worsens brain damage
and functional outcome in ischemic stroke. Nat Med. 12:621–623.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Grandbarbe L, Michelucci A, Heurtaux T,
Hemmer K, Morga E and Heuschling P: Notch signaling modulates the
activation of microglial cells. Glia. 55:1519–1530. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yuan TM and Yu HM: Notch signaling: key
role in intrauterine infection/inflammation, embryonic development
and white matter damage? J Neurosci Res. 88:461–468. 2010.
|
|
26
|
Sestan N, Artavanis-Tsakonas S and Rakic
P: Contact-dependent inhibition of cortical neurite growth mediated
by notch signaling. Science. 286:741–746. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jurynczyk M, Jurewicz A, Bielecki B, Raine
CS and Selmaj K: Overcoming failure to repair demyelination in EAE:
gamma-secretase inhibition of Notch signaling. J Neurol Sci.
265:5–11. 2008. View Article : Google Scholar
|
|
28
|
Morrison SJ, Perez SE, Qiao Z, Verdi JM,
Hicks C, Weinmaster G and Anderson DJ: Transient Notch activation
initiates an irreversible switch from neurogenesis to gliogenesis
by neural crest stem cells. Cell. 101:499–510. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mizuguchi R, Kriks S, Cordes R, Gossler A,
Ma Q and Goulding M: Ascl1 and Gsh1/2 control inhibitory and
excitatory cell fate in spinal sensory interneurons. Nat Neurosci.
9:770–778. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Givogri MI, de Planell M, Galbiati F,
Superchi D, Gritti A, Vescovi A, de Vellis J and Bongarzone ER:
Notch signaling in astrocytes and neuroblasts of the adult
subventricular zone in health and after cortical injury. Dev
Neurosci. 28:81–91. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yan LH, Hou JF, Liu MG, Li MM, Cui XY, Lu
ZM, Zhang FK, An YY, Shi L and Chen J: Imbalance between excitatory
and inhibitory amino acids at spinal level is associated with
maintenance of persistent pain-related behaviors. Pharmacol Res.
59:290–299. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jazayeri SB, Firouzi M, Abdollah Zadegan
S, Saeedi N, Pirouz E, Nategh M, Jahanzad I, Mohebbi Ashtiani A and
Rahimi-Movaghar V: The effect of timing of decompression on
neurologic recovery and histopathologic findings after spinal cord
compression in a rat model. Acta Med Iran. 51:431–437.
2013.PubMed/NCBI
|
|
33
|
Decosterd I and Woolf CJ: Spared nerve
injury: an animal model of persistent peripheral neuropathic pain.
Pain. 87:149–158. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tegeder I, Costigan M, Griffin RS, Abele
A, Belfer I, Schmidt H, Ehnert C, Nejim J, Marian C, Scholz J, et
al: GTP cyclohydrolase and tetrahydrobiopterin regulate pain
sensitivity and persistence. Nat Med. 12:1269–1277. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Woolf CJ, Shortland P and Coggeshall RE:
Peripheral nerve injury triggers central sprouting of myelinated
afferents. Nature. 355:75–78. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fitzgerald M: The development of
nociceptive circuits. Nat Rev Neurosci. 6:507–520. 2005. View Article : Google Scholar : PubMed/NCBI
|