Introduction
Osteoblasts are derived from pluripotent mesenchymal
stem cells (MSCs), which are capable of differentiation into
multiple cell types, including chondrocytes, osteoblasts and
adipocytes (1). Embryonic skeletal
development and adult bone maintenance require efficient osteoblast
differentiation from MSCs, and mature osteoblasts are able to
continuously synthe-size bone matrix to build bone as required
(2). Appropriate osteoblast
differentiation requires stimulation from extracellular signaling
and the expression of osteoblast transcription factors (3). Stimulation from the cell signals,
including bone morphogenetic proteins (BMPs), Wingless-ints (Wnts)
and Notch, are considered to be critical for osteoblast
differentiation from MSCs (3). It
has been suggested that several transcription factors, including
Runt-related transcription factor 2 (Runx2), Osterix
(Osx) and activating transcription factor 4 (ATF4),
are major regulators of osteoblast differentiation (4).
Inflammatory factors may impair the differentiation
of mesenchymal precursors into mature osteoblasts (5). In rheumatoid arthritis (RA), the
overproduction of tumor necrosis factor-α (TNF-α) in the
inflammatory joints results in local bone loss, which is due to the
reduction in osteoblast differentiation (6,7). In
the case of estrogen deficiency, TNF-α is overproduced by activated
T-cells (8,9), resulting in the inhibition of
osteoblast differentiation and inducing the development of
osteoporotic diseases.
To the best of our knowledge, the first in
vitro inhibitory effect of TNF-α on osteoblast differentiation
was identified by Canalis (10) in
1987. Further investigations revealed that TNF-α inhibited the
differentiation of fetal calvarial precursor cells to mature
osteoblasts in vitro (11).
In vivo studies of TNF-α or p55 receptor gene knockout mice
indicated that TNF-α reduced mouse maximum peak bone mass and
inhibited osteoblastic bone formation through the downstream
nuclear factor-κB (NF-κB) signaling pathway (12). NF-κB signaling was demonstrated to
have an endogenous inhibitory effect on osteoblastic bone
formation, and osteoblast-specific inactivation of NF-κB signaling
rescued the bone mass in an overiectomized mouse model (13). In conclusion, TNF-α and its
downstream NF-κB signaling have a critical role in the suppression
of osteoblast differentiation and may contribute to adult bone
loss.
TNF-α has a complex cell signaling process and
influences multiple cellular activities. TNF-α binds to two
receptors, TNF receptor type I (p55/60) and TNF receptor type II
(p75/80) (14). TNF-α activates
various downstream signals, including mitogen activated protein
kinase, cell death signals and NF-κB signaling (15). NF-κB signaling was found to exert
multiple effects on bone tissue maintenance (16). NF-κB is essential for the
differentiation and maturation of osteoclasts, which are required
in bone resorption (17). In
addition, NF-κB signaling suppresses osteoblast differentiation and
bone formation (13). The
appropriate balance between bone resorption and bone formation
determines the precise levels of bone maintenance in the adult
skeleton (18). The activation of
NF-κB signaling requires the degradation of the inhibitory protein
IκBα, which binds the NF-κB complex and prevents its translocation
to the nucleus. The degradation of IκBα facilitates the entrance of
the NF-κB complex to the nucleus and induces the subsequent
transcriptional activity (19).
Gliotoxin (GTX) is a secondary metabolite, derived
from numerous fungi (20–22). GTX has been demonstrated to have
antibacterial, antiviral and immunosuppressant activities (23). GTX is considered an NF-κB signal
inhibitor, and functions by blocking IκBα degradation, thereby
preventing the NF-κB complex from entering the nucleus, which
subsequently inhibits NF-κB complex-induced transcriptional
activity (24,25). Due to its inhibition of NF-κB
signaling, GTX was later found to be a potential anti-inflammatory
agent for the treatment of immune glomerulonephritis (26). The activation of NF-κB signaling
may prevent cell apoptosis in certain types of cell; therefore, it
is considered that NF-κB inhibitor GTX is able to facilitate cell
apoptosis (27). For example, GTX
was found to enhance radiation-induced apoptosis through NF-κB
signaling inhibition (28). The
present study aimed to explore the potential role of GTX in the
inhibition of NF-κB signaling in C2C12 mesenchymal cells, and its
potential function in the regulation of osteoblast
differentiation.
Materials and methods
Cell cultures and the induction of
osteoblast differentiation
The C2C12 mesenchymal cell line was obtained from
American Type Culture Collection (Manassas, VA, USA). The monolayer
culture was maintained in growth medium containing Dulbecco’s
modified Eagle’s medium (Invitrogen Life Technologies, Carlsbad,
CA, USA), supplemented with 10% fetal bovine serum (FBS), 50 U/ml
penicillin and 50 mg/ml streptomycin (all obtained from Hyclone,
Thermo Fisher Scientific, Logan, UT, USA). The cultures were
incubated in a humidified atmosphere at 37°C with 5%
CO2. To determine the function of GTX for protecting
osteoblast differentiation from inhibition by TNF-α, C2C12 cells
were divided into various groups. The BMP-2 group was treated with
200 ng/ml recombinant human BMP-2 (R&D Systems, Rockville, MD,
USA); the TNF-α alone group was treated with 10 ng/ml TNF-α
(Peprotech, Inc., Rocky Hill, NJ, USA); the BMP-2 + TNF-α group was
treated with a combination of 200 ng/ml recombinant human BMP-2 and
10 ng/ml TNF-α; the GTX group was treated with a combination of 200
ng/ml recombinant human BMP-2 and 10 ng/ml TNF-α, as well as the
indicated quantity of GTX simultaneously. Cells were incubated in a
humidified atmosphere at 37°C and 5% O2 for 72 h. To
examine the effect of GTX in modulating BMP-2-induced osteoblast
differentiation, C2C12 cells were divided into different groups.
The BMP-2 group was treated with 200 ng/ml recombinant human BMP-2;
the GTX alone group was treated with 1 μg/ml GTX; the BMP-2
+ GTX group was treated with a combination of 200 ng/ml recombinant
human BMP-2 and 1 μg/ml GTX. Cells were incubated in a
humidified atmosphere at 37°C and 5% CO2. For the ALP
activity assay, cells were incubated for 2 days. For the polymerase
chain reaction (PCR) assay, cells were incubated for 3 days.
RNA isolation and PCR
The total RNA of C2C12 cells was isolated using
TRIzol reagent (Invitrogen Life Technologies) according to the
manufacturer’s instructions. For reverse transcription, 2 μg
total RNA was used and mixed with 5 μM OligodT, 2 μl
10X RT buffer, 0.5 mM dNTP mix, 10 units of RNase inhibitor, 100
units of reverse transcriptase and nuclease-free water was added to
a final 20 μl volume. The reaction solution was incubated in
an Eppendorf thermocycler (Mastercycler nexus X2; Eppendorf GmbH,
Hamburg, Germany) at 42°C for 1 h and 72°C for 10 min. All the
reagents were purchased from Promega Corp. (Madison, WI, USA). PCR
was performed using an ABI 7900HT system (Applied Biosystems Life
Technologies, Foster City, CA, USA) with SYBR1 PremixEx
Taq™ (Takara, Dalian, China), according to the manufacturer’s
instructions. GAPDH was used as the internal control. Each
sample was analyzed in triplicate. The primer sequences for C2C12
cells used in the present study were as follows: GAPDH
forward, 5′-GACTTCAACAGCAACTCCCAC-3′ and reverse,
5′-TCCACCACCCTGTTGCTGTA-3′; type I collagen (Col I) forward,
5′-GAGCTGGTGTAATGGGTCCT-3′, and reverse,
5′-GAGACCCAGGAAGACCTCTG-3′; bone sialoprotein (Bsp) forward,
5′-CAGGGAGGCAGTGACTCTTC-3′ and reverse, 5′-AGTGTGGAAAGTGTGGCGTT-3′;
Osteocalcin (Ocn) forward, 5′-AAGCAGGAGGGCAATAAGGT-3′ and
reverse, 5′-TTTGTAGGCGGTCTTCAAGC-3′; Runx2 forward,
5′-GACTGTGGTTACCGTCATGGC-3′ and reverse,
5′-ACTTGGTTTTTCATAACAGCGGA-3′; ATF4 forward,
5′-CCTGAACAGCGAAGTGTTGG-3′ and reverse,
5′-TGGAGAACCCATGAGGTTTCAA-3′; Osx forward,
5′-GGAAAGGAGGCACAAAGAAGC-3′ and reverse
5′-CCCCTTAGGCACTAGGAGC-3′.
Western blotting
Cells were lysed on ice for 30 min in lysis buffer
(Thermo Fisher Scientific), which was comprised of 50 mM Tris-HCl
(pH 7.4), 150 mM NaCl, 1% Nonidet P-40 and 0.1% SDS supplemented
with protease inhibitors (10 mg/ml leupeptin, 10 mg/ml pepstatin A
and 10 mg/ml aprotinin). Protein content was measured with Pierce
bicinchoninic acid (BCA) reagent (Pierce Biotechnology, Inc.,
Rockford, IL, USA) according to the manufacturer’s instructions.
Cytosolic and nuclear fractions were prepared with a Nuclear and
Cytoplasmic Protein Extraction kit (Beyotime Institute of
Biotechnology, Shanghai, China), according to the manufacturer’s
instructions. For western blot analysis, 20–40 mg of sample was
resolved on 12% SDS-PAGE and electro-transferred onto
nitrocellulose membranes (Whatman, Piscataway, NJ, USA).
Anti-GAPDH, anti-p65 and anti-Lamin B antibody (all from Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) were used at a 1:1,000
dilution and incubated with the protein samples at 4°C overnight.
The anti-GAPDH antibody (cat. no. sc-365062) was a mouse monoclonal
IgG, raised against amino acids 1-335 representing a full length
GAPDH of human origin. The anti-p65 antibody (cat. no. sc-8008) was
a mouse monoclonal IgG antibody, raised against amino acids 1-286
mapping at the N-terminus of p65 of human origin. The anti-Lamin B
antibody (cat. no. sc-365214) was used to determine the loading of
nucleus protein level and was a mouse monoclonal IgG, raised
against amino acids 559–584 at the C-terminus of lamin B1 of human
origin. The goat anti-mouse IgG horseradish peroxidase secondary
antibody (cat. no. sc-2031; Santa Cruz Biotechnology, Inc) was used
at a 1:2,000 dilution. The antigen-antibody complexes were
visualized using an enhanced chemiluminescence detection system
(EMD Millipore, Billerica, MA, USA) according to the manufacturer’s
instructions.
Alkaline phosphatase (ALP) activity and
staining
C2C12 cells were treated with 200 ng/ml BMP-2 and/or
10 ng/ml TNF-α for 72 h at 37°C. The cultured C2C12 cells were
rinsed three times with ice-cold PBS, scraped from the dishes and
suspended in double distilled H2O, prior to three cycles
of freezing and thawing. ALP activity was determined using an
Orion™ AquaMate 7000 Vis spectrophotometer (Thermo Fisher
Scientific) at 405 nm using p-nitrophenyl phosphate (pNPP;
Sigma-Aldrich) as the substrate. A 50 ml cell sample was mixed with
50 ml pNPP (1 mg/ml) in 1 M diethanolamine buffer (Sigma-Aldrich)
supplemented with 0.5 mM MgCl2 (pH 9.8; Sigma-Aldrich)
and incubated at 37°C for 15 min on a bench shaker. The reaction
was stopped by the addition of 200 ml 2 M NaOH per 200 ml reaction
mixture. Total protein content was determined by the BCA method,
using a BCA protein assay kit (Pierce Biotechnology, Inc.). ALP
activity was presented as fold-changes in activity over the normal
control group at the respective time-points. All experiments were
conducted in triplicate. For ALP staining, C2C12 cells were rinsed
three times with PBS (Sigma-Aldrich) and fixed with 4%
paraformaldehyde (Sigma-Aldrich) for 10 min at 4°C. The fixed cells
were subsequently soaked in 0.1% naphthol AS-MX phosphate
(Sigma-Aldrich) and 0.1% fast red violet LB salt (Sigma-Aldrich) in
56 mM 2-amino-2-methyl-1,3-propanediol (pH 9.9; Sigma-Aldrich) for
10 min at room temperature, washed with PBS and observed under a
digital camera (Sony DSC-HX50; Sony Corp., Tokyo, Japan).
Plasmid transfection and luciferase
activity assays
The pGL4.32[luc2P/NF-κB-RE/Hygro] vector plasmids
were transfected into C2C12 cells for the determination of NF-κB
activity. pRL Renilla luciferase (Rluc) control reporter
vector plasmids were co-transfected into C2C12 cells with
pGL4.32[luc2P/NF-κB-RE/Hygro] vector plasmids as an inner control.
The two plasmids were purchased from Promega Corp. and were
transfected into cells using Lipofectamine® 2000
(Invitrogen Life Technologies). C2C12 cells were seeded at
0.5×105 cells/well in a 24-well plate in complete medium
and were stimulated with 200 ng/ml BMP-2, 10 ng/ml TNF-α and/or 1
μg/ml GTX, 24 h after plasmid transfection. Cells were
incubated at 37°C and harvested 48 h later. Luciferase activities
were measured using a dual luciferase system (Promega Corp.).
Immunofluorescence confocal microscopic
assay for p65 localization
C2C12 cells were treated with 200 ng/ml BMP-2, 10
ng/ml TNF-α and/or 0.6 μg/ml GTX simultaneously. Following
the incubation at 37°C for 1 h, cells were washed twice with PBS
and fixed with 4% paraformaldehyde for 20 min. The fixed cells were
incubated with primary anti-p65 antibody (1:100; Santa Cruz
Biotechnology, Inc.) overnight at 4°C and subsequently incubated
with fluorescent secondary antibody for 1 h at room temperature.
The anti-p65 antibody (cat. no. sc-8008) was a mouse monoclonal IgG
antibody, raised against amino acids 1-286 mapping at the
N-terminus of p65 of human origin. The goat anti-mouse IgG-Atto 488
antibody was used as the secondary antibody (cat. no. 62197;
Sigma-Aldrich). DAPI was used to stain the nucleic DNA of the
cells. The stained cells were analyzed with a Leica TCS SP5
confocal laser scanning microscope (Leica Microsystems GmbH,
Wetzlar, Germany).
MTT and caspase-3 activity assays
Cell viability was determined via MTT assay. MTT was
purchased from Sigma-Aldrich. C2C12 cells were treated with various
doses of GTX (0, 0.1, 0.5, 1, 2, 3, 4 or 5 μg/ml) and the
normal control group was treated with dimethyl sulfoxide. Cells
were incubated for 24 or 72 h at 37°C. Subsequently, the culture
medium was removed and the cells were washed three times with PBS.
Cells were treated with 5 mg/ml MTT salts and incubated at 37°C for
2 h. Absorbance at 570 nm was measured using the Orion™ AquaMate
7000 Vis spectrophotometer (Thermo Fisher Scientific). Apoptosis
was characterized by a caspase-3 activity assay, using a caspase
fluorescent assay kit (BD Biosciences, San Jose, CA, USA) according
to the manufacturer’s instructions C2C12 cells were treated with
various doses of GTX (0, 0.1, 0.5, 1, 2, 3, 4 or 5 μg/ml)
and the normal control group was treated with DMSO. Cells were
incubated for 24 or 72 h at 37°C. The culture medium was removed
and cells were washed three times with PBS. Subsequently, cells
were lysed on ice for 30 min in lysis buffer (Thermo Fisher
Scientific). Cell lysate (50 μl) was mixed with 0.2 ml 1X
HEPES buffer and 5 μl reconstituted Ac-DEVD-AMC, incubated
at 37°C for 1 h. The AMC liberated from Ac-DEVD-AMC was measured
using the Orion™ AquaMate 7000 Vis spectrophotometer (Thermo Fisher
Scientific) at an excitation wavelength of 380 nm and an emission
wavelength range of 420–460 nm. Caspase-3 activity was presented as
fold changes over the normal control group at the respective time
points.
Statistical analysis
Each assay was performed ≥3 times. Each testing
group contained a minimum of three samples (n≥3). Values are
expressed as the mean ± standard deviation. SPSS software version
22.0 (IBM, Armonk, NY, USA) was used for statistical analysis.
Student’s t-test was used to test statistical significance between
two groups. For the comparison of more than two groups, one-way
analysis of variance was used to determine which groups were
significantly different. P<0.05 was considered to indicate a
statistically significant difference.
Results
TNF-α inhibits C2C12 cell differentiation
into osteoblasts
As previously reported, TNF-α inhibited the
differentiation of mesenchymal precursor cells into osteoblasts. In
the present study, whether TNF-α was able to inhibit osteoblast
differentiation in a C2C12 cell system was examined. Following
three days of treatment, the alkaline phosphatase (ALP) activity of
the C2C12 cells was evaluated: The group treated with 200 ng/ml
BMP-2 exhibited significantly elevated ALP activity levels compared
with the normal control group; whereas, the group treated with a
combination of 200 ng/ml BMP-2 and 10 ng/ml TNF-α exhibited a
significantly lower level of ALP activity compared with the
BMP-treated group (P<0.01), with a value almost equal to that of
the normal control group (Fig.
1A). The examination of other osteoblast specific marker genes,
including Col I, Ocn and Bsp, indicated that
the expression levels of these genes were significantly inhibited
(P<0.01, P<0.001; Fig.
1B-D), which was consistent with the results of previous
studies. TNF-α effectively inhibited BMP-2-induced osteoblast
differentiation in the C2C12 cell system.
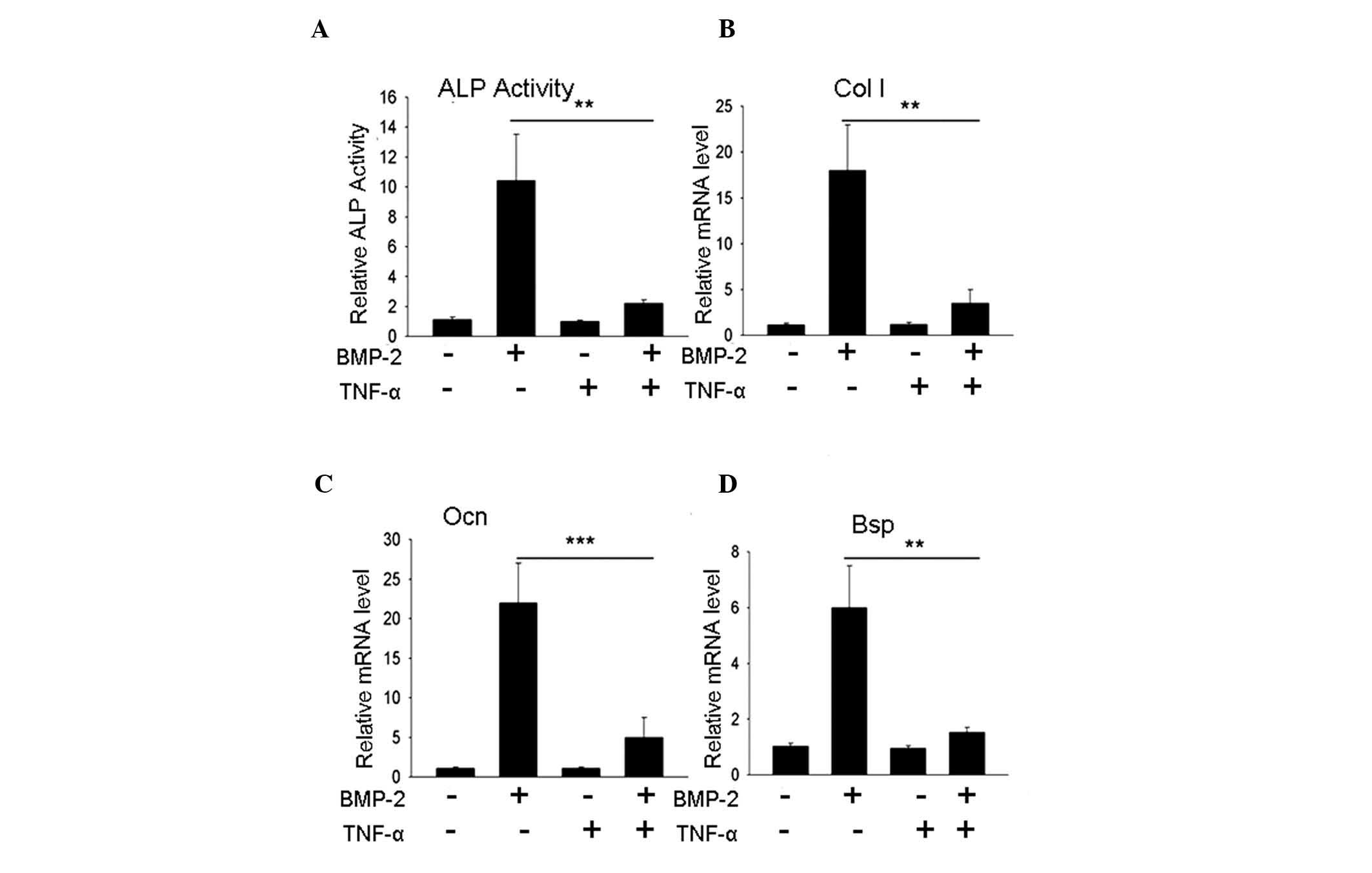 | Figure 1TNF-α inhibits BMP-2-induced
mesenchymal C2C12 cell differentiation to osteoblasts. (A) ALP
activity assay. C2C12 cells treated with 200 ng/ml BMP-2 revealed
an increase in ALP activity; however, in cells treated with a
combination of 200 ng/ml BMP-2 and 10 ng/ml TNF-α, the elevation of
ALP activity was inhibited. (B–D) Gene expression assays for Col
I, Ocn and Bsp indicated that the BMP-2-induced
increase in gene expression was inhibited by 10 ng/ml TNF-α.
**P<0.01, ***P<0.001, n>3. TNF-α,
tumor necrosis factor-α; BMP-2, bone morphogenetic protein-2; ALP,
alkaline phosphatase; NC, normal control; mRNA, messenger RNA. |
GTX blocks the inhibition of osteoblast
differentiation by TNF-α, as indicated by ALP activity and
extracellular matrix gene expression
Significant inhibition of NF-κB activity may lead to
cell apoptosis; therefore, the appropriate doses for C2C12 cell
treatment were determined. C2C12 cells were treated with various
doses of GTX (0, 0.1, 0.5, 1, 2, 3, 4 or 5 μg/ml; Fig. 2A) and cell viability was detected
following 24 and 72 h of incubation using an MTT assay. Compared
with the control group, doses of 0.1–3 μg/ml GTX did not
influence cell viability; however, 4 or 5 μg/ml GTX led to a
significant reduction in viable cell numbers (P<0.05, P<0.01;
Fig. 2B). In addition, the results
of a caspase-3 activity assay indicated a significant increase in
the levels of apoptosis in the 4 or 5 μg/ml GTX-treated
C2C12 cells; however, there were no notable changes in the groups
treated with 0.1–3 μg/ml GTX (Fig. 2C). Subsequently, whether GTX
influenced the inhibition of osteoblast differentiation by TNF-α
was examined. Significant inhibition of ALP activity was observed
in the group treated with 200 ng/ml BMP-2 and 10 ng/ml TNF-α
compared with BMP-2-treatment alone; however, ALP activity was
rescued by the addition of 1 μg/ml GTX (Fig. 2D). Further investigation indicated
that the rescued effect was GTX dose-dependent (Fig. 2E). To further confirm that GTX was
able to rescue osteoblast differentiation, the expression of three
other osteoblast marker genes, Col I, Ocn and
Bsp, were evaluated by PCR analysis. The gene expression
results demonstrated that when compared with the TNF-α inhibition
group, the expression levels of these three genes were rescued by 1
μg/ml GTX (Fig. 2F-H,
respectively), (P<0.01, Col I and Ocn; P<0.05,
Bsp).
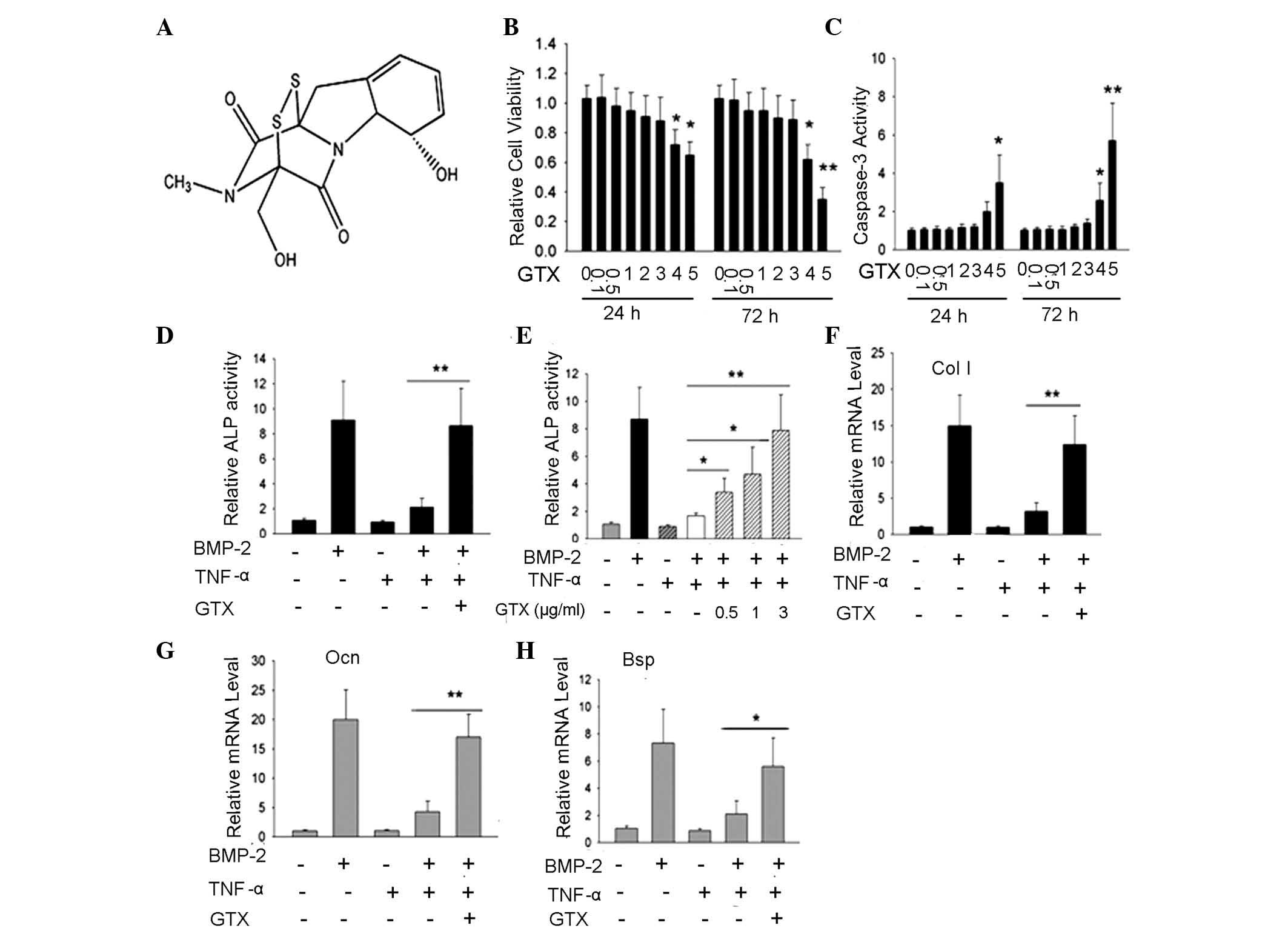 | Figure 2GTX protects osteoblast
differentiation against inhibition from TNF-α in C2C12 cells;
evidence from ALP activity, as well as Col I, Ocn and
Bsp gene expression.(A) Chemical structure of GTX. (B) MTT
cell viability assay suggested that, in the groups treated with 4
or 5 μg/ml GTX for 24 or 72 h, cell viability rate was
significantly decreased, compared with that of the DMSO-treated
control group. (C) Caspase-3 activity assay indicated that
following 4 or 5 μg/ml GTX treatment for 24 h or 72 h,
caspase-3 activity was markedly enhanced, compared with that of the
DMSO control group. (D and E) ALP activity assays performed
following 72 h of culture indicated the protective effect of GTX
for osteoblast differentiation. (D) TNF-α inhibition of
BMP-2-induced ALP activity was blocked by treatment with 1
μg/ml GTX. (E) GTX protected ALP activity in a
dose-dependent manner. (F–H) The protective effects of GTX on
osteoblast differentiation were determined via analysis of gene
expression of Col I, Ocn and Bsp.
*P<0.05, **P<0.01, n>3. GTX,
gliotoxin; TNF-α, tumor necrosis factor-α; DMSO, dimethyl
sulfoxide; ALP, alkaline phosphatase; BMP-2, bone morphogenetic
protein-2. |
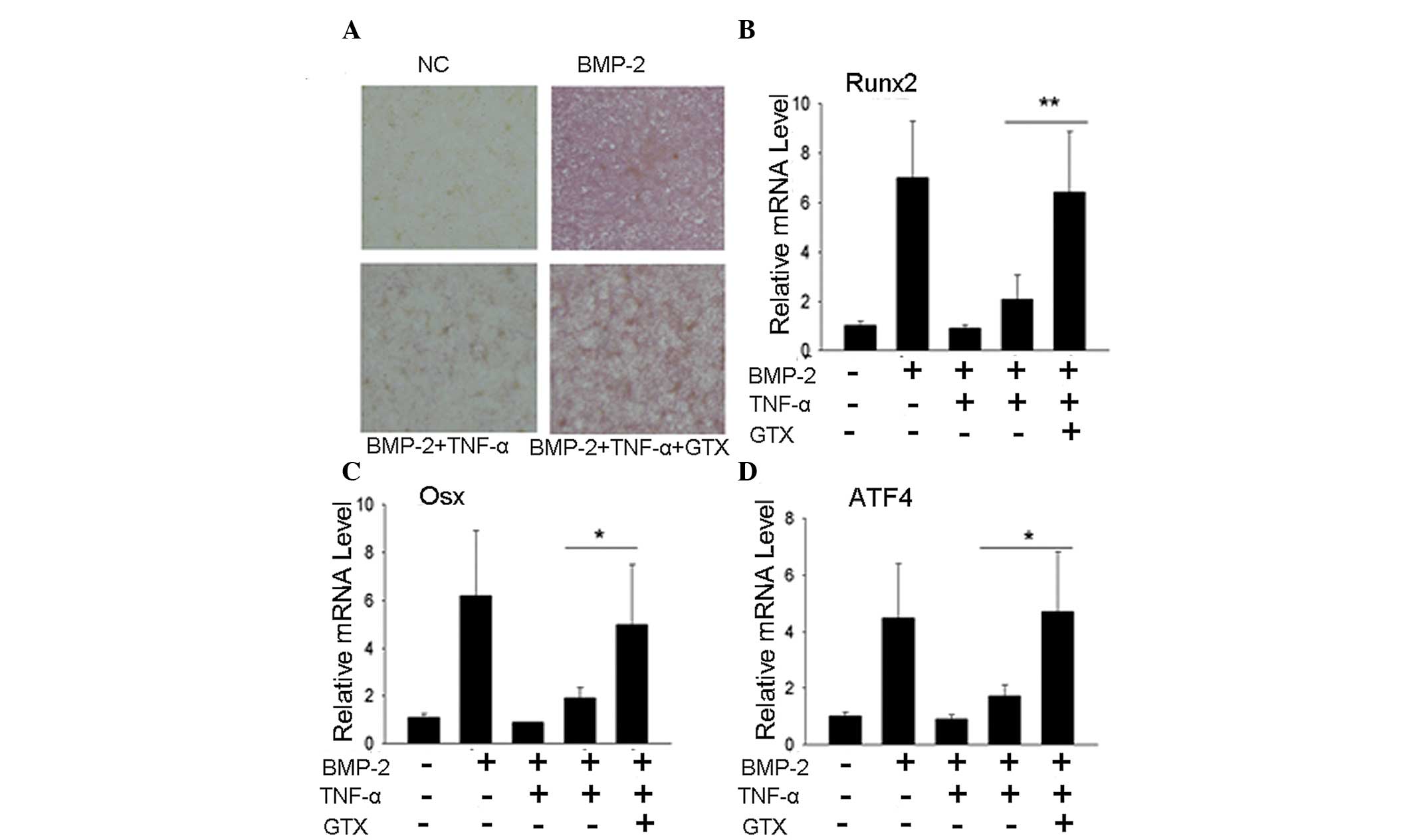
GTX blocks the inhibition of osteoblast
differentiation by TNF-α, as indicated by ALP staining and
osteoblast tran- scription factor gene expression
To further confirm that GTX was able to prevent the
inhibition of osteoblast differentiation by TNF-α, the expression
levels of other genes characteristic of osteoblast differentiation
were evaluated. An ALP staining assay revealed that the group
treated with 1 μg/ml GTX exhibited a significantly elevated
ALP level when compared with that of the TNF-α group (Fig. 3A). PCR results for transcription
factor expression indicated a similar effect. The expression levels
of Runx2, Osx and ATF4 were markedly higher in
the 1 μg/ml GTX treatment group compared with those of the
TNF-α group (Fig. 3B-D,
respectively), (P<0.01, Runx2; P<0.05, Osx and
ATF4).
GTX promotes BMP-2-induced osteoblast
differentiation of C2C12 cells
Since NF-κB signaling was identified to have
endogenous inhibitory effects on osteoblast differentiation, it was
hypothesized that blockage of NF-κB signaling may promote
osteoblast differentiation and bone formation. Whether GTX had
effects on BMP-2-induced C2C12 osteoblast differentiation was
therefore evaluated. The results indicated that 1 μg/ml GTX
was able to promote ALP activity (Fig.
4A) and expression levels of the extracellular matrix genes
Col I, Ocn and Bsp (Fig. 4B–D), as well as the transcription
factor genes Runx2 and Osx (Fig. 4E and F; P<0.05, P<0.01).
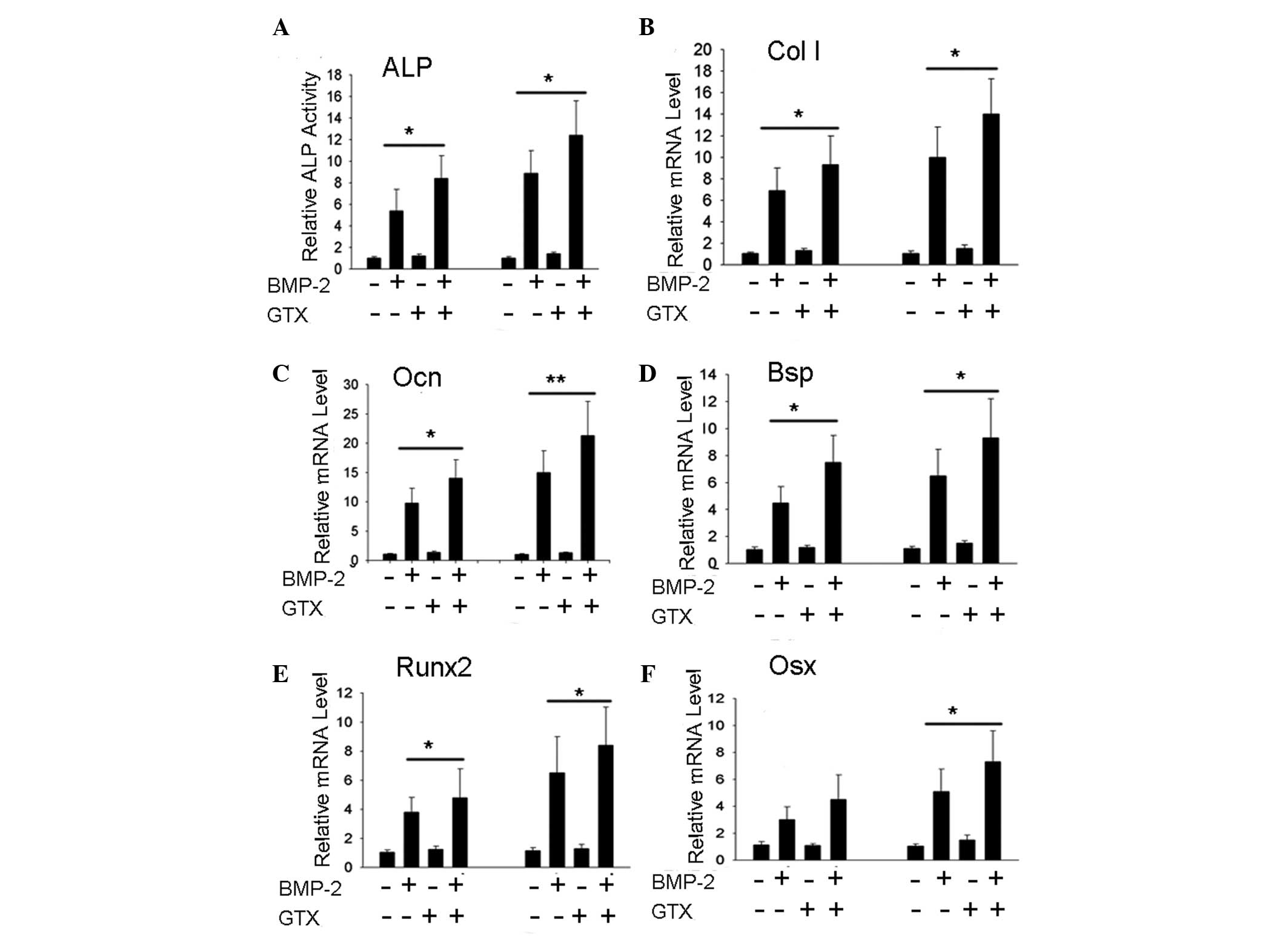 | Figure 4GTX promotes BMP-2-induced osteoblast
differentiation in C2C12 cells. (A) BMP-2-induced ALP activity was
enhanced by 1 μg/ml GTX treatment. (B–F) Gene expression
levels of Col I, Ocn, Bsp, Runx2 and
Osx were found to be enhanced by treatment with 1
μg/ml GTX + BMP-2, compared with those following treatment
with BMP-2 alone. For the ALP activity assay, cells were incubated
for 2 days and for the polymerase chain reaction assay, cells were
incubated for 3 days. *P<0.05,
**P<0.01, n>3. GTX, gliotoxin; BMP-2, bone
morphogenetic protein-2; ALP, alkaline phosphatase; mRNA, messenger
RNA. |
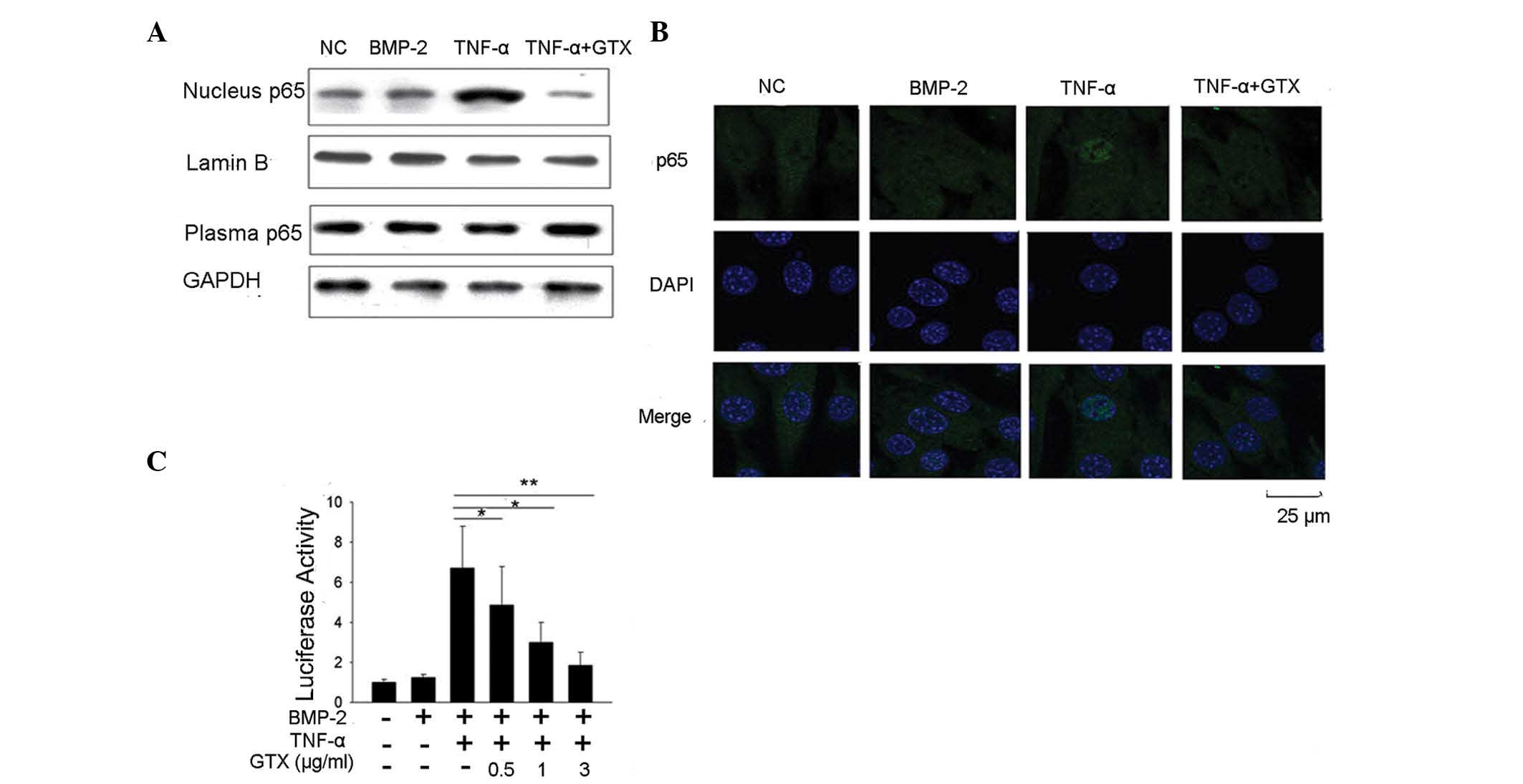
GTX modulates C2C12 osteoblast
differentiation via NF-κB signal inhibition
As indicated by our above results, GTX was able to
block TNF-α-induced inhibition of osteoblast differentiation, and
promote BMP-2-induced osteoblast differentiation. Whether GTX
possesses NF-κB signal inhibiting activity was therefore
investigated. Western blot analysis of NF-κB protein p65 was
performed to detect whether GTX prevented p65 translocation to the
nucleus. The results demonstrated that following treatment with 10
ng/ml TNF-α, p65 accumulated in the nucleus. However, no p65
accumulation was detected in the group that was also treated with 1
μg/ml GTX (Fig. 5A).
Subsequently, an immunofluorescent confocal microscopic assay for
p65 localization detection was performed, and the results concurred
that GTX prevented p65 accumulation in the nucleus (Fig. 5B). The luciferase assay method was
also used to assess whether GTX attenuated TNF-α-induced NF-κB
activity. The results suggested that 10 ng/ml TNF-α treatment
induced a marked increase in NF-κB luciferase activity in C2C12
cells. The activation of luciferase activity was significantly
reduced in the group that was treated with 0.5 μg/ml GTX
(P<0.05), suggesting that the activation of NF-κB was inhibited.
This inhibition occurred in a GTX dose-dependent manner (Fig. 5C). These results indicated that GTX
was able to markedly inhibit NF-κB signaling in C2C12 cells.
Discussion
The inflammatory factor TNF-α has been demonstrated
to have a critical role in the pathogenesis of bone-loss associated
diseases. In the case of RA, TNF-α induces bone loss in the axial
and appendicular skeleton (29).
It was initially hypothesized that the promotion of osteoclast
differentiation and bone resorption by TNF-α was significant in
such cases of inflammatory bone loss; however, the inhibition of
osteoblast differentiation by TNF-α was identified and required
further investigation (7).
Additionally, in estrogen deficiency-associated osteoporosis, TNF-α
was found to be notably overproduced and may induce bone loss due
to its biphasic effects in the promotion of bone resorption and
reduction of bone formation (8,9).
NF-κB signaling is considered to have double effects
in the development of osteoporosis, due to its bidirectional
functions in bone resorption and formation (30). NF-κB responds to stimuli from
receptor activator of nuclear factor κB ligand and directs
osteoclast differentiation (31).
Furthermore, it was identified to be an endogenous inhibitory
factor for osteoblastic bone formation (13). Normal bone maintenance requires a
balance between bone resorption and formation; therefore, the
recovery of this balance is crucial for the effective treatment of
osteoporosis. From this perspective, as biphasic factors, TNF-α and
NF-κB may represent ideal therapeutic targets, and TNF-α and NF-κB
inhibitors may represent potential therapeutic agents for the
treatment of osteoporosis.
The fungi-derived secondary metabolite GTX has been
demonstrated to be a potent NF-κB inhibitor, and is considered to
be a potential agent for the treatment of immune
glomerulo-nephritis (26). It was
also demonstrated that GTX stimulated mature rat osteoclast
apoptosis (32), and it was used
as an experimental tool to inhibit NF-κB signaling in the study of
osteoclastic gene regulations (33). However, whether GTX has a function
in the modulation of osteoblast differentiation remained to be
elucidated. In current study, the role of GTX in the regulation of
osteoblast differentiation was evaluated by determining whether GTX
was able to block the inhibition of BMP-2-induced differentiation
by TNF-α and NF-κB. Based on the C2C12 osteoblast differentiation
system, the results revealed that GTX protected ALP activity from
TNF-α-induced impairment and rescued the promotion of Col I,
Ocn and Bsp gene expression. Additional evidence from
ALP staining and the expression of the three osteoblast-associated
transcription factors, Runx2, Osx and ATF4,
indicated that these osteoblastic characteristics were protected
against the TNF-α-induced inhibition by GTX, which suggested that
GTX rescued osteoblast differentiation by blocking the inhibitory
effect exerted by TNF-α. The experiment also demonstrated that GTX
promoted BMP-2-induced osteoblast differentiation, which indicated
that GTX likely inhibited C2C12 endogenous NF-κB activity in order
to facilitate osteoblast differentiation. Whether GTX was capable
of blocking NF-κB activity in the C2C12 cell system was also
evaluated. Western blot analyses and immunofluorescence confocal
microscopic assays for the detection of p65 localization were
performed and it was revealed that GTX prevented the accumulation
of NF-κB protein p65 in the nucleus. Additionally, a luciferase
activity assay was conducted in order to confirm that GTX inhibited
the activation of NF-κB transcriptional activity, which suggested
that GTX inhibited the activation of NF-κB signaling in C2C12
cells. In conclusion, GTX potentiates osteoblast differentiation by
blocking the inhibition exerted by TNF-α and directly promotes
BMP-2-induced osteoblast differentiation; these effects may be due
to the inhibition of NF-κB signaling.
GTX was found to induce mature osteoclast apoptosis;
therefore, it was hypothesized to have a potential anti-bone
resorption function. The results of the present study suggested
that GTX has protective and promotional effects on osteoblast
differentiation and indicate that it may ultimately facilitate bone
formation. Further studies are required to examine the potential
role of GTX in regulating in vivo osteoblastic bone
formation and treating bone loss diseases. Owing to the
bidirectional function of GTX in the modulation of osteoclast and
osteoblast effects, GTX may potentially be developed as a
therapeutic agent to recover the bone resorption-formation balance
and recover bone mass.
Acknowledgments
This study was financially supported by The Wuhu Key
Science and Technology Project (grant no. 2014zd16).
References
|
1
|
Uccelli A, Moretta L and Pistoia V:
Mesenchymal stem cells in health and disease. Nat Rev Immunol.
8:726–736. 2008. View
Article : Google Scholar
|
|
2
|
Zaidi M: Skeletal remodeling in health and
disease. Nat Med. 13:791–801. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huang W, Yang S, Shao J and Li YP:
Signaling and transcriptional regulation in osteoblast commitment
and differentiation. Front Biosci. 12:3068–3092. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Karsenty G, Kronenberg HM and Settembre C:
Genetic control of bone formation. Annu Rev Cell Dev Biol.
25:629–648. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baum R and Gravallese EM: Impact of
inflammation on the osteoblast in rheumatic diseases. Curr
Osteoporos Rep. 12:9–16. 2014. View Article : Google Scholar :
|
|
6
|
Walsh NC and Gravallese EM: Bone
remodeling in rheumatic disease: a question of balance. Immunol
Rev. 233:301–312. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Walsh NC, Reinwald S, Manning CA, et al:
Osteoblast function is compromised at sites of focal bone erosion
in inflammatory arthritis. J Bone Miner Res. 24:1572–1585. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cenci S, Weitzmann MN, Roggia C, et al:
Estrogen deficiency induces bone loss by enhancing T-cell
production of TNF-alpha. J Clin Invest. 106:1229–1237. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Roggia C, Gao Y, Cenci S, et al:
Up-regulation of TNF-producing T cells in the bone marrow: a key
mechanism by which estrogen deficiency induces bone loss in vivo.
Proc Natl Acad Sci USA. 98:13960–13965. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Canalis E: Effects of tumor necrosis
factor on bone formation in vitro. Endocrinology. 121:1596–1604.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gilbert L, He X, Farmer P, et al:
Inhibition of osteoblast differentiation by tumor necrosis
factor-alpha. Endocrinology. 141:3956–3964. 2000.PubMed/NCBI
|
|
12
|
Li Y, Li A, Strait K, Zhang H, Nanes MS
and Weitzmann MN: Endogenous TNFalpha lowers maximum peak bone mass
and inhibits osteoblastic Smad activation through NF-kappaB. J Bone
Miner Res. 22:646–655. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chang J, Wang Z, Tang E, et al: Inhibition
of osteoblastic bone formation by nuclear factor-kappaB. Nat Med.
15:682–689. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Locksley RM, Killeen N and Lenardo MJ: The
TNF and TNF receptor superfamilies: integrating mammalian biology.
Cell. 104:487–501. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dempsey PW, Doyle SE, He JQ and Cheng G:
The signaling adaptors and pathways activated by TNF superfamily.
Cytokine Growth Factor Rev. 14:193–209. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Novack DV: Role of NF-κB in the skeleton.
Cell Res. 21:169–182. 2011. View Article : Google Scholar
|
|
17
|
Kular J, Tickner J, Chim SM and Xu J: An
overview of the regulation of bone remodelling at the cellular
level. Clin Biochem. 45:863–873. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Raggatt LJ and Partridge NC: Cellular and
molecular mechanisms of bone remodeling. J Biol Chem.
285:25103–25108. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu J, Wu HF, Ang ES, et al: NF-kappaB
modulators in osteolytic bone diseases. Cytokine Growth Factor Rev.
20:7–17. 2009. View Article : Google Scholar
|
|
20
|
Scharf DH, Heinekamp T, Remme N,
Hortschansky P, Brakhage AA and Hertweck C: Biosynthesis and
function of gliotoxin in Aspergillus fumigatus. Appl Microbiol
Biotechnol. 93:467–472. 2012. View Article : Google Scholar
|
|
21
|
Kupfahl C, Ruppert T, Dietz A, Geginat G
and Hof H: Candida species fail to produce the immunosuppressive
secondary metabolite gliotoxin in vitro. FEMS Yeast Res. 7:986–992.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kosalec I, Puel O, Delaforge M, et al:
Isolation and cytotoxicity of low-molecular-weight metabolites of
Candida albicans. Front Biosci. 13:6893–6904. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sutton P, Newcombe NR, Waring P and
Müllbacher A: In vivo immunosuppressive activity of gliotoxin, a
metabolite produced by human pathogenic fungi. Infect Immun.
62:1192–1198. 1994.PubMed/NCBI
|
|
24
|
Pahl HL, Krauss B, Schulze-Osthoff K, et
al: The immuno-suppressive fungal metabolite gliotoxin specifically
inhibits transcription factor NF-kappaB. J Exp Med. 183:1829–1840.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kroll M, Arenzana-Seisdedos F, Bachelerie
F, Thomas D, Friguet B and Conconi M: The secondary fungal
metabolite gliotoxin targets proteolytic activities of the
proteasome. Chem Biol. 6:689–698. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
López-Franco O, Suzuki Y, Sanjuán G, et
al: Nuclear factor-kappa B inhibitors as potential novel
anti-inflammatory agents for the treatment of immune
glomerulonephritis. Am J Pathol. 161:1497–1505. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Karin M: Nuclear factor-kappaB in cancer
development and progression. Nature. 441:431–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hur JM, Yun HJ, Yang SH, Lee WY, Joe MH
and Kim D: Gliotoxin enhances radiotherapy via inhibition of
radiation-induced GADD45a, p38 and NFkappaB activation. J Cell
Biochem. 104:2174–2184. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Goldring SR and Gravallese EM:
Pathogenesis of bone erosions in rheumatoid arthritis. Curr Opin
Rheumatol. 12:195–199. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Krum SA, Chang J, Miranda-Carboni G and
Wang CY: Novel functions for NFkappaB: inhibition of bone
formation. Nat Rev Rheumatol. 6:607–611. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Soysa NS and Alles N: NF-kappaB functions
in osteoclasts. Biochem Biophys Res Commun. 378:1–5. 2009.
View Article : Google Scholar
|
|
32
|
Ozaki K, Takeda H, Iwahashi H, Kitano S
and Hanazawa S: NF-kappaB inhibitors stimulate apoptosis of rabbit
mature osteoclasts and inhibit bone resorption by these cells. FEBS
Lett. 410:297–300. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Trebec-Reynolds DP, Voronov I, Heersche JN
and Manolson MF: VEGF-A expression in osteoclasts is regulated by
NF-kappaB induction of HIF-1alpha. J Cell Biochem. 110:343–351.
2010.PubMed/NCBI
|