Introduction
Breast cancer is the most common type of cancer in
females and is the predominant cause of female cancer-associated
mortality worldwide, accounting for 23% of the cases of newly
diagnosed cancer and 14% of the total cancer-associated mortality
(1). Current therapies using
endocrine agents, particularly selective estrogen receptor (ER)
modulators and, more recently, aromatase inhibitors, have
successfully prevented or treated ER-positive breast cancer by
interfering with estrogen signaling or production (2). However, these drugs have been
observed to reduce the incidence of breast cancer by only 50% and
had no effect in preventing ER-negative breast cancer, which
accounts for 30% of all cases of breast cancer in North America
(3,4). ER negativity is frequently combined
with high grade tumors and the proliferation and overexpression of
human epidermal growth factor receptor (HER)2/neu, resulting in a
poor prognosis (5,6). Therefore, effective novel drugs with
different molecular structures from conventional chemotherapeutic
agents, which may aid in the prevention or treatment of ER-negative
breast cancer require development.
Statins lower serum cholesterol levels by inhibiting
3-hydroxy-3-methyl glutaryl coenzyme A (HMG-CoA) reductase, the
rate-determining enzyme in the mevalonate pathway (7). This pathway produces various end
products, including cholesterol, dolichol, ubiquinone,
isopentenyladenine, geranylgeranyl pyrophosphate and farnesyl
pyrophosphate, which are critical for normal cellular functions,
including cell proliferation, differentiation and survival, in
normal and cancerous cells (8,9).
Statins are currently used as cholesterol-lowering medications and
exhibit effectiveness in the primary and secondary prevention of
heart disease and stroke (10). In
addition, statins interest for their use in cancer prevention has
increased. The anticancer function of statins is based on
preclinical evidence of their antiproliferative, pro-apoptotic and
antiangiogenic properties. A growing body of evidence suggests that
various statins possess anti-proliferative, anti-invasive,
antimetastatic and pro-apoptotic properties in various types of
cancer cell (11–14). Simvastatin, one of the HMG-CoA
reductase inhibitors, is currently used as a safe and
well-tolerated therapeutic agent for the treatment of
hypercholesterolemia, atherosclerosis and stroke (15). Simvastatin demonstrates in
vitro and in vivo antitumor actions in several human
malignancies including those of the breast, colon and prostate,
which has been attributed to cell cycle arrest, thereby inhibiting
cell proliferation and inducing apoptotic and necrotic cell death
(11,12,16).
A previous study revealed that the use of simvastatin, a highly
lipophilic statin, reduced the risk of recurrence in Danish females
with Stage I–III breast cancer, with 10 fewer cases per 100 females
over 10 years (17). In addition,
patients with breast cancer on long-term statin treatment have
proportionately fewer ER/progesterone (PR)-negative tumors, which
are of a lower grade and stage compared with patients who have
never received statin treatment (18). By contrast, Bonovas et al
concluded from a meta-analysis of seven randomized and nine
observational breast cancer trials, that treatment with statins
failed to significantly affect the risk of breast cancer (19). However, the efficacy and the
molecular mechanism underlying the effects of simvastatin on breast
cancer progression remain to be elucidated.
The requirement for alternative therapeutic
strategies is increasing and these findings, in parallel with
limited knowledge of the affect of simvastatin on breast cancer,
led to the present study evaluating its potential for therapeutic
effects in breast cancer, as the antitumor and cancer
chemopreventive effects of statins on breast cancer require further
investigation. The present study hypothesized that statin therapy
may reduce the progress of breast carcinoma by inhibiting cell
proliferation, altering the cell cycle, inducing apoptosis,
down-regulating the protein expression levels of cyclin D1 and
cyclin dependent kinases (CDKs) and decreasing the expression of
matrix metalloproteinase (MMP)-9. MDA-MB-231 cells were used in
vitro to confirm this.
Materials and methods
Cell culture
The human MDA-MB-231 breast cancer cell line was
kindly provided by the Laboratory of Molecular Biology of Anhui
Medical University (Anhui, China). The cells (1×105/ml)
were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Hyclone,
Logan, UT, USA), supplemented with 10% heat inactivated fetal
bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin
and 2 mM L-glutamine, which were all purchased from Sijiqing
Biological Engineering Materials (Hangzhou, China), and maintained
in a humidified atmosphere of 5% CO2 at 37°C.
Drugs
Simvastatin (Sigma-Aldrich, St. Louis, MO, USA) was
dissolved in dimethylsulfoxide (DMSO; Sigma-Aldrich) at a stock
concentration of 1 mM and stored at −20°C. The final concentrations
of simvastatin were 3.125, 6.25, 12.5, 25, 50, 100 μM. The
final concentration of DMSO in the DMEM was maintained at <0.1%.
An equal volume of solvent was added to cells as a control.
Cell proliferation assay
The proliferation rate of the MDA-MB-231 cells was
evaluated using a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT;
Sigma-Aldrich) assay. The exponentially growing cells were plated
at a density of 1×104 cells/well into 96-well plates,
cultured overnight at 37°C and subsequently treated with various
concentrations of simvastatin for 72 h. Following incubation with
simvastatin for 72 h, 20 μl MTT solution (5 mg/ml) was added
to each well and the plates were incubated for a further 4 h at
37°C. The colored formazan product was dissolved using 150
μl DMSO. The 96-well plates were then placed on a shaker for
10 min at room temperature to thoroughly dissolve the MTT product.
The half maximal inhibitory concentration (IC50) value
was determined as the concentration resulting in 50% cell growth
inhibition following 72 h exposure to simvastatin compared with the
untreated control cells. Six replicate wells were used for each
drug concentration and the assessment was performed independently
in triplicate.
Cell cycle analysis by flow
cytometry
The cells (2×105/well) were plated into
6-well dishes and treated with simvastatin at the IC50
concentration (7.979±0.201 μM) for 72 h. The adherent cells
were harvested by trypsinization (Sijiqing Biological Engineering
Materials), washed twice with phosphate-buffered saline (PBS;
Sijiqing Biological Engineering Materials) and fixed overnight in
70% ethanol (Sijiqing Biological Engineering Materials) at 4°C. The
ethanol was removed and the cells were washed twice in PBS, prior
to being resuspended in 1 ml propidium iodide (PI;
Sigma-Aldrich)/Triton X-100 (Sigma-Aldrich) staining solution,
containing PBS, 0.1% Triton X-100, 200 μg/ml RNAse A
(Sigma-Aldrich) and 50 μg/ml PI in the dark for 30 min at
37°C. The cell cycle was measured by flow cytometry (BD
Biosciences, San Jose, CA, USA) and analyzed using Cell Quest
WinMDI 2.9 software (BD Biosciences). The cell cycle profiles,
including the G0/G1, S and G2/M
phases, were calculated using ModFit LT™ 4.0 software.
Flow cytometric analysis of
apoptosis
The cells were plated in the exponential growth
phase into six-well plates, allowed to attach overnight at 37°C and
treated with simvastatin at the IC50 concentration for
72 h. Following treatment, the adherent and floating cells were
collected, washed twice with precooled (4°C) PBS and resuspended in
400 μl binding buffer (10 mM HEPES/NaOH pH 7.4; 140 mM NaCl;
KCl; MgCl2; and 2.5 mM CaCl2). The cells were
incubated with 5 μl annexin V-fluorescein isothiocyanate
(BestBio, Shanghai, China) at room temperature in the dark for 15
min and then with 10μl PI (40 μg/ml) at room
temperature in the dark for 5 min. The cell suspensions were
transferred to flow cytometric analysis tubes and detected using
flow cytometry. Cells without drug treatment were used as a
control.
Western blotting
The MDA-MB-231 cells, growth with or without
simvastatin, were washed with ice-cold PBS solution and scraped in
lysis buffer (50 mM Tris-HCl pH7.4; 250 mM NaCl; 0.5% Triton X100;
10% glycerol; 1 mM dichlorodiphenyltrichloroethane; and 1 mM
phenylmethylsulfonyl fluoride). The lysates were centrifuged at
16,853 × g for 30 min at 4°C and the supernatant was collected.
Briefly, the protein concentration of each sample was determined
using a Bicinchoninic Acid Protein Assay kit (Beyotime Institute of
Biotechnology, Inc., Shanghai, China). Equal quantities of protein
from each sample were loaded onto 10% SDS-polyacrylamide minigels
(HyClone Laboratories, Inc.) and electrophoresed. The proteins were
transblotted onto polyvinylidene difluoride (PVDF) membranes
(Millipore, Billerica, MA, USA) and then blocked with a solution of
PBS, containing 5% non-fat milk and 0.1% Tween 20 (HyClone
Laboratories, Inc.) for 2 h. The PVDF membranes were probed with
specific primary antibodies against anti-B cell lymphoma-2 (Bcl-2;
rabbit monoclonal; 1:2,000; Cell Signaling Technologies, Inc.,
Danvers, MA, USA), anti-Bcl-2 associated X protein (Bax; rabbit
monoclonal; 1:2,000; Cell Signaling Technologies, Inc.), rabbit
X-linked inhibitor of the apoptosis protein antibody (Xiap; rabbit
monoclonal; 1:500; Santa Cruz Biotechnology, Inc., Carlsbad, CA,
USA), anti-cyclin D1 (1:1,000; rabbit monoclonal; Abcam, Cambridge,
MA, USA), rabbit CDK2 (1:1,000; rabbit monoclonal; Abcam),
anti-caspase-3 (1:1,000; mouse monoclonal; Abcam), caspase-8
(1:1,000; mouse monoclonal; Abcam) and caspase-9 (1:1,000; mouse
monoclonal; Abcam), rabbit MMP-2 antibody (1:1,500; rabbit
monoclonal; Cell Signaling Technologies, Inc.), rabbit nuclear
factor-κB antibody (NF-κB p65; mouse monoclonal; 1:1,500; Cell
Signaling Technologies, Inc.) and anti-β-actin (1:1,500; rabbit
monoclonal; Cell Signaling Technologies, Inc.). Following washing
with Tris-buffered saline (Cell Signaling Technologies, Inc.),
containing 0.1% Tween-20 three times, the PVDF membranes were
incubated with horseradish peroxidase-conjugated secondary antibody
(1:5,000) at room temperature for 1 h. Positive bands were detected
using enhanced chemilluminescence reagents (Millipore) and β-actin
was used as a loading control.
Statistical analysis
The data were analyzed from three independent
experiments and are expressed as the mean ± standard deviation.
One-way analysis of variance and Student’s t-test were performed to
determine the statistical significance of any differences between
the control and treatment groups. All statistical analyses were
performed using GraphPad Prism 5.0 software (GraphPad, Inc., La
Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
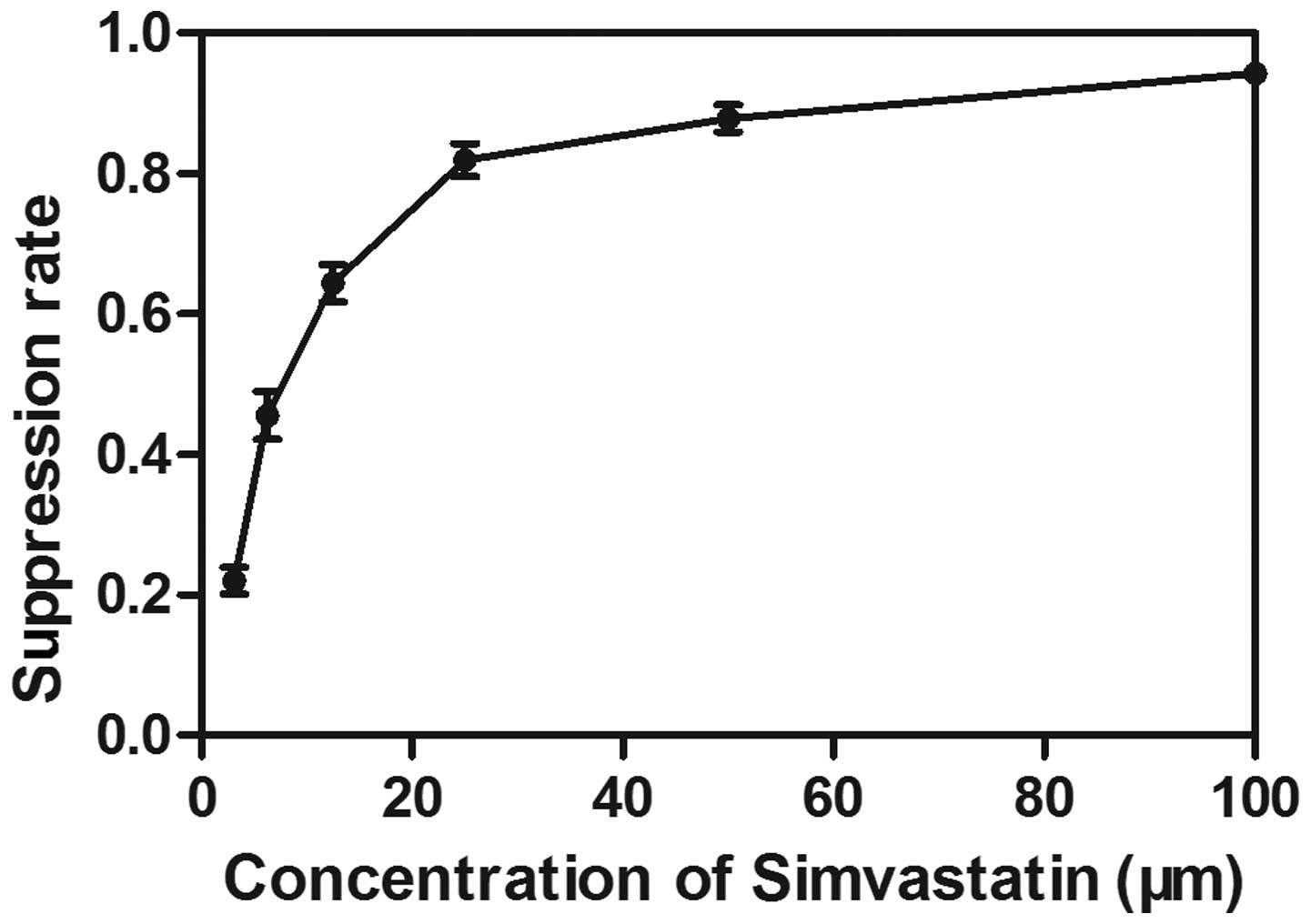
Dose-dependent antiproliferative effects
of simvastatin in the human breast cancer cell line
The effects of simvastatin on the proliferation of
MDA-MB-231 cells were determined using an MTT assay. The MDA-MB-231
cells were treated with different doses of simvastatin (3.125–100
μM) for 72 h. A dose-dependent decrease in the cell
viability was observed following treatment with simvastatin,
exhibiting an IC50 value of 7.979±0.201 μM
following exposure for 72 h (Fig.
1).
Effects of simvastatin treatment on Bcl-2
and Bax
The Bcl-2 and Bax family are important in the
regulation of apoptosis, proliferation and invasion of tumor cells
(20). The present study examined
the protein expression levels of Bcl-2 and Bax using western blot
analysis. To assess the effects of time and dose on response, the
MDA-MB-231 cells were cultured with different concentrations of
simvastatin for different durations. The most marked effects were
observed following treatment with simvastatin at the
IC50 for 72 h (P<0.05). In addition, treatment with
20 μm simvastatin significantly downregulated the protein
expression of Bcl-2 and upregulated the protein expression of Bax
in the MDA-MB-231 cells compared with the other concentrations
(P<0.05). These results demonstrated that the effect of
simvastatin on MDA-MB-231 cells occurred in a time- and
dose-dependent manner (Fig.
2).
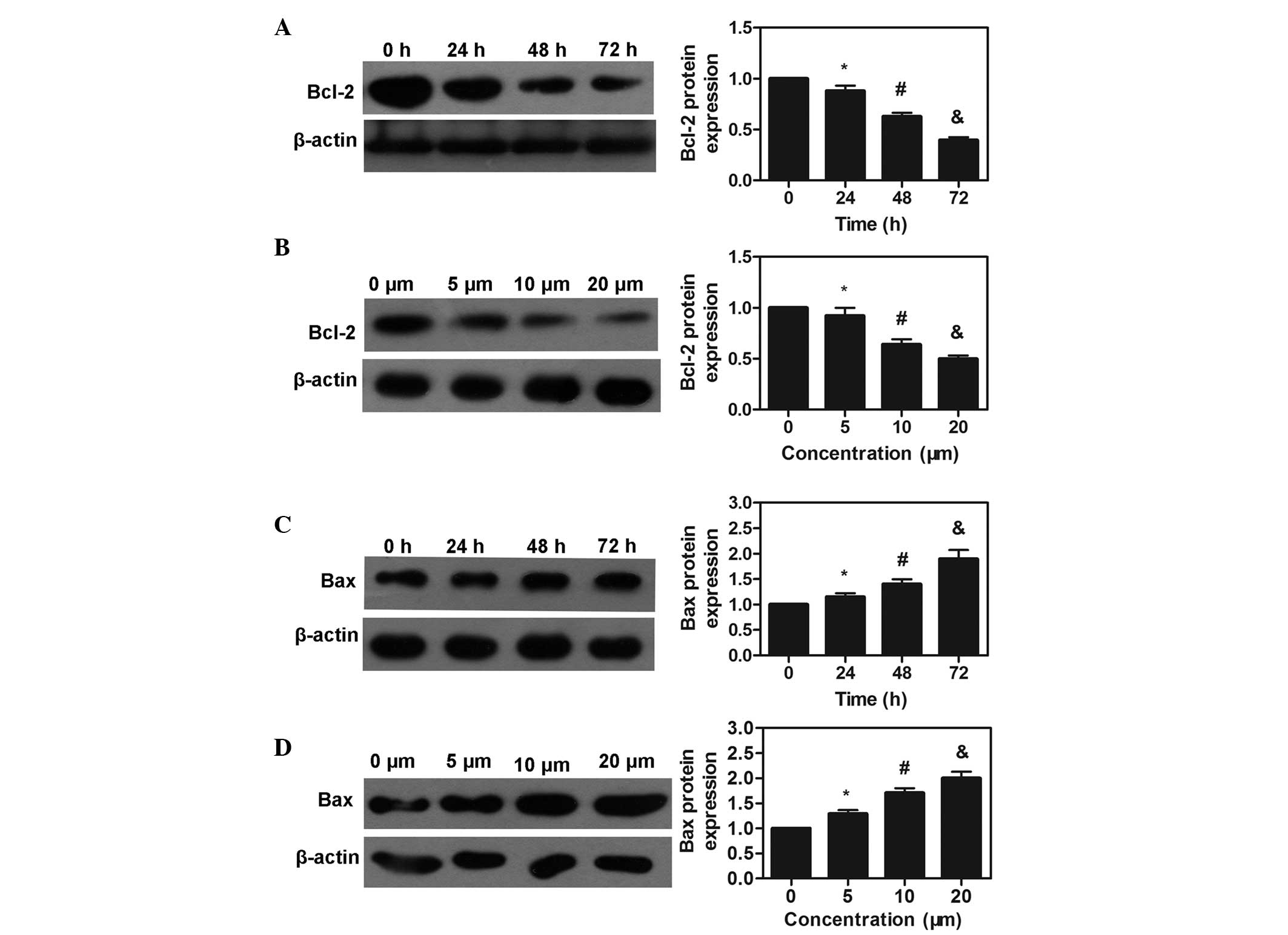 | Figure 2Protein expression levels of Bcl-2
and Bax were detected by western blotting in the MDA-MB-231 cells.
(A) Protein expression of Bcl-2 following treatment with
simvastatin for different durations (24, 48 and 72 h) at the IC
concentration, with quantitative analysis (*P<0.05,
vs. control group; #P<0.05, vs. 24 h group;
&P<0.05, vs. 48 h group). (B) Protein expression
of Bcl-2 protein following treatment with simvastatin at different
concentrations (5, 10 and 20 μm) for 72 h, with quantitative
analysis (*P<0.05, vs. control group;
#P<0.05, vs. 5 μm group;
&P<0.05, vs. 10 μm group). (C) Protein
expression of Bax following treatment with simvastatin for
different durations at its IC concentration, with quantitative
analysis (*P<0.05, vs. control group;
#P<0.05, vs. 48 h group; &P<0.05,
vs. 48 h group). (D) Protein expression of Bax following treatment
with simvastatin at different concentrations (5, 10 and 20
μm) for 72 h, with quantitative analysis
(*P<0.05, vs. control group; #P<0.05,
vs. 5 μm group; &P<0.05, vs. 10 μm
group). Values are expressed as the mean ± standard deviation. Bcl,
B-cell lymphoma; Bax, Bcl-2 associated X protien; IC 50, half
maximal inhibitory concentration. |
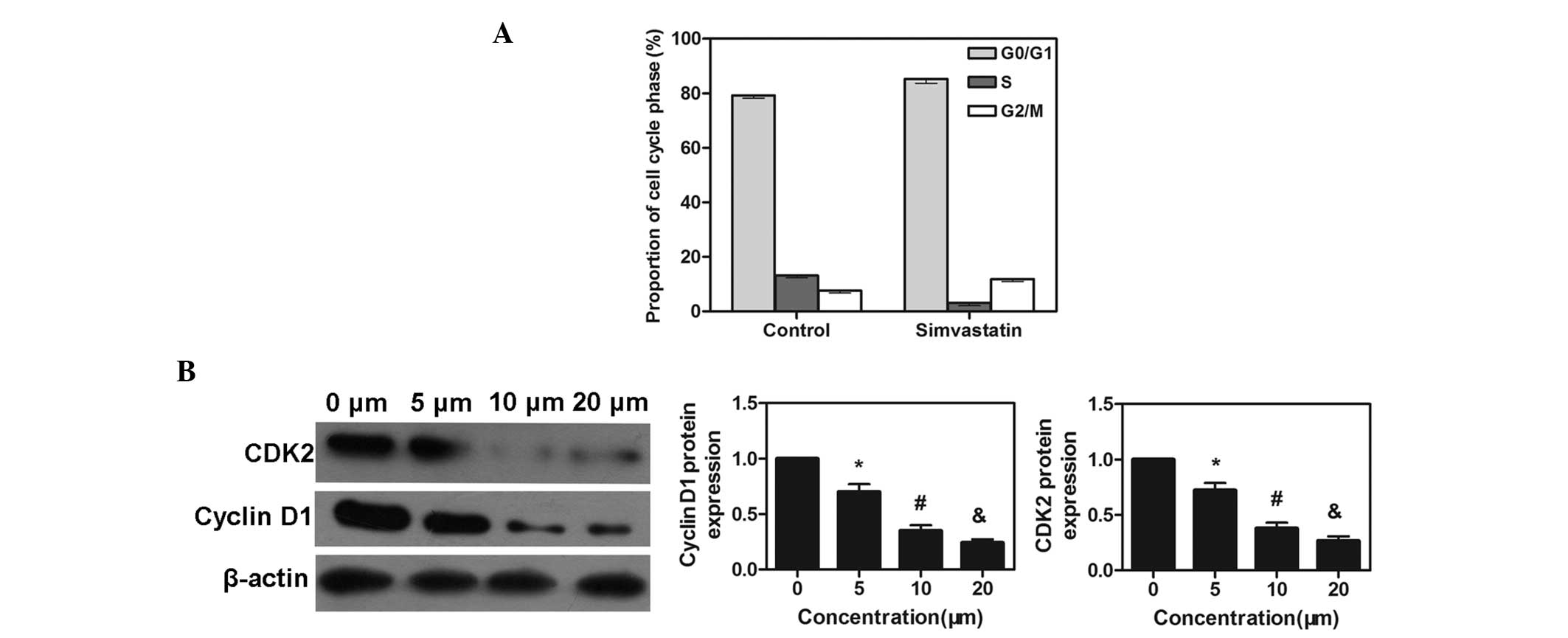
Cell cycle effects of simvastatin
The cell cycle distribution of the cells exposed to
simvastatin at the IC50 concentration for 72 h was
assessed by flow cytometry. The percentage of
G0/G1 phase cells was markedly increased
following simvastatin treatment compared with the control group
(P<0.05; Fig. 3A). This finding
suggested that simvastatin arrested the cells at the
G0/G1 phase of the cell cycle, which may be a
mechanism underlying its antitumor effect. In addition, the cell
cycle checkpoint proteins, cyclin D1 and CDK2, which are associated
with distributional change, were also assessed. The protein
expression levels of cyclin D1 and CDK were markedly decreased
following pretreatment with simvastatin for 72 h, and occurred in a
dose-dependent manner (P<0.05; Fig.
3B).
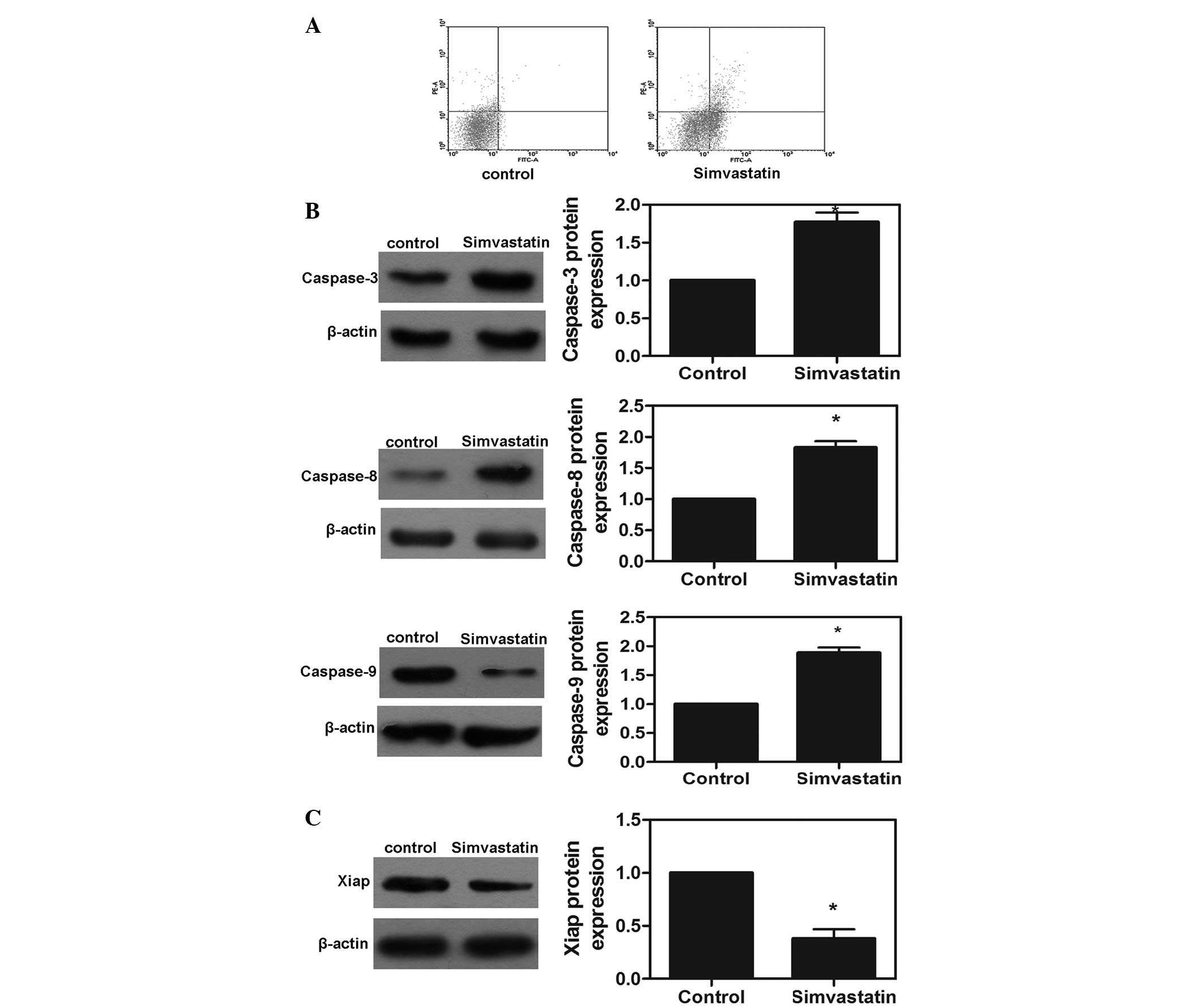
Effects of simvastatin on cell
apoptosis
To examine whether the observed growth inhibition
was caused by increased apoptosis, the present study investigated
the apoptotic response of the MDA-MD-231 cell line treated with the
IC50 of simvastatin using an annexin V/PI assay. As
shown in Fig. 4A, the apoptotic
rates induced by simvastatin in the MDA-MB-231 cells after 72 h
were 9.54%. Furthermore, protein expression of caspase-3, -8 and -9
was detected following treatment with the IC50
concentration of simvastatin for 72 h. Notably, the protein
expression levels of caspase-3, -8 and -9 were significantly
increased in the simvastatin-treated MDA-MB-231 cells compared with
the control group (P<0.05; Fig.
4B). These results demonstrated that simvastatin activated the
caspase cascade reaction and was, therefore, important in the
apoptotic response of MDA-MB-231 cells. In addition, Xiap is
important in the regulation of tumor cell apoptosis. The present
study measured the protein expression of Xiap in the MDA-MB-231
cells and found that simvastatin significantly downregulated the
protein expression of Xiap (P<0.05; Fig. 4C).
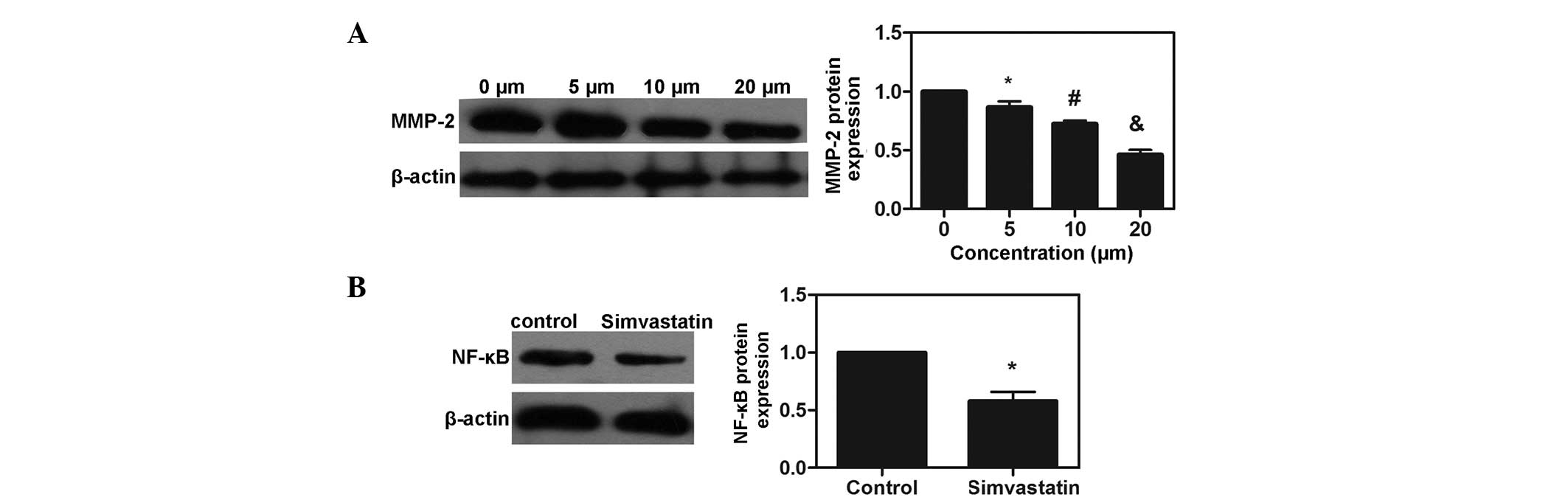
Simvastatin suppresses the expression of
MMP-2 and the activation of NF-κB in MDA-MB-231 cells
The protein expression of MMP-2 was examined by
western blot analysis, which revealed that treatment with
simvastatin decreased the protein expression of MMP-2 in a
dose-dependent manner (P<0.05). p65 is a major component of
NF-κB, and the levels of NF-κB p65 were also examined. Following
co-culture with simvastatin at the IC50 for 72 h, the
expression of NF-κB p65 was significantly suppressed in the
simvastatin-treated group (P<0.05). The results demonstrated
that simvastatin suppressed the expression of MMP-2 and the
activation of NF-κB in the MDA-MB-231 cells (Fig. 5).
Discussion
Breast cancer is one of the most life-threatening
types of cancer among female individuals worldwide (1). Statins are widely used
cholesterol-lowering drugs, and the use of statins has been
observed to significantly lower the risk of cancer (7,17).
Although an increasing quantity of evidence suggests that statins
may have useful activity in breast cancer prevention and/or therapy
(21), the molecular mechanisms
underlying the neoplastic development and progression of statins in
the breast remain to be elucidated. In the present study, the
effect of simvastatin on MDA-MB-231 breast cancer cells was
observed and the underlying mechanisms were investigated.
The present study demonstrated that simvastatin
significantly inhibited the proliferation of the breast cancer
cells. The acceleration of the cell cycle is an initial factor in
tumor growth, and control of cell cycle progression in cancer cells
is a potentially effective strategy for the control of tumor growth
(22,23). The results revealed that
simvastatin arrested the cells at the G1/S cell cycle
transition and directly induced G1/S phase arrest in the
MDA-MB-231 cells. Cell cycle progression is regulated by CDKs and
cyclin-dependent kinase inhibitors, whose activity is highly
controlled and coordinated by their association with cyclins
(24). CDK inhibitors interact
with active CDK-cyclin complexes and exert tumor-suppressive
functions that downregulate cell cycle progression (25,26).
A previous study demonstrated that simvastatin induced cell cycle
arrest at G0/G1 by downregulating the
expression of CDKs and cyclins, which was accompanied by apoptosis
and reduced cell proliferation (16). The present study demonstrated that
simvastatin significantly decreased the protein expression levels
of cyclin D1 and CDK2 in breast tumor cells, which revealed that
simvastatin-induced cell cycle arrest at
G0/G1 was associated with downregulation in
the protein expression levels of cyclin D1 and CDK2. This may be a
direct mechanism of simvastatin against the growth of breast cancer
cells.
Apoptosis is a fundamental cellular activity and is
crucial for eliminating genetically damaged cells, which is key in
the pathogenesis of cancer, and the proteins associated with this
process have been a focus of interest in investigations of cancer
onset and progression (27). The
upregulation of pro-apoptotic gene expression and downregulation of
anti-apoptotic gene expression induce the initiation of apoptosis,
and the progression of cancer depends predominantly on the balance
between pro-apoptotic proteins, including Bax, and anti-apoptotic
proteins, including Bcl-2 (20).
Several studies have suggested that the survival or death of human
breast cancer cells is determined by an altered balance between
pro-apoptotic and anti-apoptotic proteins, including the ratio of
Bcl-2 to Bax (28,29). Increased expression levels of
mitochondrial anti-apoptotic proteins contribute to augmented
survival of several types of cancer cells, including breast cancer
(30). The present study
demonstrated that simvastatin increased the expression of Bax and
downregulated the expression of Bcl-2, suggesting that the
simvastatin-induced apoptosis of MDA-MB-231 cells was associated
with modulation of the expression levels of Bax and Bcl-2.
Caspases are also involved in the execution of
apoptosis associated with these two signaling pathways. Bcl-2 and
Bax activate the caspase cascade reaction and are important in the
regulation of the intrinsic pathway of apoptosis (31). As demonstrated in previous studies,
Bcl-2 prevents the activation of caspase-3 in response to a variety
of apoptotic signals (32,33). In the caspase family, either
caspase-8 or -9 and the subsequent effector, caspase-3, are crucial
in the apoptotic process and initiation of a caspase cascade
triggers the proteolytic activation of executioner caspases,
including caspase-3, to perform the final steps in the apoptotic
process (34). Inhibition of the
expression of Xiap promotes the development of apoptosis,
therefore, Xiap suppresses apoptosis through the inhibition of
caspases (35,36). In the present study, simvastatin
increased the expression levels of caspase-3, -8 and -9, and
downregulated the expression of Xiap. This suggested that the
simvastatin-induced MDA-MB-231 apoptosis was also associated with
activation of the caspase signaling pathway and modulation of the
expression of Xiap.
The MMP family consists of 23 zinc-dependent
endopeptidases, which are all involved in the degradation of the
extracellular matrix. MMPs are upregulated in almost every type of
cancer and their expression is often associated with a poor
prognosis for patients (37).
Based on their unique ability to degrade gelatinases, a major
constituent of the basement membrane, MMP-2 and MMP-9, are the most
important MMPs involved in tumor invasion and metastasis (38). It has been reported that the
expression levels and activities of MMPs are associated with an
advanced stage of breast cancer, increased invasion of tumor cells
and building of metastatic formations (39). Atorvastatin, a member of the statin
drug family, suppresses the expression levels of MMP-2 and MMP-9 in
human endothelial cells (40). In
addition, it also inhibits the RhoA-JNK-c-Jun-MMP2 cascade,
resulting in a decrease in osteosarcoma cell invasion (41). The present study demonstrated that
simvastatin inhibited the expression levels of MMPs, potentially
inhibiting the invasion and metastasis of breast cancer.
The NF-κB complex, an essential cell mediator, is
composed of a family of inducible transcription factors, expressed
in almost all cell types (42).
The overexpression of NF-κB implies an aggressive tumor in breast
cancer and can predict tumors, which are likely to have a poor
prognosis (43). A previous study
revealed that the expression of NF-κB is necessary for the
maintenance of the malignant phenotype, and provides a therapeutic
approach for the treatment of cancer (44). The activation of NF-κB upregulates
the expression of the anti-apoptotic protein, Bcl-2, and regulates
the expression levels of cyclin D1 and MMPs (45,46).
In the present study, simvastatin inhibited the expression of
NF-κB, and this may be an important mechanism underlying the
anticancer effects of simvastatin in breast cancer.
There is increasing interest in cancer prevention
and in the drugs that, used in low doses, either alone or in
combination, which have different modes of action and low toxicity,
act as chemopreventive agents (47). Therefore, the present study
investigated other molecules, which are used for the treatment of
well-known pathological diseases and have effects on cancer cell
proliferation. Statins sensitize cancer cells to chemotherapeutic
drugs, and evidence indicates that treatment with simvastatin
increases the antitumor activity of cisplatin and docetaxel, common
chemotherapeutic agents used against a wide range of types of
cancer (48). Therefore, the
present study demonstrated the antiproliferative and
anticarcinogenic effects of simvastatin in a breast cancer cell
line, the results of which suggested that simvastatin may be
promising as a therapeutic approach for the treatment of
cancer.
Acknowledgments
This study was supported by the Anhui Provincial
Science and Technology Agency Foundation of China (nos. 1301042214,
12070403072 and KJ2012A157). The authors would like to thank Dr
Yuan Yuan, The Central Laboratory of Binhu Hospital and The Third
Affiliated Hospital of Anhui Medical University, for their
assistance.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Williams C and Lin CY: Oestrogen receptors
in breast cancer: basic mechanisms and clinical implications.
Ecancermedicalscience. 7:3702013.PubMed/NCBI
|
|
3
|
Fisher B, Costantino JP, Wickerham DL, et
al: Tamoxifen for prevention of breast cancer: report of the
National Surgical Adjuvant Breast and Bowel Project P-1 Study. J
Natl Cancer Inst. 90:1371–1388. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vogel VG, Costantino JP, Wickerham DL, et
al: Effects of tamoxifen vs raloxifene on the risk of developing
invasive breast cancer and other disease outcomes: the NSABP Study
of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA. 295:2727–2741.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baqai T and Shousha S: Oestrogen receptor
negativity as a marker for high-grade ductal carcinoma in situ of
the breast. Histopathology. 42:440–447. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Provenzano E, Hopper JL, Giles GG, Marr G,
Venter DJ and Armes JE: Biological markers that predict clinical
recurrence in ductal carcinoma in situ of the breast. Eur J Cancer.
39:622–630. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Istvan ES and Deisenhofer J: Structural
mechanism for statin inhibition of HMG-CoA reductase. Science.
292:1160–1164. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang FL and Casey PJ: Protein
prenylation: molecular mechanisms and functional consequences. Annu
Rev Biochem. 65:241–269. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jackson SM, Ericsson J and Edwards PA:
Signaling molecules derived from the cholesterol biosynthetic
pathway. Subcell Biochem. 28:1–21. 1997.PubMed/NCBI
|
|
10
|
Hebert PR, Gaziano JM, Chan KS and
Hennekens CH: Cholesterol lowering with statin drugs, risk of
stroke, and total mortality. An overview of randomized trials.
JAMA. 278:313–321. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kochuparambil ST, Al-Husein B, Goc A,
Soliman S and Somanath PR: Anticancer efficacy of simvastatin on
prostate cancer cells and tumor xenografts is associated with
inhibition of Akt and reduced prostate-specific antigen expression.
J Pharmacol Exp Ther. 336:496–505. 2011. View Article : Google Scholar
|
|
12
|
Ghosh-Choudhury N, Mandal CC, Ghosh
Choudhury N and Ghosh Choudhury G: Simvastatin induces derepression
of PTEN expression via NFkappaB to inhibit breast cancer cell
growth. Cell Signal. 22:749–758. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sassano A and Platanias LC: Statins in
tumor suppression. Cancer Lett. 260:11–19. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pelaia G, Gallelli L, Renda T, et al:
Effects of statins and farnesyl transferase inhibitors on ERK
phosphorylation, apoptosis and cell viability in non-small lung
cancer cells. Cell Prolif. 45:557–565. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Goldstein JL and Brown MS: Regulation of
the mevalonate pathway. Nature. 343:425–430. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Relja B, Meder F, Wilhelm K, Henrich D,
Marzi I and Lehnert M: Simvastatin inhibits cell growth and induces
apoptosis and G0/G1 cell cycle arrest in hepatic cancer cells. Int
J Mol Med. 26:735–741. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ahern TP, Pedersen L, Tarp M, et al:
Statin prescriptions and breast cancer recurrence risk: a Danish
nationwide prospective cohort study. J Natl Cancer Inst.
103:1461–1468. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kumar AS, Benz CC, Shim V, Minami CA,
Moore DH and Esserman LJ: Estrogen receptor-negative breast cancer
is less likely to arise among lipophilic statin users. Cancer
Epidemiol Biomarkers Prev. 17:1028–1033. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bonovas S, Filioussi K, Tsavaris N and
Sitaras NM: Use of statins and breast cancer: a meta-analysis of
seven randomized clinical trials and nine observational studies. J
Clin Oncol. 23:8606–8612. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Adams JM and Cory S: The Bcl-2 apoptotic
switch in cancer development and therapy. Oncogene. 26:1324–1337.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Riemsma R, Forbes CA, Kessels A, et al:
Systematic review of aromatase inhibitors in the first-line
treatment for hormone sensitive advanced or metastatic breast
cancer. Breast Cancer Res Treat. 123:9–24. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mork CN, Faller DV and Spanjaard RA: A
mechanistic approach to anticancer therapy: targeting the cell
cycle with histone deacetylase inhibitors. Curr Pharm Des.
11:1091–1104. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Meeran SM and Katiyar SK: Cell cycle
control as a basis for cancer chemoprevention through dietary
agents. Front Biosci. 13:2191–2202. 2008. View Article : Google Scholar :
|
|
24
|
Murray AW: Recycling the cell cycle:
cyclins revisited. Cell. 116:221–234. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Graña X and Reddy EP: Cell cycle control
in mammalian cells: role of cyclins, cyclin dependent kinases
(CDKs), growth suppressor genes and cyclin-dependent kinase
inhibitors (CKIs). Oncogene. 11:211–219. 1995.PubMed/NCBI
|
|
26
|
Pavletich NP: Mechanisms of
cyclin-dependent kinase regulation: structures of Cdks, their
cyclin activators, and Cip and INK4 inhibitors. J Mol Biol.
287:821–828. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lefranc F, Facchini V and Kiss R:
Proautophagic drugs: a novel means to combat apoptosis-resistant
cancers, with a special emphasis on glioblastomas. Oncologist.
12:1395–1403. 2007. View Article : Google Scholar
|
|
28
|
Korsmeyer SJ, Shutter JR, Veis DJ, Merry
DE and Oltvai ZN: Bcl-2/Bax: a rheostat that regulates an
anti-oxidant pathway and cell death. Semin Cancer Biol. 4:327–332.
1993.PubMed/NCBI
|
|
29
|
Oltvai ZN, Milliman CL and Korsmeyer SJ:
Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that
accelerates programmed cell death. Cell. 74:609–619. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Renault TT and Chipuk JE: Death upon a
kiss: mitochondrial outer membrane composition and organelle
communication govern sensitivity to BAK/BAX-dependent apoptosis.
Chem Biol. 21:114–123. 2014. View Article : Google Scholar :
|
|
31
|
Fang Y, Elahi A, Denley RC, Rao PH,
Brennan MF and Jhanwar SC: Molecular characterization of permanent
cell lines from primary, metastatic and recurrent malignant
peripheral nerve sheath tumors (MPNST) with underlying
neurofibro-matosis-1. Anticancer Res. 29:1255–1262. 2009.PubMed/NCBI
|
|
32
|
Boulakia CA, Chen G, Ng FW, et al: Bcl-2
and adenovirus E1B 19 kDA protein prevent E1A-induced processing of
CPP32 and cleavage of poly(ADP-ribose) polymerase. Oncogene.
12:529–535. 1996.PubMed/NCBI
|
|
33
|
Chinnaiyan AM, Orth K, O’Rourke K, Duan H,
Poirier GG and Dixit VM: Molecular ordering of the cell death
pathway. Bcl-2 and Bcl-xL function upstream of the CED-3-like
apoptotic proteases. J Biol Chem. 271:4573–4576. 1996.PubMed/NCBI
|
|
34
|
Zou H, Henzel WJ, Liu X, Lutschg A and
Wang X: Apaf-1, a human protein homologous to C. elegans CED-4,
participates in cytochrome c-dependent activation of caspase-3.
Cell. 90:405–413. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dai Y, Qiao L, Chan KW, et al: Loss of
XIAP sensitizes rosiglitazone-induced growth inhibition of colon
cancer in vivo. Int J Cancer. 122:2858–2863. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sasaki H, Sheng Y, Kotsuji F and Tsang BK:
Down-regulation of X-linked inhibitor of apoptosis protein induces
apoptosis in chemoresistant human ovarian cancer cells. Cancer Res.
60:5659–5666. 2000.PubMed/NCBI
|
|
37
|
Yadav L, Puri N, Rastogi V, Satpute P,
Ahmad R and Kaur G: Matrix metalloproteinases and cancer - roles in
threat and therapy. Asian Pac J Cancer Prev. 15:1085–1091. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li HC, Cao DC, Liu Y, et al: Prognostic
value of matrix metalloproteinases (MMP-2 and MMP-9) in patients
with lymph node-negative breast carcinoma. Breast Cancer Res Treat.
88:75–85. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lin ZM, Zhao JX, Duan XN, et al: Effects
of tissue factor, PAR-2 and MMP-9 expression on human breast cancer
cell line MCF-7 invasion. Asian Pac J Cancer Prev. 15:643–646.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Izidoro-Toledo TC, Guimaraes DA, Belo VA,
Gerlach RF and Tanus-Santos JE: Effects of statins on matrix
metalloproteinases and their endogenous inhibitors in human
endothelial cells. Naunyn Schmiedebergs Arch Pharmacol.
383:547–554. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fromigué O, Hamidouche Z and Marie PJ:
Blockade of the RhoA-JNK-c-Jun-MMP2 cascade by atorvastatin reduces
osteosarcoma cell invasion. J Biol Chem. 283:30549–30556. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Karin M and Lin A: NF-kappaB at the
crossroads of life and death. Nat Immunol. 3:221–227. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jana D, Das S, Sarkar DK, Mandal S, Maji A
and Mukhopadhyay M: Role of nuclear factor-κB in female breast
cancer: a study in Indian patients. Asian Pac J Cancer Prev.
13:5511–5515. 2012. View Article : Google Scholar
|
|
44
|
Prajoko YW and Aryandono T: Expression of
nuclear factor kappa B (NF-κB) as a predictor of poor pathologic
response to chemotherapy in patients with locally advanced breast
cancer. Asian Pac J Cancer Prev. 15:595–598. 2014. View Article : Google Scholar
|
|
45
|
Gallogly MM, Shelton MD, Qanungo S, et al:
Glutaredoxin regulates apoptosis in cardiomyocytes via NFkappaB
targets Bcl-2 and Bcl-xL: implications for cardiac aging. Antioxid
Redox Signal. 12:1339–1353. 2010. View Article : Google Scholar :
|
|
46
|
Hoesel B and Schmid JA: The complexity of
NF-κB signaling in inflammation and cancer. Mol Cancer. 12:862013.
View Article : Google Scholar
|
|
47
|
Hu LX, Du YY, Zhang Y and Pan YY:
Synergistic effects of exemestane and aspirin on MCF-7 human breast
cancer cells. Asian Pac J Cancer Prev. 13:5903–5908. 2012.
View Article : Google Scholar
|
|
48
|
Stoehr M, Mozet C, Boehm A, Aigner A,
Dietz A and Wichmann G: Simvastatin suppresses head and neck
squamous cell carcinoma ex vivo and enhances the cytostatic effects
of chemotherapeutics. Cancer Chemother Pharmacol. 73:827–837. 2014.
View Article : Google Scholar : PubMed/NCBI
|