Introduction
Alzheimer’s disease (AD), the most common form of
dementia in the aged population, presents an increasing clinical
challenge in terms of diagnosis and treatment (1). The molecular and cellular mechanisms
triggering AD remain to be elucidated. Neurodegeneration is one of
the hallmarks of AD, which consequently induces cognitive
impairment (2). The effects of
several potential disease-modifying drugs for AD on neuroprotection
have been investigated in clinical trials (3), suggesting that neuroprotection is
important in AD therapeutics.
Brain-derived neurotrophic factor (BDNF), a
neuropro-tective factor, has been implicated in neuronal survival
and proliferation (4). Lower
levels of BDNF have been reported in the cerebrospinal fluid (CSF)
of patients with AD, compared with age-matched healthy individuals
(5). However, the underlying
mechanism causing the altered BDNF levels in the CSF of patients
with AD remains to be elucidated.
It has been suggested that epigenetic mechanisms
affect gene expression in AD, as DNA methylation levels are
associated with protein expression in the postmortem AD brain
(6). The methylation of histone H3
lysine 9 (H3K9) has been demonstrated to increase with age in a
triple transgenic mouse model of AD, in combination with a reduced
gene expression of BDNF (7). Thus,
BDNF methylation is a possible mechanism responsible for the low
expression of BDNF in patients with AD, however, further
investigation is required to confirm this hypothesis. DNA
methyltransferases (DNMTs) are responsible for the establishment of
de novo genomic DNA methylation patterns, involved in normal
development and age-accociated diseases, including AD (8). Altered expression levels of DNMT have
been observed in patients with AD (9,10).
Thus, DNMT may contribute to the BDNF methylation, which is
involved in neuroprotection in AD. In addition, epigenetic
modifications, including a combination of microRNAs (miRNAs/miRs)
and DNA methylation, have been suggested as regulatory mechanisms
in the control of neuronal survival (11). Several lines of evidence have also
suggested a role for miRNAs, including miR-29, in AD (12–14).
miRNAs, act as regulators of gene expression at a
post-transcriptional level and are important in governing DNMTs
(15). Specific miRNAs targeting
DNMT transcripts lead to the demethylation and transcriptional
activation of numerous protein coding gene sequences, thereby
contributing to gene expression. In addition, DNMTs are important
in controlling the expression of specific miRNAs (16). This cooperative action among DNMTs,
miRNAs and DNA methylation indicates that miRNAs and DNMTs may be
involved in the pathogenesis of AD.
The present study aimed to investigate the role of
miR-29c in AD and to further examine the possible epigenetic
mechanisms underlying AD. The expression of miR-29c, DNMT3, and
BDNF were detected in CSF samples collected from patients with AD
and from age-matched normal control individuals. DNMT3, an
inhibitor of BDNF, was identified as a target of miR-29c using a
dual luciferase reporter assay. Gain and loss of function
experiments were used to examine the role of miR-29c in hippocampal
neurons.
Materials and methods
CSF samples
The present study was approved by the ethics
committee of Southern Medical University (Guangzhou, China). A
total of 60 CSF samples (3 ml) were obtained by lumbar puncture
from patients with AD and from age-matched normal control
individuals from the Department of Neurology, Haikou People’s
Hospital (Haikou, China). All samples were collected according to
the legislation and ethical boards of Haikou People’s Hospital.
Written informed consent was obtained from all of the patients. The
samples were stored at −80°C until use.
Cell culture and treatment
Primary hippocampal neurons were prepared from rat
embryos (Laboratory Animal Centre, Southern Medical University) at
embryonic day 18. The hippocampi were mechanically dissociated from
the brains of the embryos and treated with trypsin (Sigma-Aldrich,
St. Louis, MO, USA) for 15 min at 37°C in phosphate-buffered saline
(PBS). The cell suspensions were maintained in glial-conditioned
medium (Invitrogen Life Technologies, Carlsbad, CA, USA) in 100 mm
dishes at 37°C under 5% CO2 until use. The experimental
protocol was approved by the Animal Care and Use Committee of
Haikou People’s Hospital, and was in compliance with the National
Institutes of Health Guide for the Care and Use of Laboratory
Animals (Bethesda, MD, USA). Ectopic expression of miR-29c was
achieved in the cells via transfection with miR-29c mimics or
inhibitors using Lipofectamine 2000 (Invitrogen Life Technologies),
according to the manufacturer’s instructions.
Treatment with the DNMT inhibitor
(DNMTI)
DNMTI, 5-azacytidine (AZC), was purchased from
Sigma-Aldrich. A quantity of 30 mg/l DNMTI was added to the primary
hippocampal neurons and the culture continued for 3 days at 37°C in
an atmosphere containing 5% CO2 and 95% air, followed by
three washes with PBS. The culture was then continued for a further
2 days in Dulbecco’s modified Eagle’s medium, prior to
experimentation.
Reverse
transcription-quantitative-polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the tissues and cells,
according to the manufacturer’s instructions (CWBio, Beijing,
China). RNA was reverse transcribed using the Rever-tAid first
strand cDNA synthesis kit (Thermo Scientific, Waltham, MA, USA). A
total of 2 μl cDNA was analyzed by PCR. The mRNA expression
of DNMT3 was detected using a SYBR green qPCR assay (CWBio). The
expression of β-actin was used as an endogenous control. The
specific primers used in the present study were as follows: DNMT3,
forward 5′-CTGGGTCATGTGGTTCGG-3′ and reverse
5′-TCTA-ATAACTACTCGCGTGT-3′; and β-actin, forward
5′-CATTAAGGAGAAGCTGTGCT-3′ and reverse 5′-GTTGAAGGTAGTTTCGTGGA-3′
(Sangon Biotech Co., Ltd., Shanghai, China). An miScript SYBR-Green
PCR kit (RiboBio, Guangzhou, China) was used for RT-qPCR to detect
the expression of miR-29c. The specific primer sets for miRNA-29c
and U6 were purchased from GeneCopoeia (Rockville, MD, USA). The
sequences of the miRNA-29c and U6 primers were as follows:
miRNA-29c, forward 5′-ACACTC-CAG CTG G GTGACCGAT T TCTCCTG-3′,
reverse 5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGA GAGGGATTC-3′; and
U6, forward 5′-CTCGCTTCGGCA GCACA-3′ and reverse
5′-AACGCTTCACGAATTTGCGT-3′. The expression of U6 was used as an
endogenous control. RT-qPCR was performed using the following
conditions: 95°C for 5 min; 40 cycles of 95°C for 30 sec, 58°C for
30 sec and 72°C for 30 sec and 72°C for 10 min using the CFX96
Real- Time system thermal cycler (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Data were analyzed using the 2−ΔΔCT
method (17).
ELISA determination of the expression
levels of BDNF, DNMT1 and DNMT3
The EpiQuik human BDNF, DNMT1 and DNMT3 immunoassay
kits (Epigentek, Brooklyn, NY, USA) were used to determine the
expression levels of BDNF, DNMT1 and DNMT3 in the CSF samples and
in the primary hippocampal neurons. The CSF samples and primary
hippocampal neurons were centrifuged at 3,000 × g for 15 min at
room temperature. According to the manufacturer’s instructions, the
supernatants of the CSF samples or the primary hippocampal neurons
were used to measure the total protein quantity of each sample.
Briefly, the samples and the BDNF, DNMT1 or DNMT3 antibody were
incubated overnight at 4°C. Subsequently, they were incubated with
horseradish peroxidase-labeled anti-rabbit antibody for 30 min at
room temperature. The samples were then developed using
tetramethylbenzidine reagent (100 μl) in the dark and the
absorbance was measured at 450 nm (Synergy™ Mx; BioTek, Winooski,
VT, USA).
Measurement of the BDNF promoter CpG
island methylation status using bisulfite genomic sequencing PCR
(BSP) and methylation-specific PCR (MSP)
Genomic DNA was extracted using a genomic DNA
isolation kit (BioVision, Milpitas, CA, USA). The genomic DNA (1
μg per sample) was modified with bisulfite using an Epitect
Bisulfite kit (Epigentek), according to the manufacturer’s
instructions, and the modified DNA was amplified using the
following primers: BDNF, forward 5′-CTGTATCAAAAGGCCAACTGAA-3′ and
reverse 5′-GTGTCTATCCTTATGAATCGCCA-3′. The PCR products were gel
extracted (Qiagen, Hilden, Germany) to confirm that a single band
had been obtained. The DNA band was excised from the gel and then
dissolved in buffer QXI and QIAEX II at 50°C for 10 min. The sample
was then centrifuged at 12,000 × g for 30 sec. the pellet was
washed and air-dried, then dissolved in 20 μl sterile water.
The supernatant containing the purified DNA was then sequenced by
Invitrogen Life Technologies.
MSP was performed on the bisulfate-treated DNA. The
unmethylated and methylated BDNF primers were designed and
synthesized by Sangon Biotech Co., Ltd. The annealing temperature
was 60°C for the methylated-PCR and 55°C for the unmethylated-PCR,
with 27 cycles used for each.
Western blotting
The total protein was extracted from the cells using
cold radioimmunoprecipitation assay lysis buffer. A Bicinchoninic
Acid Protein Assay kit (Thermo Fisher Scientific, Rockford, IL,
USA) was used to determine the protein concentration. The proteins
were then separated with 10% SDS-PAGE and transferred onto a
nitrocellulose membrane (Wuhan Boster Biological Technology Ltd.,
Wuhan, China). Following blocking in 5% nonfat dried milk in PBS
for 4 h, the membrane was incubated with the indicated primary
antibodies (TrkB, mouse; cat. no. 4603s; 1:2,000; Cell Signaling
Technology, Danvers, MA, USA; phosphorylated (p) Erk and Erk;
rabbit; cat. nos. 9101S and 9102S, respectively; 1:1,000; Cell
Signaling Technology) overnight at 4°C. The membrane was
subsequently washed with tris-buffered saline containing Tween and
incubated with horseradish peroxidase-conjugated goat anti-rabbit
(1:5,000; cat. no. A12004-1) and goat anti-mouse (1:5,000; cat. no.
A12003-1) immunoglobulin G secondary antibodies (Epigentek,
Farmingdale, NY, USA) for 2 h at room temperature. Enhanced
chemiluminescence reagent (Wuhan Boster Biological Technology Ltd.)
was used to detect the signal on the membrane. The data were
analyzed via densitometry using Image-Pro plus software 6.0 (Media
Cybernetics, Rockville, MD, USA) and normalized to the expression
of the internal control.
Dual luciferase reporter assay
The wild type (wt) and mutant (mut) 3′-untranslated
region (UTR) of DNMT3 were constructed and inserted into the dual
luciferase reporter vector. To generate the wt-DNMT3-3′-UTR plasmid
the DNMT3-3′-UTR was cloned into the XbaI (cat. no. ER0683;
Thermo Scientific) site of the pGL3-control vector (Promega
Corporation, Madison, WI, USA) downstream of the lucif-erase gene.
The mut-DNMT3-3′-UTR plasmid was generated from the wt-DNMT3-3′-UTR
by mutating the binding site for miR-29c via site-directed
mutagenesis of the DNMT3-3′-UTR performed by Genecopeoia
(Genecopeoia, Guangzhou, China). For the luciferase assay, 100,000
cells were cultured, at 37°C in an atmosphere containing 5%
CO2 and 95% air, to reach ~70% confluence in 24-well
plates. Subsequently, the cells were co-transfected with the
miR-29c mimic and the wt or mut 3′-UTR of DNMT3 dual luciferase
reporter vector, respectively. Following a 5 h incubation with a
Lipofectamine 2000 (Invitrogen Life Technologies; 2 μl)/DNA
complex at 37°C in an atmosphere containing 5% CO2 and
95% air, the medium were replaced with fresh medium containing 10%
fetal bovine serum (FBS). At 48 h after transfection, a dual
luciferase reporter gene assay kit (BioVision) was used to
determine the luciferase activities in each group, using a
luminometer (Elecsys 2010; Roche Diagnostics, Basel, Germany). The
activity of Renilla luciferase was normalized to that of
firefly luciferase.
MTT assay
For all groups, 3,000 cells per well were seeded
into a 96-well plate. Following treatment, the plates were
incubated for 0, 12, 24, 48 or 72 h at 37°C in 5% CO2.
To assess cell proliferation, an MTT assay was performed, according
to the manufacturer’s instructions. MTT reagent (10 μl; 5
mg/ml) in 100 μl FBS-free medium was added to each well and
incubated for 4 h at 37°C. Subsequently, the medium was removed and
150 μl dimethyl sulfoxide was added. The absorbance was
detected at 490 nm using a microplate reader (Elecsys 2010; Roche
Diagnostics). The assay was repeated three times in triplicate
wells.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism 5 software (Graphpad Software, Inc., La Jolla, CA, USA) and
the data are presented as the mean ± standard deviation. An
unpaired two-tailed Student’s t-test was used to analyze the data.
P<0.05 was considered to indicate a statistically significant
difference.
Results
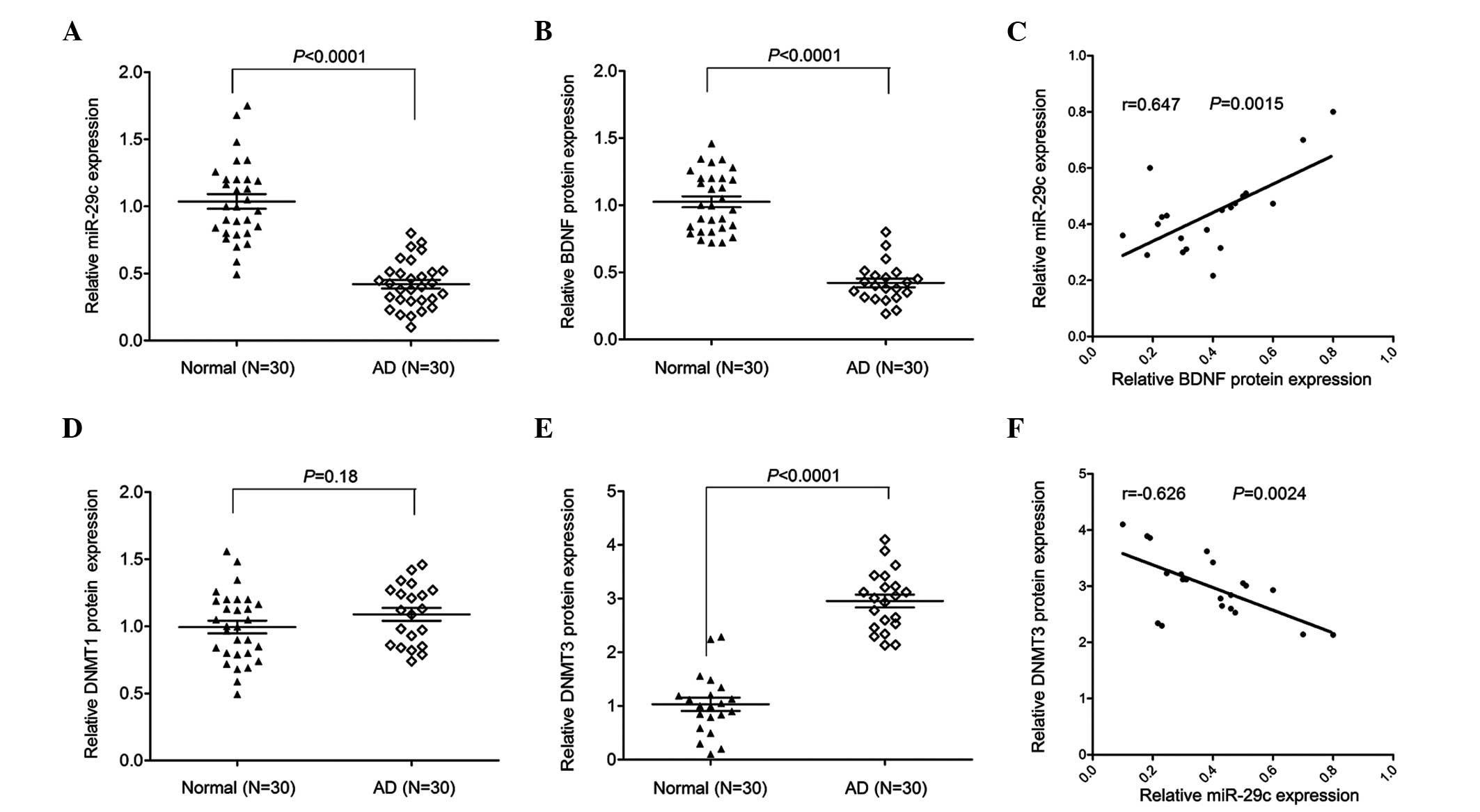
Levels of miR-29c are positively
correlated with the protein expression of BDNF and negatively
correlated with the protein expression of DNMT3 in the CSF of
patients with AD
The expression level of miR-29c was detected using
SYBR green RT-qPCR analysis. In the sample of 30 patients with AD
and control individuals, the results revealed that miR-29c was
significantly decreased in the CSF of patients with AD, compared
with that in the paired normal control individuals (Fig. 1A). In addition, a significant
decrease was observed in the protein expression of BDNF in the CSF
of patients with AD compared with the control (Fig. 1B). Subsequently, an ELISA assay was
used to measure the protein expression levels of DNMT1 and DNMT3.
As shown in Fig. 1D and E,
compared with the normal control individuals, the protein
expression of DNMT3, but not DNMT1 protein, was significantly
increased in the CSF of patients with AD compared with the normal
controls. Furthermore, the expression of miR-29c was positively
correlated with the protein expression of BDNF and negatively
correlated with the protein expression of DNMT3 in the CSF of
patients with AD (Fig. 1C and
F).
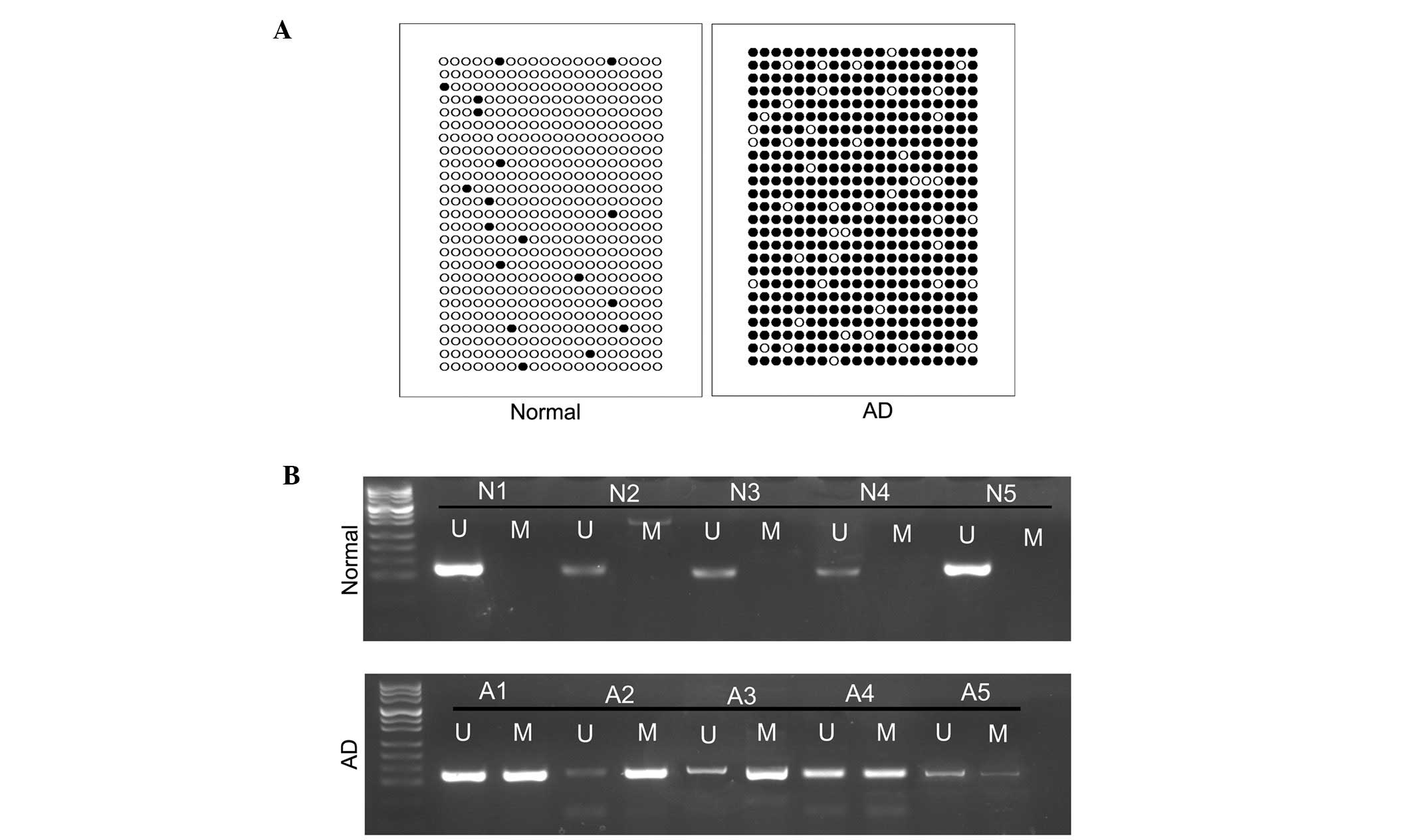
Methylation of the BDNF promoter is
increased in the CSF of patients with AD
The methylation status of the miR-196b CpG island in
the CSF of patients with AD and normal control individuals was
examined using BSP. The results indicated that methylation was
significantly increased in the CSF of patients with AD, compared
with the normal control group (Fig.
2A). MSP was used to detect the methylation status of BDNF in
five patients with AD and five normal control individuals (Fig. 2B). A significantly higher level of
methylation was observed in the patients with AD compared with the
normal control individuals.
miR-29c regulates the expression of DNMT3
by directly targeting its 3′UTR
To investigate whether miR-29c targets the 3′UTR of
DNMT3, the 3′UTR of DNMT3 was cloned downstream of a luciferase
reporter gene (wt-DNMT3). A mutant version (mut-DNMT3), with the
binding site mutagenesis, was also constructed (Fig. 3A). The wt-DNMT3 vector and
pre-miR-29c mimics or scramble control were co-transfected into
primary hippocampal neurons. The luciferase activities of the
pre-miR-29c and wt-DNMT3 co-transfected cells were significantly
reduced compared with the scramble control cells (Fig. 3B). Additionally, to assess whether
miR-29c regulated the expression of DNMT3 at a transcriptional or
translational level, the pre-miR-29c or anti-miR-29c were
transfected into primary hippocampal neurons. As shown in Fig. 3C, the transfection efficiency was
satisfactory for further analysis. The present results revealed
that the overexpression of miR-29c significantly reduced the
expression of DNMT3 at the mRNA and protein levels, whereas
downregulation of miR-29c increased the expression of DNMT3 at the
mRNA and protein levels (Fig. 3D and
E). In addition, the protein expression of BDNF was detected in
the cells treated with pre-miR-29c or anti-miR-29c alone, or
combined with DNMTI. The protein expression of BDNF was decreased
by upregulation of miR-29c and increased by downregulation of
miR-29c, and was also induced by treatment with DNMTI alone. Higher
levels of BDNF were observed in the pre-miR-29c + DNMTI treatment
group compared with the pre-miR-29c or the DNMTI alone treatment
groups. In addition, a lower level of BDNF was observed in the
anti-miR-29c + DNMTI treatment group compared with the DNMTI alone
group, but was marginally higher than that of the group treated
with anti-miR-29c alone (Fig. 3F).
These results suggested that miR-29c regulated the expression of
DNMT3 at the transcriptional level by directly targeting its 3′UTR,
consequently affecting the expression of BDNF.
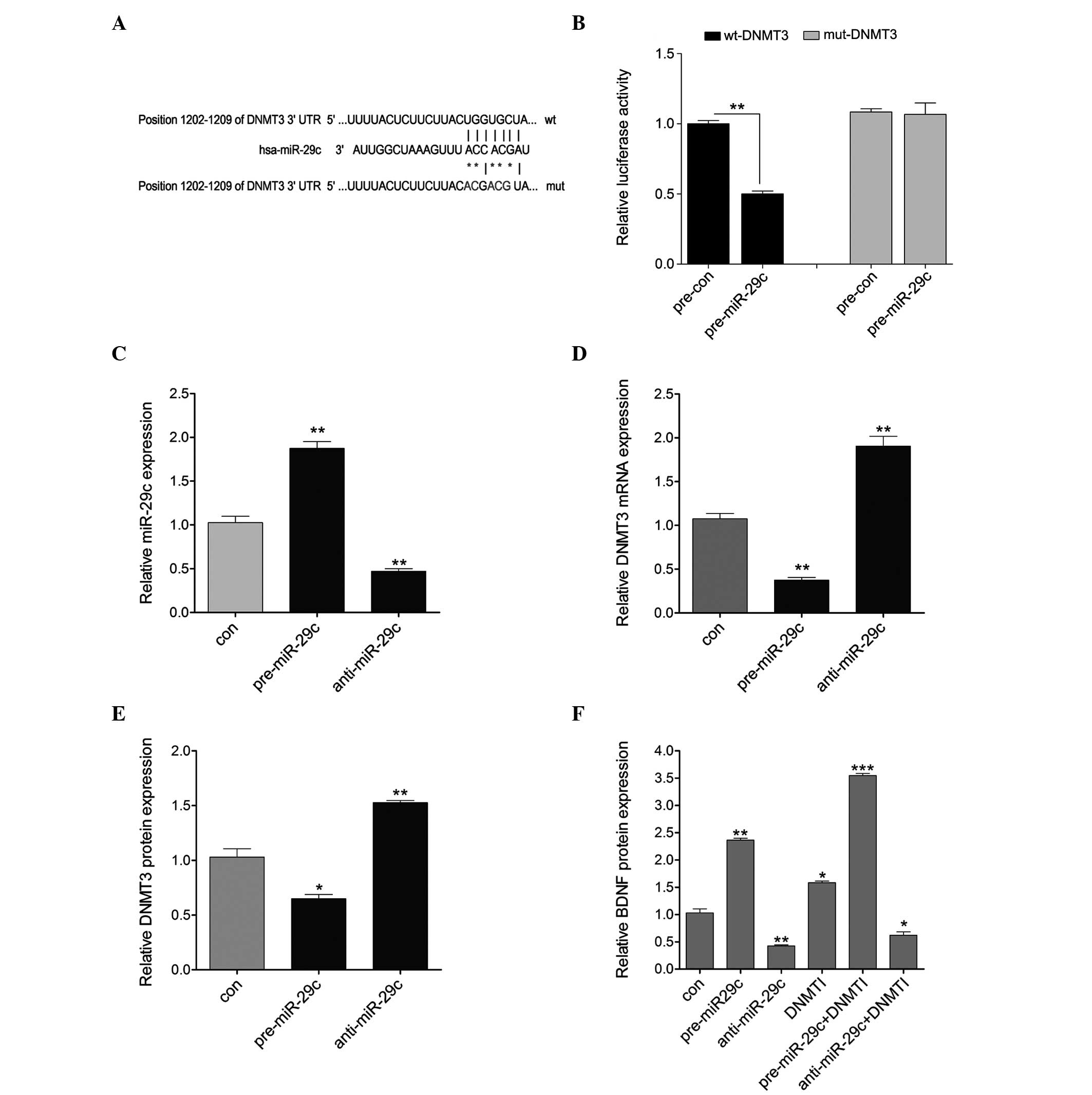 | Figure 3miR-29c directly targets the 3′UTR of
DNMT3. (A) Predicted position of the 3′UTR of DNMT3 targeted by
miR-29c, obtained from TargetScan (http://www.targetscan.org/). The mutant form was
constructed (indicated by asterisks). (B) Repression of luciferase
activity by the DNMT3 3′UTR was dependent on miR-29c. The mutated
DNMT3 3′UTR abrogated the miR-29c-mediated repression of luciferase
activity. (C) RT-qPCR detected the expression of miR-29c following
pre-miR-29c or anti-miR-29c treatment in primary hippocampal
neurons. (D) Reverse transcription-quantitative polymerase chain
reaction was used to detect the mRNA expression of DNMT3 following
pre-miR-29c or anti-miR-29c treatment. (E) An ELISA assay detected
the protein expression of DNMT3 following pre-miR-29c or
anti-miR-29c treatment. (F) An ELISA assay detected the protein
expression of BDNF in the primary hippocampal neurons following the
indicated treatment. Data are presented as the mean ± standard
deviation. *P<0.05; **P<0.01 and
***P<0.001, vs. control. DNMT3, DNA
meth-yltransferase 3; BDNF, brain-derived neurotrophic factor; miR,
microRNA; UTR, untranslated region; RT-qPCR, ; wt, wild-type; mut,
mutant; con, control. |
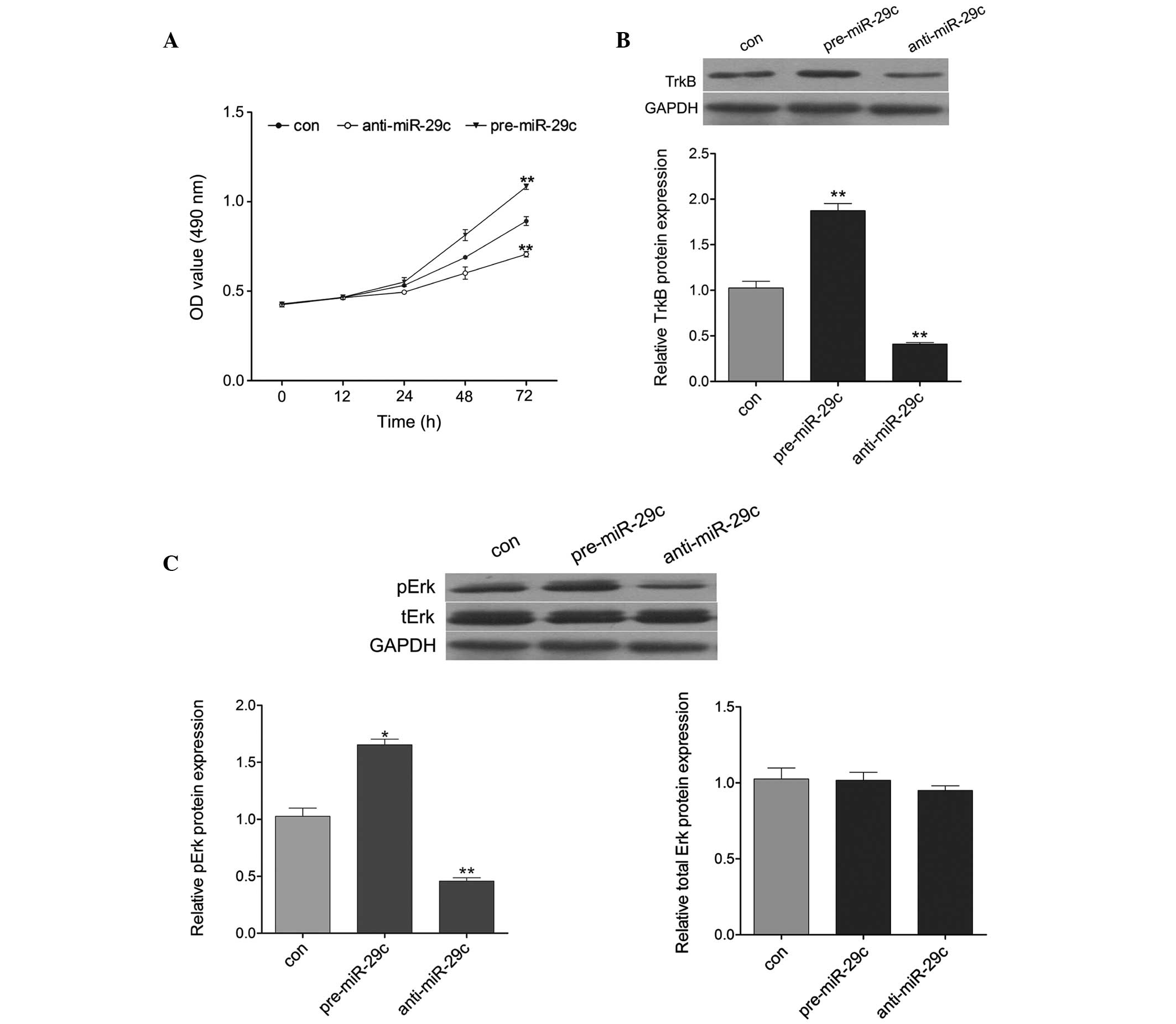
miR-29c promotes cell proliferation via
TrkB/Erk signaling
To assess whether miR-29c regulated neuronal
proliferation, an MTT assay was performed by transfecting
pre-miR-29c or anti-miR-29c into primary hippocampal neurons.
Compared with the control group, pre-miR-29c transfection
significantly promoted cell proliferation, whereas anti-miR-29c
transfection significantly inhibited cell proliferation (Fig. 4A). To further investigate the
molecular mechanisms of miR-29c on the promotion of cell growth,
the expression levels of molecules associated with TrkB signaling
were detected. As shown in Fig.
4B, the protein expression of TrkB was increased by
upregulation of miR-29c, but decreased by downregulation of
miR-29c. In addition, the expression of pErk was induced markedly
by pre-miR-29c transfection, and reduced by anti-miR-29c
transfection (Fig. 4C). These
results suggested that miR-29c promoted neuronal proliferation by
activating TrkB/Erk signaling.
Discussion
The clinical assessment of AD is key in patient
management and clinical trials, however, this evaluation has
several limitations, such as the inability to clearly predict
cortical changes to cognitive functioning (18). Thus, CSF biomarkers are useful
tools to diagnose AD promptly and accurately, enabling effective
prognosis and treating patients with AD accordingly (19). Improved understanding of the system
biomarkers may assist in identifying the most efficient drug
targets for developing optimal therapeutic strategies for AD.
Levels of BDNF are reduced in specific brain regions, including the
cortex and hippocampus, in AD, and BDNF gene polymorphisms have
been suggested to affect the risk of developing AD and memory
(20). Low serum levels of BDNF
are associated, in patients with AD, with rapid cognitive decline
and exhibit a significant correlation with the rate of cognitive
decline (21). The BDNF level in
the CSF is also lower in patients with AD compared with age-matched
normal control individuals (5).
Therefore, the levels of BDNF in the serum and CSF are consistent
with that of the brain in patients with AD, suggesting that the
level of BDNF in the CSF may reflect the status of the brain in
patients with AD. In the present study, the levels of BDNF in the
CSF were lower in patients with AD compared with age-matched normal
control individuals, consistent with previous findings (22).
In addition to the MSP and BSP investigations, the
present study demosntrated that BDNF methylation was higher in
patients with AD compared with age-matched normal control
individuals. In addition, levels of DNMT3 in the CSF were increased
significantly in patients with AD compared with the age-matched
normal control individuals. In an AD animal model, lower gene
expression levels of Bdnf is associated with increased H3K9
methylation (7). The
hypermethylation of BDNF has been observed to correlate with
elevated levels of neuroinflammation and altered pro-apoptotic
factors in the postmortem frontal cortex of patients with AD
(6). DNMT1 is the major enzyme
responsible for maintenance of the DNA methylation pattern, and the
levels of DNMT3 are often increased in various cancer tissues and
cell lines (23). Thus, DNMT3 may,
in part, account for the hypermethylation of the CpG-rich promoter
regions of genes, including BDNF. It has been reported that
prenatal stress induces a decrease in the expression of BDNF, which
may be mediated by increased levels of DNMTs levels, in rats
(24). In the present study, the
inhibition of DNMT activity with AZC increased the protein
expression of BDNF in the hippocampal neurons. Previous findings
have indicated that the activation of stress-associated signaling
pathways result in the increased transcription of amyloid precursor
protein and β-secretase 1 through DNMT-dependent hypomethylation,
thus leading to the overproduction of amyloid-β (25). However, it is reasonable that DNMTs
may simultaneously regulate the expression of genes, which either
promote or protect against AD. Further evidence is required to
confirm this hypothesis.
Furthermore, the upstream molecules, which regulate
the expression levels of DNMT3 and BDNF were also examined. It was
observed that the levels of miR-29c in the CSF were markedly
decreased in patients with AD compared with normal control
individuals. In addition, the levels of miR-29c in the CSF were
positively correlated with the protein expression of BDNF and
negatively correlated with the protein expression of DNMT3 in
patients with AD. The results also revealed that miR-29c regulated
the expression of DNMT3 at the transcriptional level by directly
targeting its 3′UTR. miRNAs are involved in development of AD.
Previous studies have revealed that circulating miRNAs in the CSF
and blood serum of patients with AD can be used as biomarkers in
the diagnosis of AD (1,26,27).
Downregulation of the miR-29 family is correlated with the density
of diffuse amyloid plaques, detected in the adjacent tissue in
human cerebral cortical gray matter (28). Additionally, DNMTs are targets of
miR-29 (29). The present study
demonstrated that the protein expression of BDNF was regulated by
treatment with miR-29c and DNMTI, which suggested that miR-29c
regulated the expression of DNMT3 at the transcriptional level by
directly targeting its 3′UTR, which consequently affected the
expression of BDNF.
BDNF acts as a neuroprotective factor, which is
important in neuronal survival and proliferation (30). It has been demonstrated that
upregulation of miR-29c significantly promotes cell proliferation,
whereas downregulation of miR-29c impairs neuronal growth
capability. According to the results of the present study,
biochemically, miR-29c regulated neuronal growth by affecting
BDNF-associated downstream molecular pathways. The present data
suggested that miR-29c may elicit a neuroprotective effect via
TrkB/Erk signaling. BDNF and TrkB have a central role in the
nervous system by providing trophic support to neurons, and
BDNF/TrkB signaling is impaired in AD (31). A previous study demonstrated that
BDNF may protect retinal neurons from hyperglycemia via the
TrkB/ERK/mitogen-activated protein kinase pathway (32). Thus, the present study hypothesized
that miR-29c exerts a neuroprotective effect via the BDNF/TrkB/Erk
signaling pathway.
In conclusion, the present study demosntrated that
low levels of miR-29c in the CSF were positively correlated with
the protein expression of BDNF and negatively correlated with the
protein expression of DNMT3 in patients with AD. miR-29c regulated
the expression of BDNF, possibly through an epigenetic mechanism,
by directly targeting DNMT3. This neuroprotective effect mediated
by miR-29c may offer a potential therapeutic target in the
treatment of AD.
References
|
1
|
Kim DH, Yeo SH, Park JM, et al: Genetic
markers for diagnosis and pathogenesis of Alzheimer’s disease.
Gene. 545:185–193. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Morales I, Guzmán-Martínez L,
Cerda-Troncoso C, Farías GA and Maccioni RB: Neuroinflammation in
the pathogenesis of Alzheimer’s disease A rational framework for
the search of novel therapeutic approaches. Front Cell Neurosci.
8:1122014. View Article : Google Scholar
|
|
3
|
Dunkel P, Chai CL, Sperlágh B, Huleatt PB
and Mátyus P: Clinical utility of neuroprotective agents in
neurodegenerative diseases: current status of drug development for
Alzheimer’s, Parkinson’s and Huntington’s diseases and amyotrophic
lateral sclerosis. Expert Opin Investig Drugs. 21:1267–1308. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
García-Mesa Y, Pareja-Galeano H,
Bonet-Costa V, et al: Physical exercise neuroprotects
ovariectomized 3xTg-AD mice through BDNF mechanisms.
Psychoneuroendocrinology. 45:154–166. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li G, Peskind ER, Millard SP, et al:
Cerebrospinal fluid concentration of brain-derived neurotrophic
factor and cognitive function in non-demented subjects. PLoS One.
4:e54242009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Keleshian VL, Modi HR, Rapoport SI and Rao
JS: Aging is associated with altered inflammatory, arachidonic acid
cascade and synaptic markers, influenced by epigenetic
modifications, in the human frontal cortex. J Neurochem. 125:63–73.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Walker MP, LaFerla FM, Oddo SS and Brewer
GJ: Reversible epigenetic histone modifications and Bdnf expression
in neurons with aging and from a mouse model of Alzheimer’s
disease. Age (Dordr). 35:519–531. 2013. View Article : Google Scholar
|
|
8
|
Mastroeni D, Chouliaras L, Grover A, et
al: Reduced RAN expression and disrupted transport between
cytoplasm and nucleus; a key event in Alzheimer’s disease
pathophysiology. PLoS One. 8:e533492013. View Article : Google Scholar
|
|
9
|
Bihaqi SW, Huang H, Wu J and Zawia NH:
Infant exposure to lead (Pb) and epigenetic modifications in the
aging primate brain: implications for Alzheimer’s disease. J
Alzheimers Dis. 27:819–833. 2011.
|
|
10
|
Fuso A, Nicolia V, Cavallaro RA and Scarpa
S: DNA methylase and demethylase activities are modulated by
one-carbon metabolism in Alzheimer’s disease models. J Nutr
Biochem. 22:242–251. 2011. View Article : Google Scholar
|
|
11
|
Imai S, Saeki M, Yanase M, et al: Change
in microRNAs associated with neuronal adaptive responses in the
nucleus accumbens under neuropathic pain. J Neurosci.
31:15294–15299. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pereira P, Sousa Â, Queiroz J, Correia I,
Figueiras A and Sousa F: Purification of pre-miR-29 by
arginine-affinity chromatography. J Chromatogr B Analyt Technol
Biomed Life Sci. 951–952:16–23. 2014. View Article : Google Scholar
|
|
13
|
Zhang J, Hu M, Teng Z, Tang YP and Chen C:
Synaptic and cognitive improvements by inhibition of 2-AG
metabolism are through upregulation of microRNA-188–3p in a mouse
model of Alzheimer’s disease. J Neurosci. 34:14919–14933. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kong Y, Wu J and Yuan L: MicroRNA
expression analysis of adult-onset Drosophila Alzheimer’s disease
model. Curr Alzheimer Res. 11:882–891. 2014.
|
|
15
|
Pogue AI, Hill JM and Lukiw WJ: MicroRNA
(miRNA): Sequence and stability, viroid-like properties and disease
association in the CNS. Brain Res. 1584:73–79. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Spencer P, Fry RC and Kisby GE: Unraveling
50-year-old Clues linking Neurodegeneration and cancer to Cycad
Toxins: Are microRNAs common mediators? Front Genet. 3:1922012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
18
|
Harvey PD: Clinical applications of
neuropsychological assessment. Dialogues Clin Neurosci. 14:91–99.
2012.PubMed/NCBI
|
|
19
|
Lista S, Zetterberg H, Dubois B, Blennow K
and Hampel H: Cerebrospinal fluid analysis in Alzheimer’s disease:
technical issues and future developments. J Neurol. 261:1234–1243.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Peng S, Wuu J, Mufson EJ and Fahnestock M:
Precursor form of brain-derived neurotrophic factor and mature
brain-derived neurotrophic factor are decreased in the pre-clinical
stages of Alzheimer’s disease. J Neurochem. 93:1412–1421. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Laske C, Stellos K, Hoffmann N, et al:
Higher BDNF serum levels predict slower cognitive decline in
Alzheimer’s disease patients. Int J Neuropsychopharmacol.
14:399–404. 2011. View Article : Google Scholar
|
|
22
|
Laske C, Stransky E, Leyhe T, et al: BDNF
serum and CSF concentrations in Alzheimer’s disease, normal
pressure hydrocephalus and healthy controls. J Psychiatr Res.
41:387–394. 2007. View Article : Google Scholar
|
|
23
|
Subramaniam D, Thombre R, Dhar A and Anant
S: DNA Methyltransferases: a novel target for prevention and
therapy. Front Oncol. 4:802014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Boersma GJ, Lee RS, Cordner ZA, et al:
Prenatal stress decreases Bdnf expression and increases methylation
of Bdnf exon IV in rats. Epigenetics. 9:437–447. 2014. View Article : Google Scholar :
|
|
25
|
Guo X, Wu X, Ren L, Liu G and Li L:
Epigenetic mechanisms of amyloid-beta production in
anisomycin-treated SH-SY5Y cells. Neuroscience. 194:272–281. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang J, Sokal I, Peskind ER, et al: CSF
multianalyte profile distinguishes Alzheimer and Parkinson disease.
Am J Clin Pathol. 129:526–529. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kiko T, Nakagawa K, Tsuduki T, Furukawa K,
Arai H and Miyazawa T: MicroRNAs in plasma and cerebrospinal fluid
as potential markers for Alzheimer’s disease. J Alzheimers Dis.
39:253–259. 2014.
|
|
28
|
Wang WX, Huang Q, Hu Y, Stromberg AJ and
Nelson PT: Patterns of microRNA expression in normal and early
Alzheimer’s disease human temporal cortex: white matter versus gray
matter. Acta Neuropathol. 121:193–205. 2011. View Article : Google Scholar :
|
|
29
|
Pandey M, Sultana S and Gupta KP:
Involvement of epigenetics and microRNA-29b in the urethane induced
inception and establishment of mouse lung tumors. Exp Mol Pathol.
96:61–70. 2014. View Article : Google Scholar
|
|
30
|
Tong L, Balazs R, Thornton PL and Cotman
CW: Beta-amyloid peptide at sublethal concentrations downregulates
brain-derived neurotrophic factor functions in cultured cortical
neurons. J Neurosci. 24:6799–6809. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jeronimo-Santos A, Vaz SH, Parreira S, et
al: Dysregulation of TrkB receptors and BDNF function by Amyloid-β
Peptide is mediated by Calpain. Cereb Cortex. 2014. View Article : Google Scholar
|
|
32
|
Liu Y, Tao L, Fu X, Zhao Y and Xu X: BDNF
protects retinal neurons from hyperglycemia through the
TrkB/ERK/MAPK pathway. Mol Med Rep. 7:1773–1778. 2013.PubMed/NCBI
|