Introduction
Esophageal cancer has garnered attention in the past
three decades, due to its increasing prevalence and poor prognosis
(1). Radiotherapy is an effective
treatment option in curing or controlling esophageal cancer.
Ionizing radiation kills tumor cells by inducing various types of
DNA damage, including double-strand and single-strand breaks, base
damage and DNA-DNA or DNA-protein cross-linking (2). However, normal tissue dose
constraints and tumor radioresistance are considered major
obstacles to the success of radiotherapy. Previous attempts have
been made to reduce resistance to radiotherapy and enhance its
therapeutic effectiveness. It has previously been shown that local
tumor control may be improved when radiation therapy is combined
with chemotherapy or thermotherapy (3,4);
however, the effect of current therapies in improving the survival
of patients with esophageal cancer remains unsatisfactory (5).
Autophagy is an evolutionarily conserved catabolic
process, which targets cellular organelles and cytoplasmic
constituents to lysosomes for degradation. Numerous studies have
indicated the importance of autophagy in the pathogenesis,
development and treatment of cancer (6–8).
However, the role of autophagy in cancer remains controversial
(9,10). A basal level of constitutive
autophagy maintains homeostasis and cellular health, through the
removal of excess or damaged intracellular components and microbial
invaders, thus suppressing cancer initiation and progression
(11). However, once cells have
turned cancerous, autophagy may aid cancer cell survival through
degradation and recycling of unnecessary, injured or aged proteins
and organelles of normal cells (12). Therefore, appropriate modification
of autophagy, such as inhibition of cytoprotective autophagy, may
be an appropriate therapeutic strategy for the treatment of
established cancers. The association between autophagy and
angiogenesis is complex, and there are various conflicting reports
regarding the role of autophagy in the process of angiogenesis.
Previous studies have demonstrated that autophagy inhibits
angiogenesis (13,14), whereas other studies have suggested
that autophagy promotes cancer, and inhibition of autophagy
prevents angiogenesis (15,16).
However, whether autophagy affects angiogenesis in esophageal
cancer remains poorly understood.
A preliminary study demonstrated that inhibition of
autophagy enhanced the cytotoxicity of radiotherapy in the TE-1
esophageal cancer cell line (17).
The present study further validated these previous observations and
also aimed to investigate the correlation between autophagy and
tumor angiogenesis using in vitro and in vivo assays.
Mechanistic studies were performed using flow cytometry,
immunohistochemistry and western blot analysis, and a xenograft
model of esophageal cells was treated with radiation and an
autophagy inhibitor followed by histological and western blot
analysis. The present study provided proof that the inhibition of
autophagy may improve the outcomes of radiation therapy of human
esophageal squamous cell carcinoma.
Materials and methods
Cell culture
The EC9706 human esophageal squamous cell carcinoma
cell line was obtained from the Type Culture Collection of the
Chinese Academy of Sciences (Beijing, China). The cells were
cultured in RPMI-1640 medium supplemented with 10% fetal bovine
serum, 2 mM glutamine, 100 units/ml penicillin and 100 µg
streptomycin/ml (all purchased from Sigma-Aldrich, St. Louis, MO,
USA). The cells were incubated at 37°C in a humidified atmosphere
containing 95% air and 5% CO2, and were sub-cultured
every three days.
Reagents and antibodies
Autophagy inhibitor 3-methyladenine (3-MA) was
obtained from Sigma-Aldrich. Anti-microtubule-associated protein
light chain 3 (LC3; cat. no. sc-16755), anti-beclin-1 cat. no.
sc-48381), anti-vascular endothelial growth factor (VEGF; cat. no.
sc-1836), anti-cleaved caspase-3 (cat. no. sc-22171-R),
anti-cleaved caspase-9 (cat. no. sc-56073), anti-cleaved poly(ADP
ribose) polymerase (PARP; cat. no. sc-23461-R), anti-proliferating
cell nuclear antigen (PCNA; cat. no. sc-56), anti-Ki-67 (cat. no.
sc-15402), anti-B-cell lymphoma 2 (Bcl-2; cat. no. sc-7382),
anti-Bcl-2-associated X protein (Bax; cat. no. sc-6236) and
anti-CD31 (cat. no. sc-71873) antibodies were all purchased from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Anti-actin
antibody, and goat anti-rabbit and goat anti-mouse immunoglobulin
(Ig)G (cat. no. sc-66931) secondary antibodies were obtained from
Santa Cruz Biotechnology, Inc.
Irradiation
The exponentially growing cells were exposed at room
temperature and irradiated with a Varian 600 CD X-ray linear
accelerator (Varian Medical Systems, Inc., Palo Alto, CA, USA), at
a dose rate of 2.5 Gy/min.
Cell viability and colony formation
assay
The exponentially growing cells were exposed at room
temperature and a total dose of 6 Gy radiation was delivered in
three fractions over three days. In the experimental study groups,
5 or 10 mM 3-MA was added to the cells for 2 h prior to
irradiation. Six hours after the treatments, all of the cells were
detached by trypsinization (Sigma-Aldrich) and the number of viable
cells was counted. For the colony formation assay, the cells were
seeded into six-well plates containing Dulbecco’s modified Eagle’s
medium supplemented with 10% fetal bovine serum, and incubated for
14 days. The cells were fixed with ethanol (Cusabio Biotech Co.,
Ltd., Wuhan, China) and stained using 0.5% crystal violet
(Sigma-Aldrich), while only colonies containing ≥50 cells were
considered surviving colonies. The sensitizing enhancement ratio
(SER) was calculated, according to the D0 values (dose
of radiation producing a 37% survival rate), using the following
formula: SER=D0 untreated cells/D0 treated
cells.
Cell cycle analysis
Flow cytometry was performed following DNA staining
with propidium iodide (PI; Santa Cruz Biotechnology, Inc.),
according to the manufacturer’s instructions. Briefly, the cells
were harvested, washed with phosphate-buffered saline (PBS) and
treated with 100 mg/ml RNase A (Santa Cruz Biotechnology, Inc.) for
30 min at room temperature. The cells were then stained with PI (1
mg/ml) solution at 4°C and incubated in the dark for 30 min. The
cell cycle distribution was evaluated using a BD FACSArray™
Bioanalyzer system (BD Biosciences, San Jose, CA, USA).
Apoptosis detection
The Annexin V-fluorescein isothiocyanate (FITC)
Apoptosis Detection kit (Santa Cruz Biotechnology, Inc.) was used
to assess the rate of cell apoptosis. Briefly, the untreated and
treated cells were seeded in six-well plates and incubated for 24
h. The cells were then harvested, washed twice in PBS and stained
with Annexin V-FITC and PI, according to the manufacturer’s
instructions. Annexin V binds to apoptotic cells with exposed
phosphatidylserine (early apoptosis), whereas PI labels cells with
membrane damage (late apoptosis). The resulting fluorescence was
detected by flow cytometry using CellQuest™ version 5.2.1 (BD
Biosciences) analysis software.
Western blot analysis
Protein extraction and tissue sample homogenization
were performed as previously described (18,19).
The cells were scraped into ice-cold PBS and centrifuged at 400 × g
for 5 min at 4°C. The pelleted cells were then lysed in 50 ml
boiling SDS solution (Sigma-Aldrich) and centrifuged at 4,350 × g
for 5 min. Membrane protein was extracted from the resulting
supernatant using Mem-PER Eukaryotic Membrane Protein Extraction
kit (Pierce Biotechnology, Inc., Rockford, IL, USA). Protein
concentrations were measured by NanoDrop 1000 (Thermo Fisher
Scientific, Waltham, MA, USA) using bovine serum albumin (Santa
Cruz Biotecnology, Inc.) as a standard. An equal amount of protein
(8 µg) was separated using 15% SDS-PAGE and transferred to
polyvinylidene fluoride membranes. The membranes were then blocked
with 5% skimmed milk (Cusabio Biotech Co., Ltd.) for 1 h and
incubated overnight at 4°C with the following primary antibodies:
anti-LC3-I/II (diluted 1:2,000), anti-beclin-1 (diluted 1:800),
anti-VEGF (diluted 1:400), anti-caspase-3 (diluted 1:400),
anti-caspase-9 (diluted 1:500), anti-PARP (diluted 1:400),
anti-PCNA (diluted 1:1,000), anti-Ki-67 (diluted 1:800), anti-Bax
(diluted 1:1,000) and anti-Bcl-2 (diluted 1:1,000). The membranes
were then incubated with horseradish peroxidase-conjugated IgG
secondary antibodies for 2 h at 37°C. The immunoreactive bands were
visualized using an enhanced chemiluminescence system (Pierce
Biotechnology, Inc.). To quantify equal loading, the membranes were
re-probed with a primary antibody targeting β-actin.
Xenograft experiment
The experiments of the present study followed the
Declaration of Helsinki, and were approved by the institutional
review board of Zhengzhou University Affiliation Cancer Hospital
(Zhengzhou, China). The EC9706 cells (2.0×106 cells)
were injected subcutaneously into the lower right side flank of
male athymic nude BALB/C-nu/nu mice (5–6 weeks-old; weighing 18–20
g), which were purchased from Keli China Experimental Animal Center
(Beijing, China). The care and treatment of the mice were in
accordance with institutional guidelines. When the tumor volume
reached 100 mm3, six mice/group were treated with 3-MA
(30 mg/kg intraperitoneally 1 h prior to radiation) and/or
radiation (2 Gy/fraction, five fractions per week to equal a total
dose of 20 Gy). The mice were divided into four treatment groups
(n=12/group): Untreated, 3-MA alone, radiation alone and radiation
combined with 3-MA. At the end of the experiments all of the mice
were sacrificed by cervical dislocation. Tumor diameters were
measured with calipers and tumor volume (V) was calculating using
the following formula for a rotational ellipsoid:
V=A×B2/2 (A, axial diameter; B, rotational
diameter).
Histological analysis of tumors
Tumor tissues derived from the four groups were
fixed in 10% formalin (Cusabio Biotech Co., Ltd.), embedded in
paraffin and cut into 5-µm sections. Hematoxylin and eosin
(H&E; Santa Cruz Biotechnology, Inc.) staining was performed on
the tumor tissue for general morphological analysis. The samples
were assayed for DNA fragmentation [terminal deoxynucleotidyl
transferase dUTP nick end labeling (TUNEL) assay] using the in
situ Cell Death Detection kit (Roche Molecular Biochemicals,
Indianapolis, IN, USA). Briefly, following deparaffinization and
dehydration, the tissue sections were incubated in proteinase K
(DAKO North America, Inc., Carpinteria, CA, USA) for 15 min, washed
with PBS, incubated in equilibration buffer and then in terminal
deoxynucleotidyl transferase enzyme solution. The sections were
subsequently rinsed in PBS, incubated with streptavidin-peroxidase
conjugate (Sigma-Aldrich) and visualized using diaminobenzidine
(Sigma-Aldrich), according to the manufacturer’s instructions.
Measurement of tumor angiogenesis
Specific staining for endothelial cells was
conducted using the neo-angiogenesis marker CD31. The slides were
fixed using cold acetone (Cusabio Biotech Co., Ltd.) for 20 min.
Following two washes with PBS, the tissue sections were incubated
with 3% hydrogen peroxide (Santa Cruz Biotechnology, Inc.) in
methanol for 30 min, in order to block endogeneous peroxidase
activity. Primary antibody incubation was conducted at 37°C for 2
h, the slides were then incubated with goat anti-mouse IgG
secondary antibody for 30 min, and with streptavidin
biotin-peroxidase complex (Santa Cruz Biotechnology, Inc.) for 40
min. Following incubation with diaminobenzidine chromogen (Santa
Cruz Biotechnology, Inc.), the tissue sections were re-stained with
hematoxylin. Vessel density was determined by counting the number
of microvessels per high-power field (Olympus IX 70; Olympus
Corporation, Tokyo, Japan).
Statistical analysis
All values are presented as the mean ± standard
error of the mean. Statistical significance was analyzed by one-way
analysis of variance with post hoc Dunnett’s test, using
SPSS version 16.0 (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Inhibition of autophagy increases
radiosensitization of tumor cells in vitro
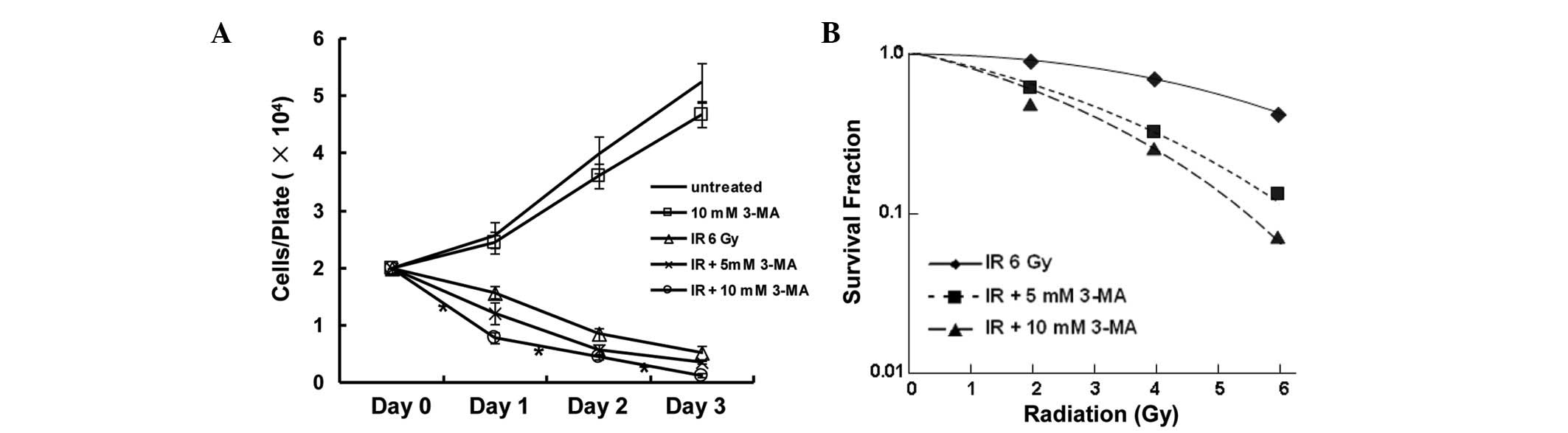
In order to demonstrate the enhancing effect of
autophagy inhibition on radiosensitivity, cells were treated with
3-MA, which is a well-known inhibitor of autophagy in mammalian
cells. Cell proliferation and colony formation were investigated in
the EC9706 esophageal squamous carcinoma cell line. Treatment with
10 mM 3-MA alone led to a slight inhibition of cell growth. The
viability of the cells was decreased in response to radiation,
whereas a combination of radiation and 3-MA treatment markedly
decreased the number of surviving cells (Fig. 1A). The radiosensitizing potential
of 3-MA was ascertained by a clonogenic survival assay, and the SER
was calculated based on the D0 values extrapolated from
from the survival curves. The SER reached 1.76 when the cells were
treated with a combination of 10 mM 3-MA and ionizing radiation
(Fig. 1B). These results indicated
that autophagy inhibition exhibits a radiosensitization potential
in vitro.
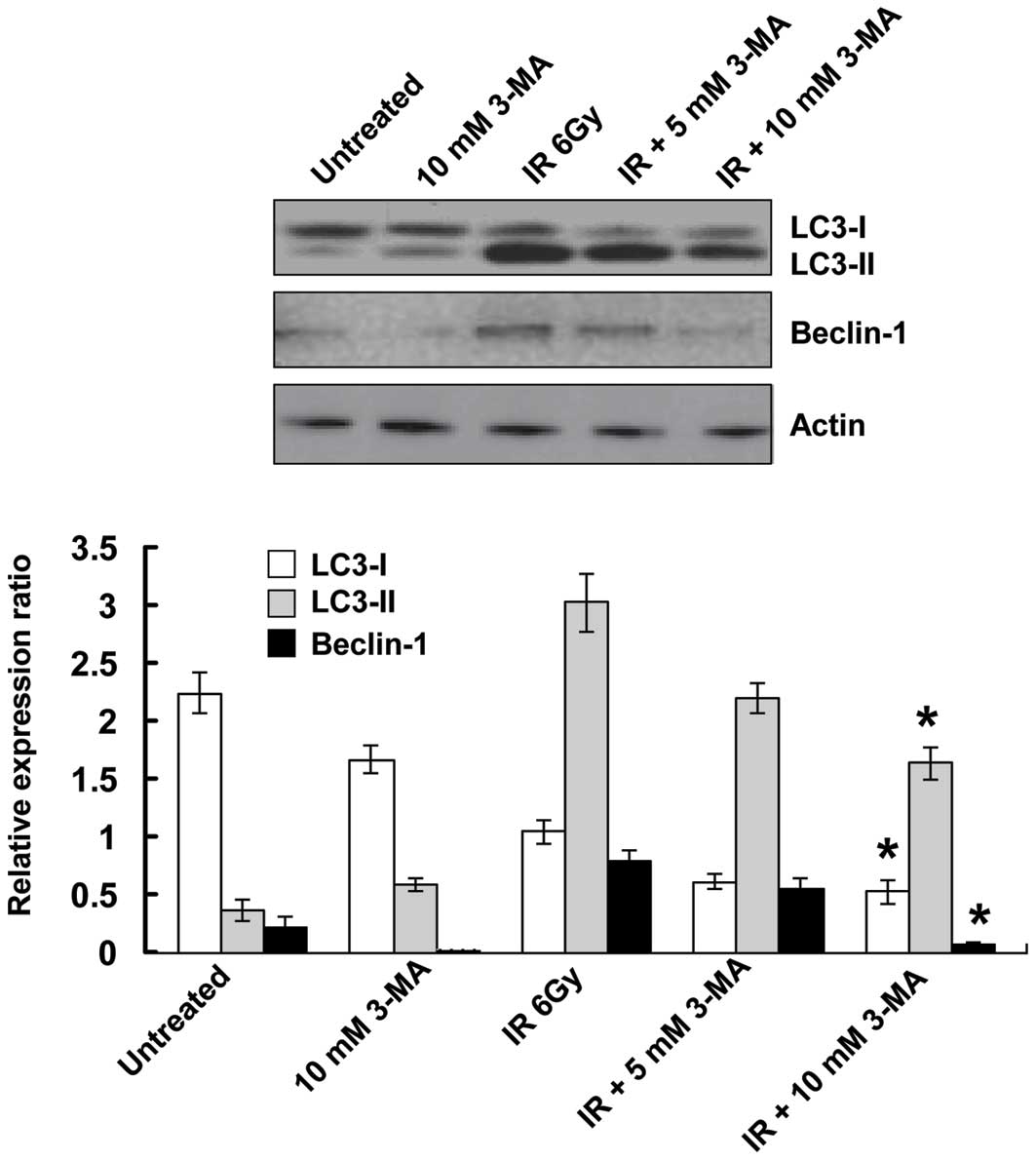
Irradiation induces autophagy in tumor
cells
Autophagosome formation and expression of two
essential autophagy-associated proteins, LC3 and beclin-1, were
detected in the present study, in order to assess autophagy. During
autophagy, phosphatidylethanolamine conjugates to the cytosolic
form of LC3 (LC3-I), resulting in the formation of LC3-II. The
amount of LC3-II is a commonly used indicator of autophagy
(20). Beclin-1 participates in
the early stages of autophagy, where it promotes the nucleation of
the autophagic vesicle and recruits proteins from the cytosol
(21). The present study detected
an upregulation of beclin-1 protein expression levels and a
downregulation of the conversion of LC3-I/II in EC9706 cells 24 h
after co-treatment with radiation and 3-MA, thus indicating an
increase in autophagic activity (Fig.
2). Following co-treatment of the cells with 3-MA, autophagic
activity was downregulated; furthermore, decreased protein
expression levels of beclin-1 and LC3-II were observed by western
blotting. The expression levels of beclin-1 and LC3-II partly
reverted to their original levels when the cells were treated with
a combination of radiation and 10 mM 3-MA. These results provided
evidence for the effective inhibitory effect of 3-MA on
autophagy.
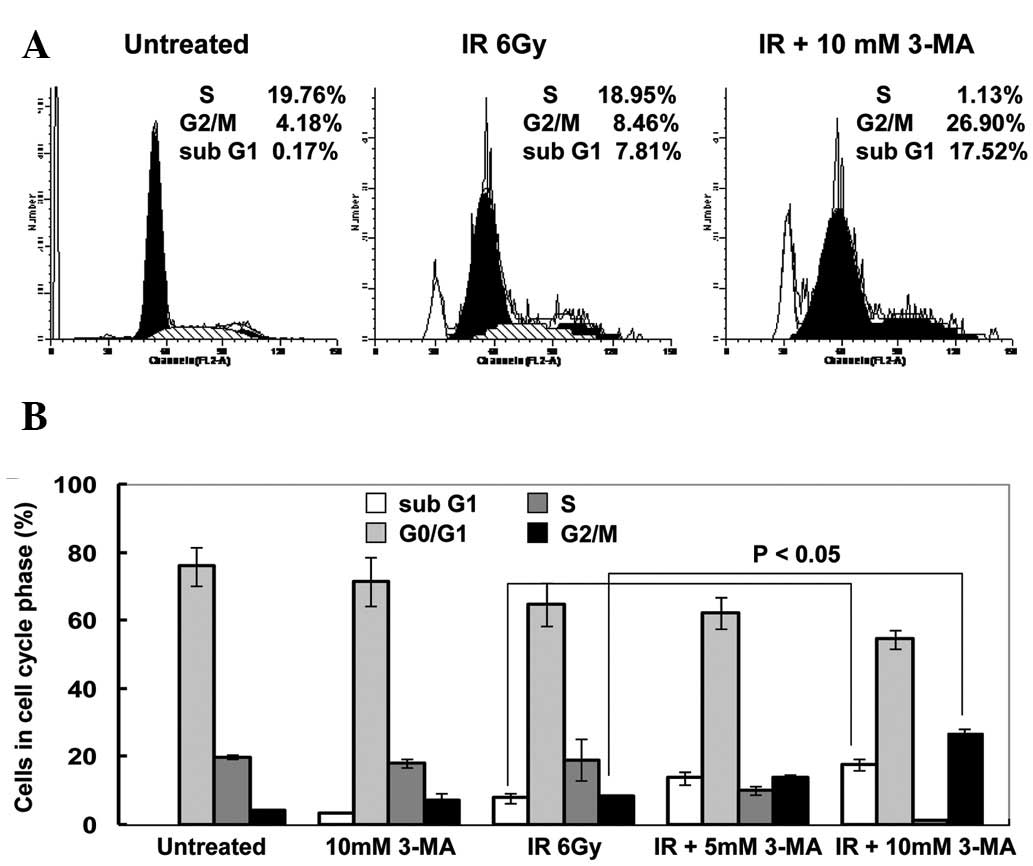
Autophagy inhibition induces cell cycle
arrest
The possible effects of autophagy inhibition on the
cell cycle distribution were investigated in the EC9706 cells
treated with 3-MA, radiation, or their combination. Treatment of
the cells with radiation (6 Gy) alone slightly affected the
percentage of cells in each phase. In the presence of 3-MA, there
was an increase in the number of EC9706 cells in G2/M
phase by 65.7% and 218.0% following treatment with 5.0 and 10 mM of
3-MA, respectively. These results suggested that treatment with
3-MA resulted in a cell cycle arrest in G2/M phase,
which is important for radiation sensitivity. Concomitant with the
G2/M arrest was an elevation in the sub-G1
population, which is an indicator of apoptotic cell death (Fig. 3A and B). These results indicated
that the effects of autophagy inhibition on radiation sensitization
may be attributed to the induction of G2/M phase arrest
and apoptosis.
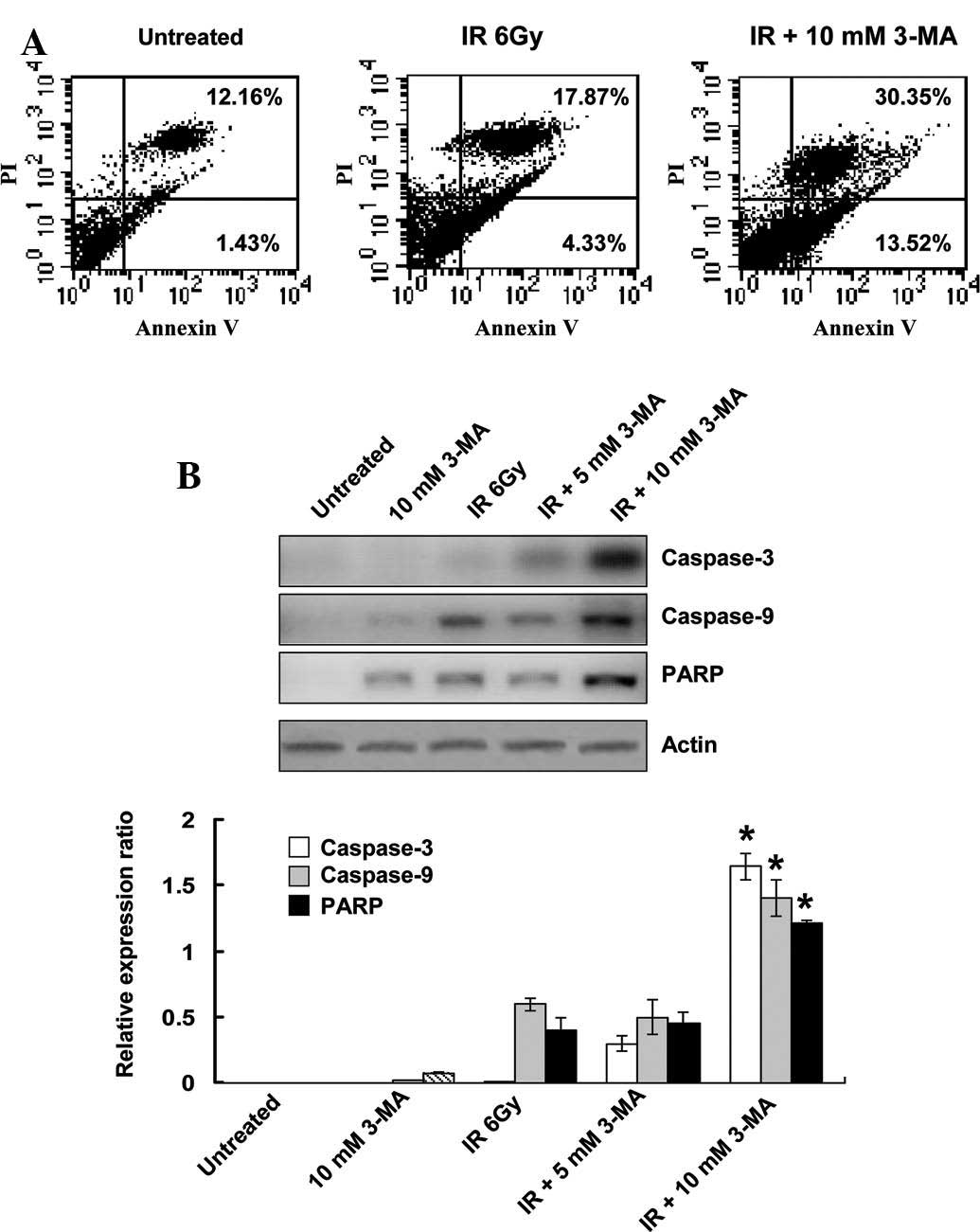
Autophagy inhibition increases cell
apoptosis
To determine whether inhibition of autophagy was
able to induce apoptosis, Annexin V-FITC and PI staining was
conducted. Flow cytometry detected apoptotic cells 24 h after
EC9706 cells were treated with radiation. When the irradiated cells
were co-treated with 3-MA for 24 h, the number of Annexin V- and
Annexin V/PI-positive cells significantly increased, as compared
with that of cells treated with radiation alone (Fig. 4A). To further confirm the increase
in apoptosis, the protein expression levels of the executioner
caspases, caspase-3, caspase-9 and PARP, were determined. In the
radiation-treated groups, caspase-3 and PARP were cleaved into
their specific active forms, and their activity in the combined
treatment group was significantly higher as compared with that in
the cells treated with radiation alone (Fig. 4B). These results indicated that
radiation-induced autophagy has a protective role in tumor cells
against apoptosis, and inhibition of autophagy subsequently
enhances the rate of apoptosis in the cells.
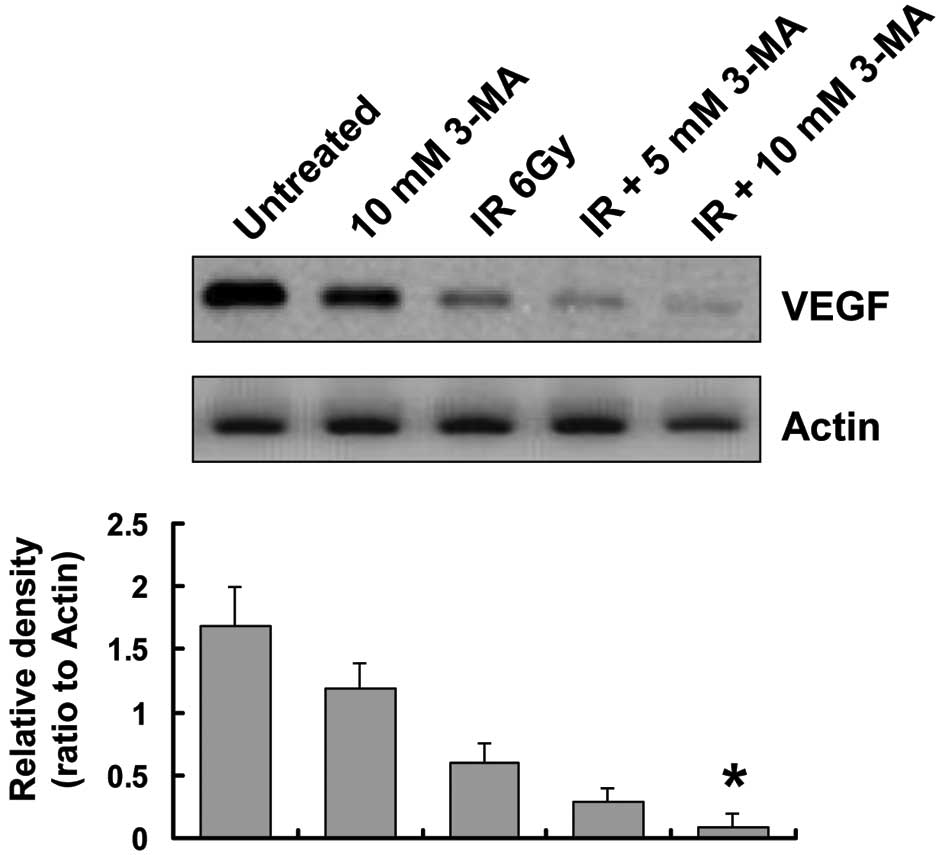
Autophagy inhibition decreases VEGF
protein expression levels
To explore other potential mechanisms underlying the
positive effects of autophagy inhibition on radiation in esophageal
cancer, the role of autophagy in tumor angiogenesis was examined.
VEGF is currently regarded as the most potent pro-angiogenic factor
(22); therefore, the present
study assessed VEGF protein expression levels in EC9706 cells. The
protein expression levels of VEGF were reduced in EC9706 cells
treated with 3-MA, as compared with those in the untreated cells.
Of note, when irradiated cells were co-treated with 3-MA for 24 h,
the protein expression levels of VEGF were significantly lower as
compared with those in the radiation group (Fig. 5).
Radiosensitizing effects of autophagy
inhibition in vivo
The present study also investigated whether
inhibition of autophagy was able to affect the tumor response to
radiotherapy. The response of the EC9706 xenografts to radiation
plus 3-MA was significantly enhanced, as compared with the that of
the untreated, 3-MA alone and radiation alone groups (P<0.01,
Fig. 6A). Treatment with 3-MA
alone reduced the mean tumor volume by 28.2% (2,138.5±247.7
mm3, as compared with 2,977.3±352.2 mm3 in
the untreated group), and treatment with radiation alone reduced
the mean tumor volume by 68.29% (925.6±127.3 mm3, as
compared with 2,977.3±352.2 mm3 in the untreated group).
However, co-treatment of 3-MA with radiation resulted in
significantly smaller tumors, with the tumor volume reduced by
93.9% (181.7±97.3 mm3, as compared with 2,977.3±352.2
mm3 in the untreated group; P<0.01).
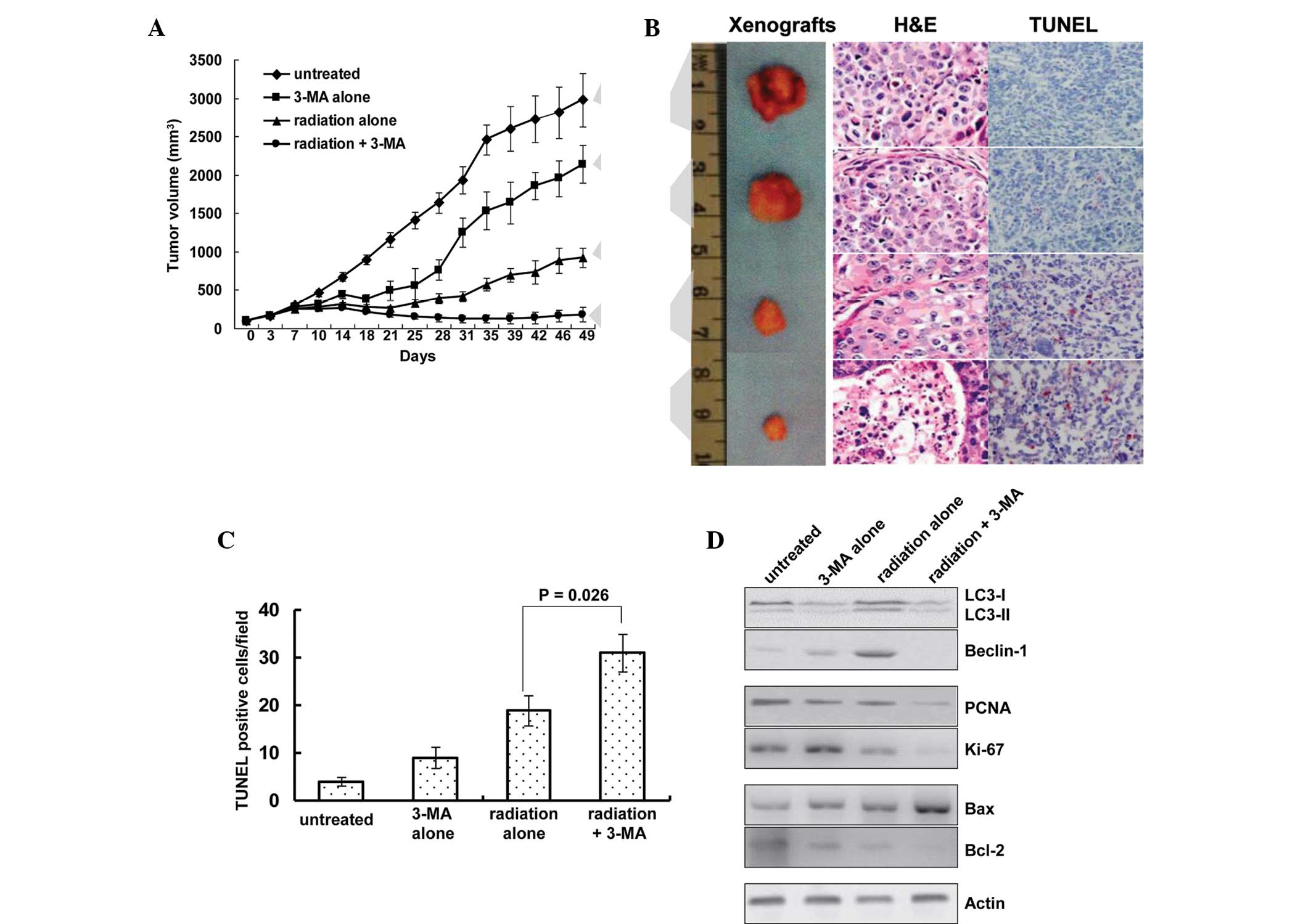 | Figure 6Autophagy inhibition sensitizes
tumors to radiation in vivo. (A) Treatment with a
combination of radiation and 3-MA reduced the growth of EC9706
human esophageal squamous cell carcinoma cell line xenografts. (B)
Representative images of tumors and tumor slides subjected to TUNEL
as well as H&E staining (magnification, ×400). Tumor size was
decreased following treatment with 3-MA and radiation alone, and
tumor size was smallest in the radiation + 3-MA group. (C)
TUNEL-positive cells were quantified, showing increased numbers of
apoptotic cells in single treated and significantly higher
increases in the number of apoptotic cells in the combined
treatment group. Results are expressed as the number of
TUNEL-positive cells/field counted (five random fields per slide
from a total of five slides per study group). (D) Western blot
analysis of autophagic indicators LC-3 and beclin-1; proliferative
indicators PCNA and Ki-67; and Bax and Bcl-2 apoptotic proteins in
the xenografts. 3-MA, 3-methyladenine; IR, ionizing radiation; LC3,
microtubule-associated protein light chain 3; PCNA, proliferating
cell nuclear antigen; Bcl-2; B-cell lymphoma 2; Bax,
Bcl-2-associated X protein; H&E, hematoxylin & eosin;
TUNEL, terminal deoxynucleotidyl transferase dUTP nick end
labeling. |
Of note, the in vitro results were concordant
with the findings of the in vivo experiments in the present
study. A TUNEL assay was used to measure levels of apoptosis, and
increased apoptosis was detected in the radiation plus 3-MA
treatment group (Fig. 6B and C).
The number of TUNEL-positive apoptotic cells per field was 4.1±0.9
in the untreated group, 9.3±2.2 in the 3-MA group, 18.9±3.1 in the
radiation group and 31.2±3.9 in the co-treatment group
(P<0.05).
Western blot analysis showed that radiation
increased the protein expression levels of beclin-1 and
LC3-II/LC3-I in the xenografts, whereas treatment with 3-MA
inhibited the expression levels of LC3-II and beclin-1 (Fig. 6D). In addition, the protein
expression levels of the proliferative markers PCNA and Ki-67 were
assessed; in the tumors treated with radiation and 3-MA combined,
the levels were decreased to a level that was barely detectable.
Furthermore, the expression levels of the pro-apoptotic protein Bax
and the anti-apoptotic protein Bcl-2 were detected. A significant
upregulation in the protein expression levels of Bax and
downregulation in the protein expression levels of Bcl-2 was
detected in the tumors treated with radiation and 3-MA combined
(Fig. 6C).
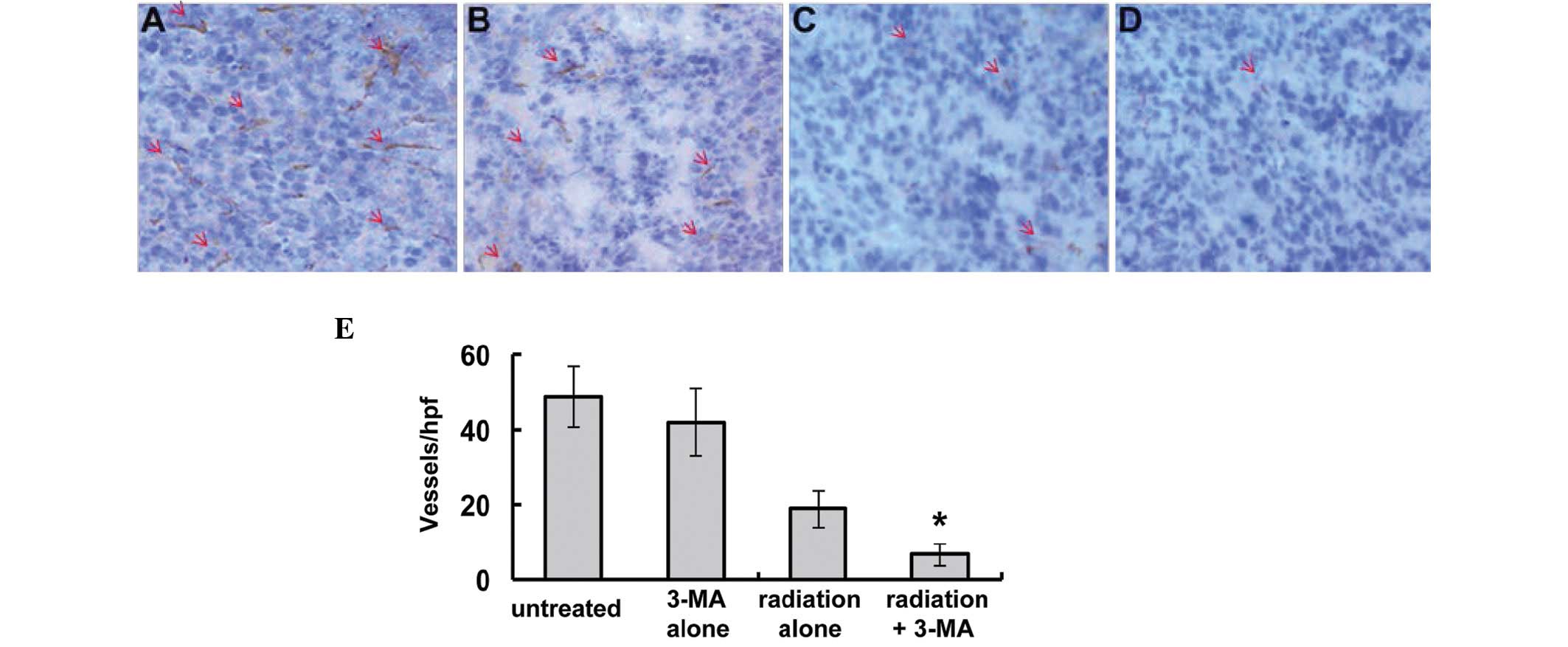
Inhibition of tumor angiogenesis
To investigate the effects of autophagy inhibition
on tumor angiogenesis, immunohistochemical staining of frozen tumor
tissue was performed using antibodies targeting CD31. Furthermore,
angiogenesis within the tumor sections was evaluated by counting
the number of microvessels in each section. Inhibition of autophagy
by 3-MA suppressed angiogenesis, and mice in the radiation plus
3-MA treatment group exhibited a decreased microvessel density
within the tumor, as compared with that in the radiation only group
(Fig. 7; P<0.05).
Discussion
Esophageal cancer is the eighth most common
neoplastic malignancy worldwide, with 455,784 novel cases and
400,156 mortalities estimated in 2012, making it the sixth most
common cause of cancer-associated mortality (23). Definitive radiotherapy and
chemoradiation are the standard therapeutic approaches for the
treatment of patients with esophageal cancer who are not suitable
for surgery, due to the advanced stage of disease or significant
co-morbidity (24). However, local
treatment failure remains a major concern, with persistent or
recurrent disease being reported in ~46–68% of patients, and most
failures of local treatment occur when tumors are gross (25,26).
Therefore, improvements in local control may translate into
increased effectiveness in long-term cures. The clinical efficacy
of radiotherapy is considered to be limited by normal tissue
tolerance and inherent tumor radioresistance. Therefore, the
development of novel radiosensitizing agents, which specifically
sensitize tumor cells whilst protecting normal tissue function, is
required.
Autophagy is an intracellular bulk degradation
system, which is found ubiquitously in eukaryotes. Autophagy may
lead to autophagic cell death through excessive self-digestion and
degradation of essential cellular constituents under certain
conditions (27). However, it has
been suggested that the main role of autophagy is the assistance of
cells in managing stressful metabolic environments, and thereby
promoting cell survival (28). A
family of autophagy-associated genes (ATG) is directly involved in
the process of autophagy; LC3 is often used as a key molecule to
monitor autophagosome formation in mammalian systems and beclin-1
is an essential modifier of the autophagic process (29). Furthermore, esophageal cancer cells
may exploit autophagy to cope with the cytotoxicity of anti-cancer
therapy. O’Donovan et al (18) investigated the cell-death
mechanisms induced in esophageal cancer cells in response to the
chemotherapeutic drugs 5-fuorouracil and cisplatin. In response to
treatment, chemosensitive cell lines exhibited apoptosis, whereas
chemoresistant cells exhibited autophagy. Inhibition of autophagy
induction using small interfering (si)RNA targeted to beclin-1 and
ATG7 significantly enhanced the effects of chemotherapeutic drugs,
and reduced the recovery of drug-treated cells. Autophagy is
frequently observed in cancer cells following exposure to ionizing
radiation, and inhibition of autophagy has been shown to
precipitate radiation-induced cell death (30). Lomonaco et al (31) previously demonstrated that
γ-radiation activated autophagy and inhibition of autophagy
significantly increased the radiosensitivity of glioma cells and
glioma stem cells. Apel et al (32) investigated the effects of autophagy
on the clonogenic survival of irradiated cancer cells, and showed
that inhibition of autophagy-associated genes by specific
target-siRNA oligonucleotides, led to enhanced cytotoxicity of
radiotherapy in five types of human cancer cell lines. These
results indicated that activation of autophagy under therapeutic
stress contributes to the survival of cancer cells.
Whether autophagy contributes to tumor cell death or
represents a radiation resistance mechanism in esophageal cancer
has yet to be elucidated. The present study examined the
contribution of radiation-induced autophagy using in vitro
as well as in vivo models of esophageal cancer. Induction of
autophagosome formation was confirmed by the protein expression of
reliable markers of autophagy: LC3-II and beclin-1. Furthermore,
treatment with the autophagy inhibitor 3-MA, which is a specific
inhibitor of the early stage of the autophagic process, inhibited
radiation-induced autophagy. Of note, treatment with a combination
of radiation and 3-MA increased the therapeutic efficacy of
radiation in human esophageal squamous cell carcinoma. Although the
anti-cancer effects were limited in response to treatment with the
various doses of radiation and 3-MA alone, cancer cell
proliferation and tumor progression were markedly inhibited in the
xenograft mouse model when the treatments were combined. These
results suggested that autophagy represents a mechanism of
resistance to radiation-mediated cell death.
The present study also aimed to determine the
mechanisms underlying the effects of autophagy inhibition on
radiosensitization in esophageal squamous cell carcinoma. The
results of the present study demonstrated that treatment with 3-MA
induced G2/M phase cell cycle arrest. It is well known
that cancer cells are typically sensitive to radiation in
G2/M phase. A key contributor to radiation resistance in
autophagic cancer cells is their failure to engage in apoptosis
(33). The flow cytometry results
of the present study demonstrated that direct inhibition of
autophagy by 3-MA significantly increased radiation-induced cell
apoptosis, and this process was initiated through activation of
caspases in esophageal squamous cell carcinoma cells. A TUNEL assay
conducted on tumor tissue from xenografts showed that enhanced
apoptosis was most pronounced in the radiation plus 3-MA treatment
group. These findings were further confirmed by western blotting
results, which demonstrated a significant upregulation in the
protein expression levels of the pro-apoptotic protein Bax and
downregulation in the protein expression levels of the
anti-apoptotic protein Bcl-2. Furthermore, cellular proliferation
was evaluated by measuring the expression levels of PCNA and Ki-67;
decreased protein expression levels of PCNA and Ki-67 were most
significant in the radiation plus 3-MA-treated tumor samples. These
findings suggested that autophagy inhibition may enhance
radiosensitization through increasing the rate of apoptosis and
reducing tumor cell proliferation.
The complex association between autophagy and
angio-genesis is currently poorly defined. Du et al
(15) previously investigated the
role of autophagy in angiogenesis. Treatment with 3-MA and siRNA
targeting ATG5 were used to inhibit autophagy induced by nutrient
deprivation of cultured bovine aortic endothelial cells. Inhibition
of autophagy by 3-MA or siRNA targeting ATG5 suppressed
angiogenesis, including VEGF-induced angiogenesis. Conversely,
induction of autophagy by overexpression of ATG5 was able to
promote angiogenesis in endothelial cells. It has previously been
demonstrated that radiation-induced endothelial cell dysfunction
may lead to impaired angiogenesis (34). The present study demonstrated that
VEGF protein expression levels were decreased when autophagy was
inhibited in esophageal squamous cell carcinoma cells following
treatment with 3-MA, and analysis of tumor microvessels stained
with rabbit anti-mouse CD31 antibody revealed that combining
autophagy inhibition with radiation significantly reduced tumor
microvessel density in vivo. These data indicated that
autophagy inhibition synergistically enhances the anti-tumor
activity of radiation through inhibition of tumor angiogenesis.
Autophagy inhibition has garnered attention as a
novel anti-cancer therapeutic strategy, and inhibitors of autophagy
have been reported to act as potent anti-cancer drugs and to
sensitize cancer cells to anti-cancer therapy (35). The present study demonstrated that
inhibition of autophagy was able to markedly enhance the
anti-cancer effects of radiotherapy by promoting apoptotic cell
death and downregulating angiogenesis. These results indicated that
the use of anti-autophagy agents may improve the treatment outcomes
of human esophageal squamous cell carcinoma.
Acknowledgments
The present study was supported by a grant-in-aid
from the National Natural Science Foundation of China (grant no.
U1204816).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Overgaard J: Hypoxic modification of
radiotherapy in squamous cell carcinoma of the head and neck – a
systematic review and meta-analysis. Radiother Oncol. 100:22–32.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cooper JS, Guo MD, Herskovic A, et al:
Chemoradiotherapy of locally advanced esophageal cancer: Long-term
follow-up of a prospective randomized trial (RTOG 85-01). Radiation
Therapy Oncology Group. JAMA. 281:1623–1627. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhu HW, Huo XD, Chen LY, Wang HH and Yu H:
Clinical experience with radio-, chemo- and hyperthermotherapy
combined trimodality on locally advanced esophageal cancer. Mol
Clin Oncol. 1:1009–1012. 2013.
|
|
5
|
Suntharalingam M: Definitive
chemoradiation in the management of locally advanced esophageal
cancer. Semin Radiat Oncol. 17:22–28. 2007. View Article : Google Scholar
|
|
6
|
Tsuchihara K, Fujii S and Esumi H:
Autophagy and cancer: Dynamism of the metabolism of tumor cells and
tissues. Cancer Lett. 278:130–138. 2009. View Article : Google Scholar
|
|
7
|
Shimizu S, Yoshida T, Tsujioka M and
Arakawa S: Autophagic cell death and cancer. Int J Mol Sci.
15:3145–3153. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Czarny P, Pawlowska E, Bialkowska-Warzecha
J, Kaarniranta K and Blasiak J: Autophagy in DNA damage response.
Int J Mol Sci. 16:2641–2662. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Eisenberg-Lerner A and Kimchi A: The
paradox of autophagy and its implication in cancer etiology and
therapy. Apoptosis. 14:376–391. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brech A, Ahlquist T, Lothe RA and Stenmark
H: Autophagy in tumour suppression and promotion. Mol Oncol.
3:366–375. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stroikin Y, Dalen H, Lööf S and Terman A:
Inhibition of autophagy with 3-methyladenine results in impaired
turnover of lysosomes and accumulation of lipofuscin-like material.
Eur J Cell Biol. 83:583–590. 2004. View Article : Google Scholar
|
|
12
|
Chen HY and White E: Role of autophagy in
cancer prevention. Cancer Prev Res (Phila). 4:973–983. 2011.
View Article : Google Scholar
|
|
13
|
Ramakrishnan S, Nguyen TM, Subramanian IV
and Kelekar A: Autophagy and angiogenesis inhibition. Autophagy.
3:512–515. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kumar S, Guru SK, Pathania AS, Kumar A,
Bhushan S and Malik F: Autophagy triggered by magnolol derivative
negatively regulates angiogenesis. Cell Death Dis. 4:e8892013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Du J, Teng RJ, Guan T, Eis A, Kaul S,
Konduri GG and Shi Y: Role of autophagy in angiogenesis in aortic
endothelial cells. Am J Physiol Cell Physiol. 302:C383–C391. 2012.
View Article : Google Scholar :
|
|
16
|
Shen W, Tian C, Chen H, Yang Y, Zhu D, Gao
P and Liu J: Oxidative stress mediates chemerin-induced autophagy
in endothelial cells. Free Radic Biol Med. 55:73–82. 2013.
View Article : Google Scholar
|
|
17
|
Chen YS, Song HX, Lu Y, Li X, Chen T,
Zhang Y, Xue JX, Liu H, Kan B, Yang G and Fu T: Autophagy
inhibition contributes to radiation sensitization of esophageal
squamous carcinoma cells. Dis Esophagus. 24:437–443. 2011.
View Article : Google Scholar
|
|
18
|
Zhou GZ, Xu SL, Sun GC and Chen XB: Novel
curcumin analogue IHCH exhibits potent anti proliferative effects
by inducing autophagy in A549 lung cancer cells. Mol Med Rep.
10:441–446. 2014.PubMed/NCBI
|
|
19
|
Yuan H, Li AJ, Ma SL, Cui LJ, Wu B, Yin L
and Wu MC: Inhibition of autophagy significantly enhances
combination therapy with sorafenib and HDAC inhibitors for human
hepatoma cells. World J Gastroenterol. 20:4953–4962. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nakatogawa H, Ichimura Y and Ohsumi Y:
Atg8, a ubiquitin-like protein required for autophagosome
formation, mediates membrane tethering and hemifusion. Cell.
130:165–178. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mac Gabbhan F, Qutub AA, Annex BH and
Popel AS: Systems biology of pro-angiogenic therapies targeting the
VEGF system. Wiley Interdiscip Rev Syst Biol Med. 2:694–707. 2010.
View Article : Google Scholar
|
|
23
|
GLOBOCAN 2012: Estimated cancer incidence,
mortality and prevalence worldwide in 2012. http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx.
Accessed October 14, 2014.
|
|
24
|
Gwynne S, Hurt C, Evans M, Holden C, Vout
L and Crosby T: Definitive chemoradiation for oesophageal cancer –
a standard of care in patients with non-metastatic oseophageal
cancer. Clin Oncol (R Coll Radiol). 23:182–188. 2011. View Article : Google Scholar
|
|
25
|
Shridhar R, Almhanna K, Meredith KL,
Biagioli MC, Chuong MD, Cruz A and Hoffe SE: Radiation therapy and
esophageal cancer. Cancer Control. 20:97–110. 2013.PubMed/NCBI
|
|
26
|
Welsh J, Settle SH, Amini A, Xiao L,
Suzuki A, Hayashi Y, Hofstetter W, Komaki R, Liao Z and Ajani JA:
Failure patterns in patients with esophageal cancer treated with
definitive chemo-radiation. Cancer. 118:2632–2640. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dewaele M, Maes H and Agostinis P:
ROS-mediated mechanisms of autophagy stimulation and their
relevance in cancer therapy. Autophagy. 6:838–854. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wilkinson S and Ryan KM: Autophagy: An
adaptable modifier of tumourigenesis. Curr Opin Genet Dev.
20:57–64. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Klionsky DJ, Abeliovich H, Agostinis P,
Agrawal DK, Aliev G, Askew DS, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy in higher
eukaryotes. Autophagy. 4:151–175. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Benzina S, Altmeyer A, Malek F, et al:
High-LET radiation combined with oxaliplatin induce autophagy in
U-87 glioblastoma cells. Cancer Lett. 264:63–70. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lomonaco SL, Finniss S, Xiang C,
Decarvalho A, Umansky F, Kalkanis SN, Mikkelsen T and Brodie C: The
induction of autophagy by gamma-radiation contributes to the
radioresistance of glioma stem cells. Int J Cancer. 125:717–722.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Apel A, Herr I, Schwarz H, Rodemann HP and
Mayer A: Blocked autophagy sensitizes resistant carcinoma cells to
radiation therapy. Cancer Res. 68:1485–1494. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Moretti L, Cha YI, Niermann KJ and Lu B:
Switch between apoptosis and autophagy: Radiation-induced
endoplasmic reticulum stress? Cell Cycle. 6:793–798. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen YH, Pan SL, Wang JC, Kuo SH, Cheng JC
and Teng CM: Radiation-induced VEGF-C expression and endothelial
cell proliferation in lung cancer. Strahlenther Onkol.
190:1154–1162. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang YP, Hu LF, Zheng HF, Mao CJ, Hu WD,
Xiong KP, Wang F and Liu CF: Application and interpretation of
current autophagy inhibitors and activators. Acta Pharmacol Sin.
34:625–635. 2013. View Article : Google Scholar : PubMed/NCBI
|